

Abstract
Aims
To examine the effect of ticlopidine administration on the activities CYP2C19 and CYP3 A in vivo using omeprazole as a model substrate.
Methods
A single dose of 40 mg omeprazole was administered orally with or without ticlopidine (300 mg daily for 6 days) to six Japanese extensive metabolisers with respect to CYP2C19. Blood samples were taken for the measurement of plasma concentrations of omeprazole, 5-hydroxyomeprazole and omeprazole sulphone.
Results
Ticlopidine administration increased omeprazole Cmax (1978±859/ 3442±569 (control phase/ticlopidine phase, nm)) and decreased the oral clearance of omeprazole (CL/F; 25.70±16.17/10.76±1.16 (control phase/ticlopidine phase, l h−1)) significantly. The 5-hydroxyomeprazole to omeprazole AUC ratio (0.817±0.448/0.236±0.053 (control phase/ticlopidine phase)) and the 5-hydroxyomeprazole to omeprazole sulphone AUC ratio (1.114±0.782/0.256±0.051 (control phase/ticlopidine phase)) were decreased significantly after ticlopidine administration. The decrease in omeprazole CL/F and the 5-hydroxyomeprazole to omeprazole AUC ratio correlated significantly with their respective absolute values when the drug was given alone. The decrease in CL/F following ticlopidine administration correlated with that in the 5-hydroxyomeprazole to omeprazole AUC ratio.
Conclusions
These findings suggest that ticlopidine inhibited the in vivo activity of CYP2C19, but not, or to a lesser extent CYP3 A4, and that the magnitude of inhibition by ticlopidine is related to the in vivo activity of CYP2C19 before inhibition.
Keywords: CYP2C19, CYP3A, drug interaction, omeprazole, ticlopidine
Introduction
Ticlopidine is a potent antiplatelet agent and is used in patients with cerebrovascular disease, peripheral arterial disease or ischaemic heart disease who are at high risk of thromboembolic events associated with platelet hyperaggregation [1]. Ticlopidine has been reported to be superior to aspirin in lowering the incidence of stroke for high risk patients [2]. However, ticlopidine is known to inhibit the metabolism of some drugs [3]. Recent case reports have shown that ticlopidine inhibits phenytoin clearance, which results in acute toxicity [4–6]. Although phenytoin is metabolized by CYP2C9 and CYP2C19 [7, 8], ticlopidine does not appear to affect CYP2C9 activity in vivo [9] as determined by S-warfarin clearance [10]. Furthermore, in human liver microsomes [11] a lower concentration of ticlopidine was required to inhibit S-mephenytoin hydroxylation, an index of CYP2C19 activity [12], than was required to inhibit tolbutamide 4-methylhydroxylation, an index of CYP2C9 activity [12]. These findings suggest that ticlopidine primarily inhibits the in vivo activity of CYP2C19. Thus, we studied the effect of ticlopidine pretreatment on the pharmacokinetics of omeprazole, a substrate of CYP2C19 [13], in subjects who were extensive metabolisers with respect to this enzyme.
Methods
Subjects
Six unrelated healthy native Japanese men with a mean age of 33.8±8.3 years (mean±s.d.; range 24–43 years) and mean weight of 64.3±7.5 kg participated in the study. No subject had taken any medication for at least 7 days before the study, and each abstained from alcohol for 3 days before the study. Each subject had normal histories, physical examination and clinical chemistry results. All were genotyped as extensive metabolisers with respect to CYP2C19 [14]. The study was approved by the institutional review board of St Marianna University School of Medicine, and each participant gave written informed consent.
Protocol
The study was performed according to an open, randomized two-period cross-over design. In the control phase, a 40 mg enteric-coated omeprazole tablet (Losec, Yuhan Co., Seoul, Korea) was given with 200 ml of water after an overnight fast. In the ticlopidine phase, a 100 mg ticlopidine tablet was given three times daily for 6 days, and an additional 100 mg ticlopidine tablet was given on the 7th day at 1 h before the administration of a 40 mg omeprazole tablet after an overnight fast. The two trials were separated by a wash-out period of 2 weeks. Blood samples were obtained before and at 0.5, 1, 1.5, 2, 3, 4, 6, and 8 h after omeprazole administration and were centrifuged (3000 g for 10 min) immediately. Plasma was stored at −20° C until assayed.
Analytical methods
Omeprazole and its two primary metabolites, 5-hydroxyomeprazole and omeprazole sulphone in plasma were measured by h.p.l.c. [15]. Analytical reference samples of the three compounds were a generous gift of Dr T. Ishizaki of International Medical Center of Japan. The mean recoveries of omeprazole and its metabolites at concentrations of 150 and 1200 nm were 93–116%. The lowest determinable concentration, defined as three times baseline noise, for each compound was 10 nm (3 ng ml−1). Coefficients of variation (interassay) for the three compounds were below 10% at a concentration of 150 nm, and below 4% at a concentration of 1200 nm.
Pharmacokinetic analysis
Pharmacokinetic parameters were calculated using noncompartmental methods. The maximum drug concentration in plasma (Cmax) and the time required to reach maximum concentration (tmax) were obtained directly from the concentration-time data. The slope of the terminal elimination phase (λz) was obtained by least-squares linear regression analysis. Elimination half-life (t1/2) was calculated as ln2/λz. The area under the plasma concentration-time curve from zero to 8 h (AUC(0,8 h)) was calculated by the trapezoidal rule. The oral clearance (CL/F) was calculated as CL/F=dose/AUC.
Statistical analysis
Data are presented as mean±s.d. Control and treatment phases were compared by Wilcoxon signed rank test. A P value of <0.05 was considered statistically significant.
Results
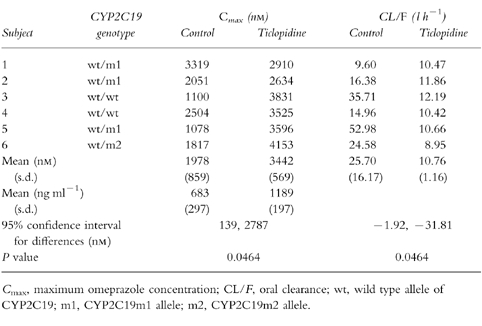
Ticlopidine administration decreased omeprazole CL/F and increased omeprazole Cmax (Table 1) as well as AUC(0,8 h) (6169±3593/10875±1224 (control phase/ticlopidine phase, nmol l−1h, P=0.0464)) significantly. There was no significant difference in tmax (2.7±1.2/ 2.8±0.8 (control phase/ticlopidine phase, h)) and t1/2 (2.8±1.2/3.8±0.8 (control phase/ticlopidine phase, h)).
Table 1.
Omeprazole (40 mg) Cmax and CL/F given alone (control) or following treatment with ticlopidine (300 mg daily for 6 days).
The 5-hydroxyomeprazole to omeprazole AUC ratio (AUC(OHOME)/AUC(OME)) and the 5-hydroxyomeprazole to omeprazole sulphone AUC ratio (AUC(OHOME)/AUC(OMESUL)) were decreased significantly in the ticlopidine phase in comparison with that in the control phase (Figure 1a and 1c respectively), whereas the omeprazole sulphone to omeprazole AUC ratio (AUC(OMESUL)/AUC(OME)) remained unchanged (Figure 1b).
Figure 1.
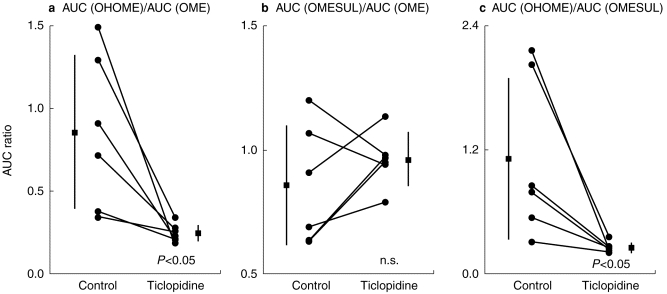
Effect of ticlopidine administration on the (a) 5-hydroxyomeprazole to omeprazole AUC ratio (AUC(OHOME)/AUC(OME)) (b) omeprazole sulphone to omeprazole AUC ratio (AUC(OMESUL)/AUC(OME)), and (c) 5-hydroxyomeprazole to omeprazole sulphone AUC ratio (AUC(OHOME)/AUC(OMESUL)). Closed boxes and bars represent means and s.d., respectively.
The decrease in CL/F and AUC(OHOME)/ AUC(OME) following ticlopidine administration were significantly correlated with CL/F (r2=0.995, P<0.001) and AUC(OHOME)/AUC(OME) (r2=0.986, P<0.001) when the drug was given alone, respectively. In addition, there was a significant correlation between the decrease in CL/F and that in AUC(OHOME)/AUC(OME) (r2=0.941, P<0.005).
Discussion
We studied the effect of the ticlopidine administration on the pharmacokinetics and metabolism of omeprazole. The administration of 300 mg ticlopidine for 6 days increased omeprazole Cmax and AUC(0,8 h) significantly. Omeprazole is metabolized by both CYP3A and CYP2C19, which catalyse the formation of omeprazole sulphone and 5-hydroxyomeprazole, respectively [13]. AUC(OHOME)/AUC(OME) and AUC(OMESUL)/ AUC(OME) are used as in vivo probes of CYP3A and CYP2C19 activity, respectively [16]. In the present study, AUC(OHOME)/AUC(OME) was decreased by ticlopidine treatment, which reflects inhibition of CYP2C19 and/or induction of CYP3A, because 5-hydroxyomeprazole is further metabolized by CYP3A4 [17]. AUC(OMESUL)/AUC(OME) remained unchanged, but inhibition or induction of CYP3A activity cannot be ruled out because this ratio will not be altered if a parallel change occurs in both CYP3A and CYP2C19 activities. The decreased AUC(OHOME)/AUC(OMESUL) ratio implies that ticlopidine inhibits CYP2C19 activity, but not, or to a lesser extent CYP3A4 activity.
The decreased AUC(OHOME)/AUC(OME) ratio and the correlation between this decrease and the reduction in CL/F suggest that inhibition of CYP2C19 by ticlopidine increases AUC(OME) and decreased omeprazole CL/F. In addition, the significant correlation betweens the decrease and the absolute values for CL/F and the AUC(OHOME)/AUC(OME) ratio in the control phase indicate that subjects with higher CYP2C19 activity show a greater decrease in omeprazole CL/F.
In a previous study of an oriental population [18], the mean omeprazole CL/F in poor metabolisers with respect to CYP2C19 was reported to be 3.90 (l h−1; mean corrected CL/F, 0.0595 = l h−1 kg−1; mean BW, 65.5 kg) and the mean AUC(OHOME)/AUC(OME) was less than 0.1. In the present study, mean CL/F was 10.8 = l h−1 and mean AUC(OHOME)/ AUC(OME) ratio was larger than 0.1 in all subject following ticlopidine administration. Therefore, we assume that ticlopidine administration substantially decreases but does not abolish the in vivo CYP2C19 activity, since subjects treated with ticlopidine still possess some activity. The dose of ticlopidine (100 mg three times daily) used in the current study is smaller than the recommended clinical dosage of 250 mg twice daily [1]. Larger doses of ticlopidine might inhibit CYP2C19 activity to a greater extent, since it has been reported that ticlopidine inhibits S-mephenytoin hydroxylation in a concentration-dependent fashion in human liver microsomes [11].
Omeprazole itself is an inhibitor of several human CYP activities [3], and is also an inducer of CYP1 A [19], which converts procarcinogens to reactive metabolites [20]. There is no published information on what isoforms of CYP catalyse the metabolism of ticlopidine, and thus it is impossible to predict how omeprazole might affect the pharmacokinetics of ticlopidine.
In the present study, all subjects were genotyped as extensive metabolisers with respect to CYP2C19. However, the subject with the lowest CL/F showed no change of omeprazole metabolism after the ticlopidine administration. Since the magnitude of inhibition by ticlopidine depended on in vivo CYP2C19 activity, ticlopidine should not affect the pharmacokinetics of omeprazole in poor metabolisers with respect to CYP2C19. Further work is required to determine whether ticlopidine administration impairs the elimination of other clinically used CYP2C19 substrates [21] and whether any increased plasma drug concentrations are associated with toxic effects.
References
- 1.McTavish D, Faulds D, Goa KL. Ticlopidine: an updated review of its pharmacology and therapeutic use in platelet-dependent disorders. Drugs. 1990;40:238–259. doi: 10.2165/00003495-199040020-00006. [DOI] [PubMed] [Google Scholar]
- 2.Hass WK, Easton JD, Adams HP, et al. A randomized trial comparing ticlopidine hydrochloride with aspirin for the prevention of stroke in high-risk patients. N Engl J Med. 1989;321:501–507. doi: 10.1056/NEJM198908243210804. [DOI] [PubMed] [Google Scholar]
- 3.Desager J-P. Clinical pharmacokinetics of ticlopidine. Clin Pharmacokinet. 1994;26:347–355. doi: 10.2165/00003088-199426050-00003. [DOI] [PubMed] [Google Scholar]
- 4.Riva R, Cerullo A, Albani F, Baruzzi A. Ticlopidine impairs phenytoin clearance: a case report. Neurology. 1996;46:1172–1173. doi: 10.1212/wnl.46.4.1172. [DOI] [PubMed] [Google Scholar]
- 5.Privitera M, Welty TE. Acute phenytoin toxicity followed by seizure breakthrough from a ticlopidine–phenytoin interaction. Arch Neurol. 1996;53:1191–1192. doi: 10.1001/archneur.1996.00550110143027. [DOI] [PubMed] [Google Scholar]
- 6.Rindon JP, Bryan GII, Ariz P. Phenytoin toxicity associated with ticlopidine administration [letter] Arch Intern Med. 1996;156:1113. [PubMed] [Google Scholar]
- 7.Veronese ME, Doecke CJ, Mackenzie PI, et al. Site-directed mutation studies of human liver cytochrome-P450 isoenzymes in the CYP2C subfamily. Biochem J. 1993;289:533–538. doi: 10.1042/bj2890533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ieiri I, Mamiya K, Urae A, et al. Stereoselective 4′-hydroxylation of phenytoin: relationship to (S)-mephenytoin polymorphism in Japanese. Br J Clin Pharmacol. 1997;43:441–445. doi: 10.1046/j.1365-2125.1997.00572.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gidal BE, Sorkness CA, McGill KA, Larson R, Levine RR. Evaluation of a potential enantioselective interaction between ticlopidine and warfarin in chronically anticoagulated patients. Ther Drug Monit. 1995;17:33–38. doi: 10.1097/00007691-199502000-00006. [DOI] [PubMed] [Google Scholar]
- 10.Kaminsky LS, Zhang ZY. Human P450 metabolism of warfarin. Pharmacol Ther. 1997;73:67–74. doi: 10.1016/s0163-7258(96)00140-4. [DOI] [PubMed] [Google Scholar]
- 11.Donahue SR, Flockhart DA, Abernethy DR, Ko J-W. Ticlopidine inhibition of phenytoin metabolism mediated by potent inhibition of CYP2C19. Clin Pharmacol Ther. 1997;62:572–577. doi: 10.1016/S0009-9236(97)90054-0. [DOI] [PubMed] [Google Scholar]
- 12.Guengerich FP. Human cytochrome P450 enzymes. In: Oritiz de Montellano PR, editor. Cytochrome P450. New York: Plenum Press; 1995. pp. 473–535. [Google Scholar]
- 13.Andersson T, Miners JO, Tassaneeyakul W, et al. Identification of human liver cytochrome P450 isoforms mediating omeprazole metabolism. Br J Clin Pharmacol. 1993;36:521–530. doi: 10.1111/j.1365-2125.1993.tb00410.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Morais SM, Wilkinson GR, Blaisdell J, et al. Identification of a new genetic defect responsible for the polymorphism of S-mephenytoin metabolism in Japanese. Mol Pharmacol. 1994;46:594–598. [PubMed] [Google Scholar]
- 15.Kobayashi K, Chiba K, Sohn D-R, Kato Y, Ishizaki T. Simultaneous determination of omeprazole and its metabolites in plasma and urine by reversed-phase high-performance liquid chromatography with an alkaline-resistant polymer-coated C18 column. J Chromatogr. 1992;579:299–305. doi: 10.1016/0378-4347(92)80395-7. [DOI] [PubMed] [Google Scholar]
- 16.Bertilsson L, Tybring G, Widén J, Chang M, Tomson T. Carbamazepine treatment induces the CYP3A4 catalysed sulphoxidation of omeprazole, but has no or less effect on hydroxylation via CYP2C19. Br J Clin Pharmacol. 1997;44:186–189. doi: 10.1046/j.1365-2125.1997.00630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andersson T, Miners JO, Veronese ME, Birkett DJ. Identification of human liver cytochrome P450 isoforms mediating secondary omeprazole metabolsim. Br J Clin Pharmacol. 1994;37:597–604. doi: 10.1111/j.1365-2125.1994.tb04310.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sohn D-R, Kobayashi K, Hiba K, et al. Disposition kinetics and metabolism of omeprazole in extensive and poor metabolizers of S-mephenytoin 4′-hydroxylation recruited from an oriental population. J Pharmacol Exp Ther. 1992;262:1195–1202. [PubMed] [Google Scholar]
- 19.Diaz D, Fabre I, Daujat M, et al. Omeprazole is an aryl hydrocarbon-like inducer of human hepatic cytochrome P450. Gastroenterology. 1990;99:737–747. doi: 10.1016/0016-5085(90)90963-2. [DOI] [PubMed] [Google Scholar]
- 20.Guengerirh FP. Roles of cytochrome P450 enzymes in chemical carcinogenesis and cancer chemotherapy. Cancer Res. 1988;48:2946–2954. [PubMed] [Google Scholar]
- 21.Goldstein JA, de Morais. Biochemistry and molecular biology of the human CYP2C subfamily. Pharmacogenetics. 1994;4:285–299. doi: 10.1097/00008571-199412000-00001. [DOI] [PubMed] [Google Scholar]