

Introduction
Individualizing a patient’s drug therapy to obtain the optimum balance between therapeutic efficacy and the occurrence of adverse events is the physician’s goal. However, achieving this goal is not always straight forward, being complicated by within and between patient variability in both pharmacokinetics and pharmacodynamics. In the early 1960s new analytical techniques became available allowing the measurement of the low drug concentrations seen in biological fluids during drug treatment. This offered the opportunity to reduce the pharmacokinetic component of variability by controlling drug therapy using concentrations in the body rather than by dose alone. This process became known as therapeutic drug monitoring (TDM) [1].
For a drug to be a suitable candidate for therapeutic drug monitoring it must satisfy the following criteria:-
✓ There should be a clear relationship between drug concentration and effect.
✓ The drug should have a narrow therapeutic index; that is, the difference in the concentrations exerting therapeutic benefit and those causing adverse events should be small.
✓ There should be considerable between-subject pharmacokinetic variability and, therefore, a poor relationship between dose and drug concentration/response.
✓ The pharmacological response of the drug should be difficult to assess or to distinguish from adverse events.
The immunosuppressive drug cyclosporin satisfies all four of these criteria and, despite over 16 years of clinical use with therapeutic drug monitoring, there is still no firm consensus on the best way to use the drug. In addition, the number of available agents for use as immunosuppressants has more than doubled in recent years and the range of diseases in which these drugs are used has also widened [2]. The purpose of this review is to examine the current strategies in use for the therapeutic drug monitoring of immunosuppressant drugs [3] and to discuss some of the factors that impinge on the monitoring of these drugs.
Azathioprine, steroids, anti-lymphocyte globulin, and OKT3
The combination of azathioprine and prednisolone was responsible for making clinical transplantation viable [4]. With the addition of anti-lymphocyte globulin [5] (ALG or ATG if human thymocytes instead of human lymphocytes are used to immunise the animal host) they formed the basis of immunosuppression in the early years of transplantation and these drugs are still in widespread use today. Monitoring the blood or plasma concentration of these drugs is not considered worthwhile as they all have relatively wide therapeutic indices. The three agents are generally given in fixed doses and are not subjected to therapeutic drug monitoring.
However, a case can be made for the measurement of the activity of the enzyme thiopurine methyltransferase (TPMT) as an adjunct to azathioprine therapy [6]. Azathioprine is not directly immunosuppressive, since it must be metabolised first to 6-mercapto-purine, then by TPMT to 6-methyl-mercapto-purine and then on to the pharmacologically active 6-thioguanine nucleotides. The expression of the enzyme TPMT is inherited in an autosomal co-dominant fashion and consequently varies widely within the population [7] with 11% of the Caucasian population heterozygous and 0.3% homozygous with respect to TPMT deficiency [8]. Potentially fatal complications could be avoided if TPMT activity was monitored in erythrocytes [9]. The therapeutic drug monitoring of azathioprine in cancer chemotherapy is outside the scope of this article but has been reviewed recently in this journal by Lennard [10].
OKT3 (muromonab-CD3) is a mouse-monoclonal antibody directed against the CD3 complex on T cells [11]. When complexed with its antigen, the antibody prevents the initiation of signal transduction and blocks all T cell function [12]. In a pilot study using OKT3 serum concentrations as a guide to therapy in kidney transplant patients excellent results were reported for the prevention of early graft rejection [13]. Although there is a correlation between OKT3 concentration and T cell killing the relationship is complicated by the patients’ antibody response to murine-derived protein [14]. In another study using flow cytometry measurements to monitor OKT3 therapy the authors not only measured OKT3 concentration but also anti-OKT3 antibody concentration and the number of CD3+ cells (the therapeutic target of OKT3) [15]. Although the authors’ conclusions were positive about the use of flow cytometry for monitoring, their over all conclusions were that ‘this treatment cannot protect against acute cellular rejection due to the presence of a dimly positive CD3+ population’. In those centres using muromonab-CD3, TDM for OKT3 is not in widespread use.
Cyclosporin
In the early days of kidney transplantation, Calne gave the 2 year graft survival rate for cadaveric renal transplantation in the USA as less than 18% [16]. Five years later, in 1974, the same author reported that graft survival at 2 years, had risen to over 50% [17]. The change was due to better surgical technique and improved patient management since the main drug therapies, azathioprine and prednisolone, had not changed. Twenty-one years later, in those patients receiving that drug combination, the 2 year graft survival was still only ≈60% [18]. However, with the discovery [19, 20] and use of cyclosporin for immunosuppression the 2 year graft survival for the majority of patients is now better than 80% [18]. Long term graft survival has also improved; after the first year from transplant, the estimated graft half-life in patients on cyclosporin alone is 30 years compared with 10.5 years in those patients maintained on azathioprine and steriods [18].
While the introduction of cyclosporin resulted in a marked improvement in graft survival, its use was not without problems. From the initial discovery of cyclosporin [21] to the present day [22], the absorption of the drug has been problematical [23]. Variable absorption and a narrow therapeutic index (the drug causes irreversible kidney damage when given in too high a dose [24]) has resulted in the measurement of cyclosporin blood concentrations so that the dose of the drug can be tailored to the patient to maximise therapeutic efficacy while minimising toxicity [25]. In the past, the utility of cyclosporin therapeutic drug monitoring has been reduced by, choice of sample matrix for monitoring [26, 27], lack of assay specificity [28], inconsistent assay performance [29], the variable absorption of the drug from the original formulation (Sandimmun®) [30] and the poor correlation between trough concentration and clinical effects [31]. Over time, the majority of these problems have been addressed [32]; blood, and not plasma or serum, is the chosen matrix for measurement [33, 34], assays are now more selective for the parent compound [35, 36], most laboratories participate in proficiency testing [37] and the drug has been re-formulated (Neoral®) to improve absorption [38–40]. However, trough whole blood concentration remains an imperfect measure of the total exposure to cyclosporin during a dose interval [3].
In the Sandimmun® era substantial efforts were made by Kahan and co-workers to characterize the relationship between area under the concentration-time curve for cyclosporin (AUC) and clinical events including patient outcome [41]. Although many acknowledge the advantages of AUC monitoring, it has failed to gain widespread acceptance because of the practical difficulties, both for the patient and clinician, in implementing AUC measurements [42]. The advent of the microemulsion formulation of cyclosporin, Neoral®, with its improved pharmacokinetic characteristics [43], provides an opportunity to simplify the measurement of AUC [42]. Previous studies with Sandimmun® have shown that the concentrations of three blood samples, drawn at specific times, can be used to derive an accurate estimate of cyclosporin AUC [44]. For Neoral® a similar degree of accuracy in the prediction can be achieved by two optimally timed blood samples [45]. Prospective studies are now underway to compare pre-dose concentration monitoring with sparse, or limited sampling AUC monitoring.
However, pre-dose, trough concentration and AUC monitoring are not the only options for the therapeutic monitoring of cyclosporin. For some time, Cantarovich and co-workers have been advocating the use of a single timed sample 6 h after dosing [46]. In a prospective study of heart transplant patients comparing pre-dose and 6 h cyclosporin concentration with control cyclosporin therapy, the use of the 6 h value resulted in a 30% lower dose of the drug with the same effectiveness in preventing rejection, and similar cardiac and renal function as seen in those dosed using the pre-dose concentration [47]. These authors have also reported a good relationship between 6 h cyclosporin concentrations and efficacy in non-infectious uveitis [48] and other auto-immune diseases [49].
Another option that is being promoted for monitoring is the use of the cyclosporin blood concentration at 2 h post-dose (C2). The rationale for this comes from the observation made during the clinical development of Neoral® that in liver transplant patients there is an inverse relationship between the incidence of rejection and the maximum blood cyclosporin concentration (Cmax) [50–52]. As yet, the use of C2 monitoring has only been tested in a small open-labeled trial but the initial results look promising [53]. The opportunities presented by new therapeutic drug monitoring strategies to optimise cyclosporin therapy have been the subject of a recent consensus meeting [54].
Tacrolimus
Tacrolimus (previously known as FK506), like cyclosporin, has been shown to be an effective immunosuppressive for the prevention of organ rejection after transplantation and, like cyclosporin, too much drug is associated with toxicity and too little with rejection. Again, like cyclosporin, whole blood concentration measurements are used for the monitoring of tacrolimus therapy [55]. Initial clinical trials did not include concentration monitoring and patients often experienced neuro- and nephrotoxicity [56].
The pharmacokinetics of tacrolimus are highly variable [57]. Since tacrolimus shares many of the pharmacokinetic and pharmacodynamic problems associated with cyclosporin the rationale for therapeutic drug monitoring is similar. An early observational study correlating concentration and effect failed to show a significant difference between the blood concentration in those kidney transplant patients who did not experience rejection and those who did [58]. However other, more statistically rigorous, studies have shown, in kidney and liver transplant patients, significant associations of low tacrolimus concentrations with rejection and of high concentrations with nephrotoxicity [59]. Although the feasibility of a limited sampling scheme to predict AUC has been demonstrated [60], as yet, trough, or pre-dose, whole blood concentration monitoring is still the method of choice. Unlike cyclosporin, there is no move towards the use of other timed samples or AUC monitoring. This may, in part, be due to the high correlation between trough concentration and Cmax or AUC [61].
Mycophenolate mofetil (MMF)
This is the morpholinethylester of mycophenolic acid (MPA) and acts as a pro-drug for that compound [62]. When given orally to man, MMF undergoes rapid and complete absorption and is hydrolyzed, pre-systemically, to MPA, with no MMF measurable in plasma [63]. The drug reduces both B and T cell proliferation by inhibition of de novo guanine nucleotide production [64]. Since, unlike cyclosporin and tacrolimus, both B and T cells are inhibited it has been suggested that MMF may be effective against both acute and chronic rejection [65]. In man, MPA is metabolised in the liver to MPA β-glucuronide (MPAG), an inactive metabolite which is present in plasma at approximately 40-fold higher concentrations than MPA. The MPA glucuronide was thought to be the only metabolite of MPA but, recently, this view has been challenged by comparative results of analyses using high performance liquid chromatography and enzyme immunoassay [66]. These showed a discrepancy between the assays and this was traced to previously unknown, possibly active, metabolites of MPA. The significance of these findings is being assessed [67].
Although the action of the active moiety, MPA, has been known for over 25 years [68], the amount of published data in peer reviewed journals, relating concentration to effect, is limited. Large scale multi-centre, double-blind, randomised, controlled studies in renal transplant patients have shown the effectiveness of MMF in the suppression of acute, biopsy proven, rejection when used in combination with cyclosporin and steroids [69]. Logistic regression has been used to relate the AUC and Cmax of plasma MPA to the incidence of rejection in renal transplant patients and has demonstrated a highly statistically significant relationship [63]. The results of the logistic regression and data from other trials [70] suggest that low plasma MPA AUC is a significant risk factor in developing rejection [71]. These data have been confirmed by the results of a randomised concentration-controlled study of MMF in renal patients [72]. The link between high MPA concentrations and adverse effects has not been characterised.
The rôle of TDM in MMF therapy has yet to be established. Some authors believe that the inter-individual pharmacokinetic variability is low and therefore the utility of TDM in the majority of patients would be limited [70]. Whereas others, using the same data, believe that the inter-individual pharmacokinetic variability is high and that TDM may have a worthwhile function in the control of MMF therapy [73]. Support for the latter view comes from a study of 30 de novo heart transplant patients receiving tacrolimus and MMF in which the dose of MMF was adjusted to maintain the MPA trough plasma concentration between 2.5 and 4 mg l−1 [74]. These patients were rejection free at 6 months post-transplant and their MMF dose ranged between 0.5 and 6 g day−1 to achieve trough concentrations within a target range.
Recent consensus guidelines are circumspect about recommending TDM for the control of MMF therapy [75]. The guidelines suggest that TDM should be used to establish that adequate MPA concentrations are achieved soon after surgery and that it could be useful in cases of adverse reaction to MMF. These guidelines were written from the view point of MMF as secondary immunosuppression to cyclosporin or tacrolimus. However, in a recent study it has been shown that MMF can be used successfully as a primary immunosuppressant [76] and if MMF is to be used as mono-therapy the rôle of MPA TDM may assume more importance.
Sirolimus
This is a macrolide antibiotic produced by Streptomyces hygroscopicus, a fungus isolated originally from a soil sample from Easter Island (Rapa Nui). Sirolimus (previously known as rapamycin), although similar in structure to tacrolimus, exerts its immunosuppressant effect via a different mechanism [77] and at another point in the cell cycle [78]. The initial clinical trials were with the drug in combination with cyclosporin and were concentration controlled [79]. These trials confirmed the clinical efficacy of the drug [80, 81]. A recent review of the pharmacokinetics of sirolimus suggested a therapeutic range, based on animal allo-transplant data, of 5 to 10 μg l−1 in whole blood [82]. However, the only consensus guidelines published on the therapeutic monitoring of sirolimus concluded that there was not enough information available about the clinical use of the drug to make recommendations [83].
Daclizumab and basiliximab
The binding of interleukin 2 (IL-2) to its receptor on antigen activated T cells stimulates clonal proliferation of the T cells that mediate organ allograft rejection [84]. The receptor complex is made up of at least three sub-units (α, β and γ) and, of these, only the α sub-unit is thought to be specific to IL-2 [85]. This presents a potential therapeutic target for specific immunosuppressive therapy and it has been exploited by a series of monoclonal antibodies raised against the IL-2 receptor’s α sub-unit (IL-2Rα). Two such antibodies are currently available. One is a molecularly engineered human IgG1 incorporating the antigen-binding regions of a murine monoclonal antibody (daclizumab [86]) against IL-2Rα. The other is a murine-human chimeric antibody (basiliximab [87]) to IL-2Rα. Both have been used successfully for the prevention of acute rejection following renal transplantation [88, 89]. The therapeutic index of these antibodies is wide, neither antibody being associated with major adverse effects and, since the half life of both antibodies is long, daclizumab (t1/2≈6 days [90]) is given as five, and basiliximab (t1/2>20 days [91]) as two, weight related intra-venous doses in the first 8 weeks following transplantation. Thus, there appears little to be gained from therapeutic drug monitoring for these agents [92, 93].
The ‘therapeutic’ range
In a survey of the therapeutic ranges used by 21 transplant centres to guide cyclosporin maintenance therapy in kidney transplant patients there was a six fold range in the concentrations considered effective and a three fold range in those considered toxic [94]. There was also considerable variation in the width of the therapeutic ‘window’. Although some of this variation must come from the different combinations of immunosuppressant drugs used and differences in assay methodology [35] and, perhaps, specificity [95], much of the variation is due to the empirical way that these ranges have been derived.
Early attempts to set target ranges to achieve efficacy, while avoiding toxicity, were based on simple clinical observations in patients who had undergone kidney transplantation [31]. More systematic approaches have been made by the retrospective statistical analysis of large volumes of patient data gathered over a period of years [96]. However, the ranges arrived at are biased by subjective judgments; a more objective approach has been taken by Perna et al. [97]. These authors used logistic regression to determine the most probable concentrations for the occurrence of rejection and toxicity in renal transplant patients. This approach has also been used by Nicholls [71] to determine the therapeutic range for mycophenolic acid and is applicable to the other immunosuppressant agents and their combinations.
Morris [98] has called the whole concept of the therapeutic range into question. This author suggests that the concept should be abandoned as it puts too much emphasis on achieving the desired numbers rather than treating the patient. He favours a single concentration approach in which clinicians aim, initially, to dose a patient to achieve a set target concentration. The target is then individualised for that patient based on the number of rejection episodes, occurrence of toxicity, concomitant medication, etc. This has the advantage that pharmacokinetic variability is controlled by the target concentration and pharmacodynamic variability is dealt with by tailoring the target to the patient.
Some of the problems alluded to by Morris could be addressed if the boundaries of the therapeutic ranges were not seen as yes-no cut-off points. To do this the performance characteristics of the concentrations need to be seen in terms of a diagnostic test for determining the probability of drug-effectiveness or toxicity [99]. This, again, requires better information to be available to the clinician so that the blood or plasma concentrations can be interpreted within a Bayesian statistical framework and informed decisions can be made [100].
Assay methodology
Cyclosporin
The correct measurement of cyclosporin has been the subject of many publications [26, 27, 101] and reviews [37, 95]. Currently four commercial companies are producing eight different immunoassay assay systems for the measurement of cyclosporin in whole blood (Table 1). In addition, a number of laboratories are using high performance liquid chromatography (h.p.l.c.) to measure the drug. Using h.p.l.c., it is possible to separate the parent compound from metabolites and for this reason this technique has long been considered the ‘gold standard’ in cyclosporin measurement, particularly when coupled with mass-spectrometry. However, although h.p.l.c. is specific for cyclosporin, the technique can suffer from poor precision and can give spurious results due to interference from other sources [102].
Table 1.
Available assays for the measurement of cyclosporin and abbreviations and manufacturers.

Of the eight immunoassay variants, two are non-specific and cross-react, markedly, with the metabolites of cyclosporin, (Figure 1). The Abbott TDx® Drug and Metabolite assay uses a polyclonal antibody and produces results that are approximately 3 to 5 times those of h.p.l.c. whereas the DiaSorin CYCLO-Trac-NS radioimmunoassay uses a monoclonal non-specific antibody and gives results about 5 to 7 times higher than h.p.l.c. The ratio of the non-specific assays to h.p.l.c. changes with the metabolite:parent compound ratio in the blood and, therefore, will vary with transplant type and time after transplant. The results of the non-specific assays have a poor correlation with clinical events [103].
Figure 1.

Measurement of cyclosporin in an aliquot of pooled blood samples from heart transplant patients receiving the drug. The results are shown as Box and Whisker. The line is drawn across the box at the median. The left of the box is at the first quartile (Q1), and the right is at the third quartile (Q3) value. The whiskers are the lines that extend from the left and right of the box to the adjacent values within ±1.5×(Q3−Q1), values outside the whiskers are plotted as circles. The mean result is shown as a solid circle. plots for h.p.l.c. (11 centres), TDx non-specific (19 Centres) and r.i.a. non-specific. The dotted line is at the median value, 190 μg l−1, for h.p.l.c. (data from the Cyclosporin International Proficiency Testing Scheme [114]).
The other six immunoassays are regarded as ‘specific’ for the parent drug but do, to a limited extent, cross-react with some of the metabolites of the drug and, therefore, do not necessarily give the same result for a given sample (Figure 2). Although the differences between the results of the ‘specific’ assays are due, in part, to the different cross-reactivities of the anti-bodies used, some of the differences result from incorrect calibration [104]. This can be seen clearly with the measurement of spiked samples (Figure 3). (It is interesting to note that for one of the manufacturers the results of their three different assays do not agree). These differences in measurement accuracy do not seem to affect the clinical usefulness of the assays [103] but do add to variability of reported concentrations in the literature [105] and have an impact on the local target ranges [94]. However, in clinical conditions where the cyclosporin metabolite load in blood is high, for example, in liver transplant patients immediately post-transplant, h.p.l.c. is the only method that can accurately measure the parent compound [106].
Figure 2.
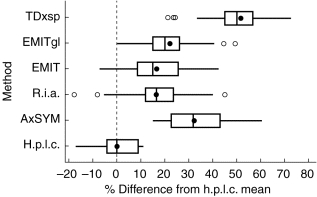
Measurement of cyclosporin in an aliquot of pooled blood samples from heart transplant patients receiving the drug. The results are shown as Box and Whisker. The line is drawn across the box at the median. The left of the box is at the first quartile (Q1), and the right is at the third quartile (Q3) value. The whiskers are the lines that extend from the left and right of the box to the adjacent values within ±1.5×(Q3−Q1), values outside the whiskers are plotted as circles. The mean result is shown as a solid circle. plots for percentage difference from the median result for h.p.l.c. (11 centres), AxSYM, r.i.a.sp, EMIT, EMITgl and TDxsp.
Figure 3.

Measurement of cyclosporin in an aliquot of drug free blood to which 150 μg l−1 of cyclosporin had been added. The results are shown as Box and Whisker. The line is drawn across the box at the median. The left of the box is at the first quartile (Q1), and the right is at the third quartile (Q3) value. The whiskers are the lines that extend from the left and right of the box to the adjacent values within ±1.5×(Q3−Q1), values outside the whiskers are plotted as circles. The mean result is shown as a solid circle. plots for h.p.l.c. (11 centres), AxSYM, r.i.a.sp, EMIT, EMITgl, TDxsp and TDxns.
Tacrolimus
The concentrations of tacrolimus seen in the blood of stable renal transplant patients is low, <30 μg l−1, and this makes the measurement of the drug difficult. In-house ELISA, commercial ELISA (DiaSorin), microparticulate enzyme immunoassays, and h.p.l.c.-MS [107] methods have been available. The majority of laboratories monitoring tacrolimus use the commercial microparticulate enzyme immunoassay (MEIA, Abbott Laboratories) which measures the drug within the range 3 to 30 μg l−1 and cross-reacts to a small degree with the metabolites of tacrolimus [108].
Sirolimus
Therapeutic drug monitoring of sirolimus is still under investigation. The assays that are in current use are based on h.p.l.c. using either ultra-violet or mass spectroscopy to detect the drug. However, a commercial MEIA (Abbott Laboratories) is being tested for widespread, routine, measurement of the drug [109].
Receptor assays
Cyclosporin, tacrolimus and sirolimus bind to a group of widely occurring proteins, the immunophilins. The two major immunophilins are cyclophilin, which binds cyclosporin, and FK-binding protein 12, which binds tacrolimus and sirolimus. Receptor assays have theoretical advantages over current immunoassays in that binding is associated with pharmacological activity so that only active drug or active metabolite concentration is measured [110]. However, this contention still remains to be proved and, as yet, these assays are not in routine clinical use.
Mycophenolic acid
Compared with the other immunosuppressant drugs in current use, the plasma concentration of MPA is much higher and this makes h.p.l.c. measurement of the drug straightforward. However, the major glucuronide metabolite of MPA complicates the assay as it elutes much later than the parent drug and this leads to extended run times. A commercial homogeneous enzyme immunoassay (Dade Behring) is available which is capable of accurate and precise measurement of the drug in the concentration range 0.5 to 15 mg l−1. This assay does not cross react with the major glucuronide of MPA but does give results marginally higher than h.p.l.c. because of cross reactivity to minor MPA metabolites [66].
Proficiency testing
The difficulties in measuring blood cyclosporin concentration led to the development of a proficiency-testing scheme for laboratories providing monitoring services for the drug [29]. The Scheme has been in existence for over 14 years and has over 400 participating centres world-wide. The Scheme circulates three blood samples per month to each participating centre and the measurements made on these samples enable laboratories to benchmark their analytical performance against currently available best practice [111]. The manufacturer of cyclosporin, Sandoz AG (now Novartis AG), initially funded the Scheme to promote measurement accuracy of the drug. However, the individual centres, the drug manufacturer and TDM reagent suppliers, now fund it.
In a similar fashion Fujisawa GmbH, the makers of tacrolimus [112], have funded a scheme for that drug and Dade Behring, manufacturer of a kit for the measurement of MPA, have funded a mycophenolate proficiency scheme [113]. These schemes have 200 and 38 members, respectively. A sirolimus proficiency testing scheme was introduced in January 1999 [114]. The rôle and rationale for proficiency testing schemes in immunosuppressive drug monitoring has recently been reviewed [115].
Pharmacodynamic monitoring
Blood or plasma drug concentration measurements are only a surrogate for effect. Their use would be unnecessary were it possible to measure the immunosuppressive action of these drugs directly. However, defining the pharmacodynamic target for monitoring is not an easy task. For example, in a recent study investigating immunological monitoring of azathioprine the authors examined multiple subsets of peripheral blood lymphocytes, natural killer activity, the serum concentrations of IgG, IgM, interferon β (IFN β), tumour necrosis factor α (TNF α), interleukin 2 (IL-2), soluble interleukin 2 receptors (sIL-2R), interleukin 6 (IL-6) and the soluble adhesion molecule sICAM-1 [116]. There is a danger that one surrogate will be replaced by another.
Cyclosporin and tacrolimus inhibit T-cell proliferation, which is thought to result from their inhibition of calcineurin [117]. This is a serine-threonine phosphatase that plays an essential rôle in intra-cellular, calcium-dependent, signal transduction [118]. Inhibition of calcineurin in activated T cells reduces the translocation of the cytoplasmic sub-unit of the nuclear factor to the nuclear sub-unit and, hence, impairs the transcription of genes for many of the cytokines essential for the rejection response [119]. For this reason calcineurin has been the primary focus of pharmacodynamic monitoring for cyclosporin. In a study of 62 renal transplant patients the measured calcineurin activity in leukocytes was half that of controls [120]. The trough cyclosporin concentration was inversely related to the calcineurin activity. Since tacrolimus acts on the same target, calcineurin would also be applicable for monitoring that drug. However, the measurement of calcineurin activity is technically challenging and a much simpler procedure would be required before it could be used in the routine setting. In any case, further studies are needed to confirm that calcineurin activity correlates better with patient response than simple blood concentrations.
Mycophenolic acid exerts its immunosuppressive action by inhibition of inosine monophosphate dehydrogenase (IMPDH) and, thereby, blocking de novo purine biosynthesis in lymphocytes [121]. An assay has been developed for the measurement of IMPDH activity in whole blood to measure drug effect rather than concentration [122]. As yet there are few published data to support the use of this assay, but initial studies suggest a correlation between IMPDH activity and clinical events [123].
Sirolimus is thought to exert its immunosuppressive activity by blocking the phosphorylation and activation of the P70 S6 kinase that is involved in cell signalling, and this prevents, or reduces, lymphocyte proliferation [124]. The measurement of P70 S6 activity is, therefore, a prime target for pharmacodynamic measurements. An assay has been developed for its measurement in whole blood and studies are ongoing to determine its utility in clinical practice [125].
Antithymocyte globulin (ATG) has been monitored in renal transplant patients using the effect of ATG on subsets of T lymphocytes [126]. In this study, the author identified the CD3+ lymphocytes as a pharmacodynamic marker of ATG response. The dose of ATG administered to patients was titrated to maintain the patients’ absolute CD3+ lymphocyte count at ≈50 cells μl−1 of blood. Forty-four patients who were treated in this way for steroid resistant rejection had significantly less serious viral infections than 10 patients who were treated on a fixed dose ATG regimen, but their 1 year graft survival was not compromised. Although not a formal pharmaco-economic study, the author also noted that there was a net saving of £1600 ($2500) per patient.
Benefits of monitoring
Evidence-based medicine is sadly lacking in the area of drug monitoring [127]. Although the perception of therapeutic drug monitoring is that it is beneficial, and aids patient management, there is little hard evidence to support that view. Outside the field of immunosuppressive drugs there are numerous articles, detailing predominantly retrospective studies, which suggest that TDM has a useful and cost effective rôle in monitoring therapy [128, 129]. However, as the authors of a recent review of clinical pharmacokinetics point out, there is little evidence to support the effect of TDM on true patient outcomes [128].
For the immunosuppressive drugs some positive evidence for TDM is given by the results of prospective concentration-controlled clinical trials [130]. A concentration-controlled trial is one in which patients are dosed to achieve pre-assigned target concentrations and, therefore, the results can be assessed in terms of drug concentration and response rather than dose and response. This type of trial has several benefits over the randomized dose-controlled trials [131] and can provide the basis for future TDM decisions.
In a study of tacrolimus, patients were randomly allocated to be targeted at low, medium or high drug blood concentrations [132]. Although there was no difference in the incidence of rejection or toxicity in the three groups in the first 42 days post transplant, logistic regression demonstrated a clear relationship between concentration and effect (Table 2). In a study of similar design using mycophenolate mofetil, patients were randomly allocated to a high, medium or low mycophenolic acid AUC [72]. Again there was a clear concentration related effect of the drug with rejection in the low, medium and high groups of 26, 9 and 6%, respectively.
Table 2.
Incidence of toxicity and rejection by whole blood tacrolimus concentration [132].

Conclusion
At present the majority of drug regimes in use for transplantation are based on cyclosporin and excellent results can be obtained in solid organ transplantation using this drug. This is also true in the treatment of autoimmune diseases [2]. However, cyclosporin’s dominance is being challenged as the number of immunosuppressive agents increases and clinical experience is gained with other drugs. We are entering an era in which combination therapy will be the norm and clinicians will tailor the immunosuppression to the characteristics of the individual patient changing dose and drugs as time progresses and conditions change [133]. In addition, generic [134] and other new formulations of cyclosporin [135] will be available in the near future and these together with the new drugs under clinical development, such as FTY720 [136] and SDZ-RAD [137], and the possibilities of xenotransplantation [138] present future challenges which will add to the complexity of TDM.
References
- 1.Spector R, Park GD, Johnson GF, Vesell ES. Therapeutic drug monitoring. Clin Pharmacol Ther. 1988;43:345–353. doi: 10.1038/clpt.1988.42. [DOI] [PubMed] [Google Scholar]
- 2.Holt DW, Johnston A. Cyclosporin monitoring: its role in autoimmune indications. J Autoimmunity. 1992;5(Suppl-82) doi: 10.1016/0896-8411(92)90032-l. pp. ? [DOI] [PubMed] [Google Scholar]
- 3.Holt DW, Johnston A. Monitoring new immunosuppressive agents. Are the methods adequate? Drug Metabolism & Drug Interactions. 1997;14:5–15. doi: 10.1515/dmdi.1997.14.1.5. [DOI] [PubMed] [Google Scholar]
- 4.Calne RY. The present position and future prospects of organ transplantation. Ann R Coll Surg Engl. 1968;42:283–306. [PMC free article] [PubMed] [Google Scholar]
- 5.Starzl TE, Marchioro TL, Porter KA, Iwasaki Y, Cerilli GJ. The use of heterologous antilymphoid agents in canine renal and liver homotransplantation and in human renal homotransplantation. Surg Gynecol Obstet. 1967;124:301–308. [PMC free article] [PubMed] [Google Scholar]
- 6.Schutz E, Gummert J, Mohr FW, Armstrong VW, Oellerich M. Should 6-thioguanine nucleotides be monitored in heart transplant recipients given azathioprine? Ther Drug Monit. 1996;18:228–233. doi: 10.1097/00007691-199606000-00002. [DOI] [PubMed] [Google Scholar]
- 7.Weinshilboum RM, Sladek SL. Mercaptopurine pharmacogenetics: monogenic inheritance of erythrocyte thiopurine methyltransferase activity. Am J Hum Genet. 1980;32:651–662. [PMC free article] [PubMed] [Google Scholar]
- 8.Loennechen T, Yates CR, Fessing MY, Relling MV, Krynetski EY, Evans WE. Isolation of a human thipurine S-methyltransferase (TPMT) complementary DNA with a single nucleotide transition A719G (TPMT*3C) and its association with loss of TPMT protein and catalytic activity in humans. Clin Pharmacol Ther. 1998;64:46–51. doi: 10.1016/S0009-9236(98)90021-2. [DOI] [PubMed] [Google Scholar]
- 9.Gummert JF, Schutz E, Oellerich M, Mohr FW, Dalichau H. Monitoring of TPMT in heart transplant recipients under immunosuppressive therapy with azathioprine. Artif Organs. 1995;19:918–920. doi: 10.1111/j.1525-1594.1995.tb02451.x. [DOI] [PubMed] [Google Scholar]
- 10.Lennard L. Therapeutic drug monitoring of antimetabolic cytotoxic drugs. Br J Clin Pharmacol. 1998;47:131–143. doi: 10.1046/j.1365-2125.1999.00884.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilde MI, Goa KL. Muromonab CD3: a reappraisal of its pharmacology and use as prophylaxis of solid organ transplant rejection. Drugs. 1996;51:865–894. doi: 10.2165/00003495-199651050-00010. [DOI] [PubMed] [Google Scholar]
- 12.Smith SL. Ten years of Orthoclone OKT3 (muromonab-CD3): a review. J Transplant Coordination. 1996;6:109–119. doi: 10.7182/prtr.1.6.3.8145l3u185493182. [DOI] [PubMed] [Google Scholar]
- 13.Abramowicz D, Goldman M, Mat O, et al. OKT3 serum levels as a guide for prophylactic therapy: a pilot study in kidney transplant recipients. Transpl Int. 1994;7:258–263. doi: 10.1007/BF00327153. [DOI] [PubMed] [Google Scholar]
- 14.Schena FP. New insights into therapy with monoclonal antibodies in allograft transplantation. Nephrol Dial Transplant. 1997;12(Supplement 1):55–58. [PubMed] [Google Scholar]
- 15.Cinti P, Cocciolo P, Evangelista B, et al. OKT3 prophylaxis in kidney transplant recipients: drug monitoring by flow cytometry. Transplant Proc. 1996;28:3214–3216. [PubMed] [Google Scholar]
- 16.Calne RY. Organ transplantation. The present position and future prospects of organ transplantation. Trans Med Soc Lond. 1969;85:56–67. [PubMed] [Google Scholar]
- 17.Calne RY. Immunosuppression and clinical organ transplantation. Transplant Proc. 1974;6(4:Suppl 1: Suppl-51) pp? [PubMed] [Google Scholar]
- 18.Opelz G. Influence of treatment with cyclosporine, azathioprine and steroids on chronic allograft failure. The Collaborative Transplant Study. Kidney International—Supplement. 1995;52:S89–S92. [PubMed] [Google Scholar]
- 19.Borel JF, Feurer C, Gubler HU, Stahelin H. Biological effects of cyclosporin A: a new antilymphocytic agent. Agents Actions. 1976;6:468–475. doi: 10.1007/BF01973261. [DOI] [PubMed] [Google Scholar]
- 20.Borel JF, Kis ZL. The discovery and development of cyclosporine (Sandimmune) Transplant Proc. 1991;23:1867–1874. [PubMed] [Google Scholar]
- 21.Borel JF, Kis ZL, Beveridge T, Meluzzi VJ, Adams J, editors. The history of the discovery and development of cyclosporine. Vol. 2. Boston, USA: Birkhäuser; 1995. The search for anti-inlammatory drugs; pp. 27–63. [Google Scholar]
- 22.Dunn SP, Cooney GF, Kulinsky A, Falkenstein K, Pierson A, Meligeni J. Reduced cyclosporine absorption preceded acute allograft rejection in a child with a liver transplant. Liver Transplantation & Surgery. 1997;3:538–540. doi: 10.1002/lt.500030510. [DOI] [PubMed] [Google Scholar]
- 23.Stahelin HF. The history of cyclosporin A (Sandimmune) revisited: another point of view. Experientia. 1996;52:5–13. doi: 10.1007/BF01922409. [DOI] [PubMed] [Google Scholar]
- 24.Shaw LM, Kaplan B, Kaufman D. Toxic effects of immunosuppressive drugs: mechanisms and strategies for controlling them. Clin Chem. 1996;42:1316–1321. [PubMed] [Google Scholar]
- 25.Holt DW, Johnston A, Thomson A, Starzl T, editors. Pharmacokinetics and monitoring of cyclosporin A. Vol. 3. London: Edward Arnold; 1994. Immunosuppressive Drugs: Developments in anti-rejection therapy; pp. 37–45. [Google Scholar]
- 26.Johnston A, Marsden JT, Holt DW. The influence of haematocrit on blood cyclosporin measurements in vivo. Br J Clin Pharmacol. 1988;25:509–513. doi: 10.1111/j.1365-2125.1988.tb03336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnston A, Marsden JT, Holt DW. Sample pretreatment to minimize interference from whole blood in the radioimmunoassay for cyclosporine. Transplantation. 1987;44:332. doi: 10.1097/00007890-198708000-00033. [DOI] [PubMed] [Google Scholar]
- 28.Holt DW, Marsden JT, Johnston A. Measurement of cyclosporine: methodological problems. Transplant Proc. 1986;18(Suppl 5):101–110. [PubMed] [Google Scholar]
- 29.Johnston A, Marsden JT, Holt DW. The United Kingdom Cyclosporin Quality Assessment Scheme. Ther Drug Monit. 1986;8:200–204. doi: 10.1097/00007691-198606000-00013. [DOI] [PubMed] [Google Scholar]
- 30.Tsang VT, Johnston A, Heritier F, Leaver N, Hodson ME, Yacoub M. Cyclosporin pharmacokinetics in heart-lung transplant recipients with cystic fibrosis. Effects of pancreatic enzymes and ranitidine. Eur J Clin Pharmacol. 1994;46:261–265. doi: 10.1007/BF00192559. [DOI] [PubMed] [Google Scholar]
- 31.Holt DW, Marsden JT, Johnston A, Bewick M, Taube DH. Blood cyclosporin concentrations and renal allograft dysfunction. Br Med J. 1986;293:1057–1059. doi: 10.1136/bmj.293.6554.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kahan BD, Shaw LM, Holt D, Grevel J, Johnston A. Consensus document: Hawk’s Cay meeting on therapeutic drug monitoring of cyclosporine. Clin Chem. 1990;36:1510–1516. [PubMed] [Google Scholar]
- 33.Shaw LM, Yatscoff RW, Bowers LD, et al. Canadian Consensus Meeting on cyclosporine monitoring: report of the consensus panel. Clin Chem. 1990;36:1841–1846. [PubMed] [Google Scholar]
- 34.Sketris I, Yatscoff R, Keown P, et al. Optimizing the use of cyclosporine in renal transplantation. Clin Biochem. 1995;28:195–211. doi: 10.1016/0009-9120(95)91341-y. [DOI] [PubMed] [Google Scholar]
- 35.Holt DW, Johnston A. Cyclosporin assay techniques. Accuracy and reproducibility variables impacting on measurements. Int J Rad Appl Instrum B. 1990;17:733–736. doi: 10.1016/0883-2897(90)90098-l. [DOI] [PubMed] [Google Scholar]
- 36.Holt DW, Johnston A, den Boer NC, van der Heiden C, Leijnse B, Souverijn JHM, editors. Practical applications of therapeutic drug monitoring: The impact of technological developments. Plenum Publishing Corporation; 1989. Clinical Chemistry; pp. 93–102. [Google Scholar]
- 37.Holt DW, Johnston A, Roberts NB, Tredger JM, Trull AK. Methodological and clinical aspects of cyclosporin monitoring: report of the Association of Clinical Biochemists task force. Ann Clin Biochem. 1994;31:420–446. doi: 10.1177/000456329403100503. [DOI] [PubMed] [Google Scholar]
- 38.Dalrymple-Hay M, Meara M, Reynolds L, et al. Changing stable heart transplant recipients from Sandimmune to Neoral. Transplant Proc. 1996;28:2285–2286. [PubMed] [Google Scholar]
- 39.Holt DW, Mueller EA, Kovarik JM, van Bree JB, Richard F, Kutz K. Sandimmun neoral pharmacokinetics: impact of the new oral formulation. Transplant Proc. 1995;27:1434–1437. [PubMed] [Google Scholar]
- 40.Holt DW, Mueller EA, Kovarik JM, van Bree JB, Kutz K. The pharmacokinetics of Sandimmun Neoral: a new oral formulation of cyclosporine. Transplant Proc. 1994;26:2935–2939. [PubMed] [Google Scholar]
- 41.Lindholm A, Kahan BD. Influence of cyclosporine pharmacokinetics, trough concentrations, and AUC monitoring on outcome after kidney transplantation. Clinical Pharmacol Ther. 1993;54:205–218. doi: 10.1038/clpt.1993.132. [DOI] [PubMed] [Google Scholar]
- 42.Holt DW, Johnston A. Cyclosporin monitoring: trough or AUC? Perspectives. 1996:49–52. [Google Scholar]
- 43.Holt DW, Johnston A. Cyclosporin microemulsion. A guide to usage and monitoring. BioDrugs. 1997;7:175–197. [Google Scholar]
- 44.Johnston A, Sketris I, Marsden JT, et al. A limited sampling strategy for the measurement of cyclosporine AUC. Transplant Proc. 1990;22:1345–1346. [PubMed] [Google Scholar]
- 45.Johnston A, Kovarik JM, Mueller EA, Holt DW. Predicting patients’ exposure to cyclosporin. Transplant International. 1996;9(Suppl-7):5305–5307. doi: 10.1007/978-3-662-00818-8_75. [DOI] [PubMed] [Google Scholar]
- 46.Cantarovich F, Bizollon C, Cantarovich D, Lefrancois N, Dubernard JM, Traeger J. Cyclosporine plasma levels six hours after oral administration. A useful tool for monitoring therapy. Transplantation. 1988;45:389–394. doi: 10.1097/00007890-198802000-00029. [DOI] [PubMed] [Google Scholar]
- 47.Cantarovich M, Besner JG, Fitchett DH, Latter DA. Efficacy and side-effects of cyclosporine dose monitoring with levels 6 h after the morning dose in heart transplant patients. Clin Transplantation. 1997;11:405. [PubMed] [Google Scholar]
- 48.Rocha G, Deschenes J, Cantarovich M. Cyclosporine monitoring with levels 6 hours after the morning dose in patients with noninfectious uveitis. Ophthalmology. 1997;104:245–251. doi: 10.1016/s0161-6420(97)30328-5. [DOI] [PubMed] [Google Scholar]
- 49.Cantarovich M, Deschenes J. Cyclosporine dose adjustment using levels obtained six hours after the morning dose: effect on side effects in patients with autoimmune diseases. Am J Nephrology. 1997;17:450–457. doi: 10.1159/000169140. [DOI] [PubMed] [Google Scholar]
- 50.Grant D, Rochon J, Levy G. Comparison of the long-term tolerability, pharmacodynamics, and safety of Sandimmune and Neoral in liver transplant recipients. Ontario Liver Transplant Study Group. Transplant Proc. 1996;28:2232–2233. [PubMed] [Google Scholar]
- 51.Levy GA, Grant D. Neoral in liver transplantation. Transplant Proc. 1996;28:1019–1021. [PubMed] [Google Scholar]
- 52.Freeman D, Grant D, Levy G, et al. Pharmacokinetics of a new oral formulation of cyclosporine in liver transplant recipients. Ther Drug Monit. 1995;17:213–216. doi: 10.1097/00007691-199506000-00001. [DOI] [PubMed] [Google Scholar]
- 53.Levy G. Two-hour cyclosporin concentration (C2) as a monitoring tool for Neoral. Oxford: Blackwell Science; 1998. New strategies for therapeutic drug monitoring of Neoral; pp. 19–22. [Google Scholar]
- 54.Keown P, Kahan BD, Johnston A, et al. Optimization of cyclosporin therapy with new therapeutic drug monitoring strategies: Report from the international Neoral TDM advisory consensus meeting (Vancouver, November 1997) Transplant Proc. 1998;30:1645–1649. doi: 10.1016/s0041-1345(98)00375-3. [DOI] [PubMed] [Google Scholar]
- 55.Jusko WJ, Thomson AW, Fung J, et al. Consensus document: therapeutic monitoring of tacrolimus (FK-506) Ther Drug Monit. 1995;17:606–614. doi: 10.1097/00007691-199512000-00011. [DOI] [PubMed] [Google Scholar]
- 56.McMaster P, Mirza DF, Ismail T, Vennarecci G, Patapis P, Mayer AD. Therapeutic drug monitoring of tacrolimus in clinical transplantation. Ther Drug Monit. 1995;17:602–605. doi: 10.1097/00007691-199512000-00010. [DOI] [PubMed] [Google Scholar]
- 57.Venkataramanan R, Swaminathan A, Prasad T, et al. Clinical pharmacokinetics of tacrolimus. Clin Pharmacokin. 1995;29:404–430. doi: 10.2165/00003088-199529060-00003. [DOI] [PubMed] [Google Scholar]
- 58.Anonymous Japanese study of FK 506 on kidney transplantation: the benefit of monitoring the whole blood FK 506 concentration. Japanese FK 506 Study Group. Transplant Proc. 1991;23:3085–3088. [PubMed] [Google Scholar]
- 59.Hedayat S, Kershner RP, Su G. Relationship of whole-blood FK506 concentrations to rejection and toxicity in liver and kidney transplants. J Biopharm Stat. 1996;6:411–424. doi: 10.1080/10543409608835153. [DOI] [PubMed] [Google Scholar]
- 60.Ku Y-M, Min DI. An abbreviated area-under-the-curve monitoring for tacrolimus patients with liver transplants. Ther Drug Monit. 1998;20:219–223. doi: 10.1097/00007691-199804000-00014. [DOI] [PubMed] [Google Scholar]
- 61.Ihara H, Shinkuma D, Ichikawa Y, Nojima M, Nagano S, Ikoma F. Intra- and interindividual variation in the pharmacokinetics of tacrolimus (FK506) in kidney transplant recipients—importance of trough level as a practical indicator. Int J Urol. 1995;2:151–155. doi: 10.1111/j.1442-2042.1995.tb00444.x. [DOI] [PubMed] [Google Scholar]
- 62.Lipsky JJ. Mycophenolate mofetil. Lancet. 1996;348:1357–1359. doi: 10.1016/S0140-6736(96)10310-X. [DOI] [PubMed] [Google Scholar]
- 63.Bullingham RE, Nicholls A, Hale M. Pharmacokinetics of mycophenolate mofetil (RS61443): a short review. Transplant Proc. 1996;28:925–929. [PubMed] [Google Scholar]
- 64.Allison AC, Eugui EM. Immunosuppressive and other effects of mycophenolic acid and an ester prodrug, mycophenolate mofetil. Immunol Rev. 1993;136:5–28. doi: 10.1111/j.1600-065x.1993.tb00652.x. [DOI] [PubMed] [Google Scholar]
- 65.Gray DW. Mycophenolate mofetil for transplantation: new drug, old problems? Lancet. 1995;346:390. doi: 10.1016/s0140-6736(95)92775-1. [DOI] [PubMed] [Google Scholar]
- 66.Schutz E, Shipkova M, Armstrong VW, et al. Therapeutic drug monitoring of mycophenolic acid: comparison of HPLC and immunoassay reveals new MPA metabolites. Transplant Proc. 1998;30:1185–1187. doi: 10.1016/s0041-1345(98)00201-2. [DOI] [PubMed] [Google Scholar]
- 67.Schutz E, Shipkova M, Armstrong VW, Niedmann PD, Oellerich M. Metabolism of MPA: Impact of metabolite patterns in MPA Monitoring. Berichte der OsterreichischenGesellschaft fur Klinische Chemie. 1999. (in press)
- 68.Brewin TB, Cole MP, Jones CT, Platt DS, Todd ID. Mycophenolic acid (NSC-129185): preliminary clinical trials. Cancer Chemother Rep. 1972;56:83–87. [PubMed] [Google Scholar]
- 69.Anonymous Placebo-controlled study of mycophenolate mofetil combined with cyclosporin and corticosteroids for prevention of acute rejection. European Mycophenolate Mofetil Cooperative Study Group. Lancet. 1995;345:1321–1325. [PubMed] [Google Scholar]
- 70.Bullingham RES, Nicholls AJ, Kamm BR. Clinical pharmacokinetics of mycophenolate mofetil. Clin Pharmacokinet. 1998;34:429–455. doi: 10.2165/00003088-199834060-00002. [DOI] [PubMed] [Google Scholar]
- 71.Nicholls AJ. Opportunities for therapeutic drug monitoring of mycophenolate mofetil dose in renal transplantation suggested by the pharmacokinetic/pharmacodynamic relationship for mycophenolic acid and suppression of rejection. Clin Biochem. 1998;31:329–333. doi: 10.1016/s0009-9120(98)00056-3. [DOI] [PubMed] [Google Scholar]
- 72.Hale MD, Nicholls AJ, Bullingham RE, et al. The pharmacokinetic–pharmacodynamic relationship for mycophenolate mofetil in renal transplantation. Clin Pharmacol Ther. 1998;64:672–683. doi: 10.1016/S0009-9236(98)90058-3. [DOI] [PubMed] [Google Scholar]
- 73.Shaw LM, Korecka M, van Breeman R, Nowak I, Brayman KL. Analysis, pharmacokinetics and therapeutic drug monitoring of mycophenolic acid. Clin Biochem. 1998;31:323–328. doi: 10.1016/s0009-9120(98)00050-2. [DOI] [PubMed] [Google Scholar]
- 74.Meiser BM, Pfeiffer M, Jagiello-Kraatz M, et al. Mycophenolate mofetil dose adjustment based on trough levels improves outcome after heart transplantion. J Heart Lung Transplant. 1998;17:85. [Google Scholar]
- 75.Shaw LM, Nicholls A, Hale M, et al. Therapeutic monitoring of mycophenolic acid: A consensus panel report. Clin Biochem. 1998;31:317–322. doi: 10.1016/s0009-9120(98)00040-x. [DOI] [PubMed] [Google Scholar]
- 76.Zanker B, Schneeberger H, Rothenpieler U, et al. Mycophenolate mofetil-based, cyclosporin-free induction and maintenance immunosuppression. Transplantation. 1998;66:44–49. doi: 10.1097/00007890-199807150-00007. [DOI] [PubMed] [Google Scholar]
- 77.Sehgal SN. Rapamune® (RAPA, rapamycin, sirolimus): Mechanism of action immunosuppressive effect results from blockade of signal transduction and inhibition of cell cycle progression. Clin Biochem. 1998;31:335–340. doi: 10.1016/s0009-9120(98)00045-9. [DOI] [PubMed] [Google Scholar]
- 78.Helderman JH. New immunologically non-specific immunosuppressive agents dictate changes in standard protocols for renal transplant management. Nephrol Dial Transplant. 1997;12(Suppl 1):51–54. [PubMed] [Google Scholar]
- 79.Brattstrom C, Holt DW, Backman L, et al. 17th World Congress. Montreal, Canada: The Transplant Society; 1998. Concentration-controlled dosing of Rapamune® in renal allograft recipients to optomise therapy. [Google Scholar]
- 80.Kahan BD. Sirolimus: a new agent for clinical renal transplantation. Transplant Proc. 1997;29:48–50. doi: 10.1016/s0041-1345(96)00008-5. [DOI] [PubMed] [Google Scholar]
- 81.Stepkowski SM, Tian L, Wang ME, Qu X, Napoli K, Kahan BD. Sirolimus in transplantation. Arch Immunol Ther Exp (Warsz) 1997;45:383–390. [PubMed] [Google Scholar]
- 82.Trepanier DJ, Gallant H, LeGatt DF, Yatscoff RW. Rapamycin: Distribution, pharmacokinetics and therapeutic range investigations: An update. Clin Biochem. 1998;31:345–351. doi: 10.1016/s0009-9120(98)00048-4. [DOI] [PubMed] [Google Scholar]
- 83.Yatscoff RW, Boeckx R, Holt DW, et al. Consensus guidelines for therapeutic drug monitoring of rapamycin: report of the consensus panel. Ther Drug Monit. 1995;17:676–680. doi: 10.1097/00007691-199512000-00022. [DOI] [PubMed] [Google Scholar]
- 84.Minami Y, Kono T, Miyazaki T, Taniguchi T. The IL-2 receptor complex: its structure, function, and target genes. Ann Rev Immunol. 1993;11:245–268. doi: 10.1146/annurev.iy.11.040193.001333. [DOI] [PubMed] [Google Scholar]
- 85.Taniguchi T, Minami Y. The IL-2/IL-2 receptor system: a current overview. Cell. 1993;73:5–8. doi: 10.1016/0092-8674(93)90152-g. [DOI] [PubMed] [Google Scholar]
- 86.Vincenti F, Lantz M, Birnbaum J, et al. A phase I trial of humanized anti-interleukin 2 receptor antibody in renal transplantation. Transplantation. 1997;63:33–38. doi: 10.1097/00007890-199701150-00007. [DOI] [PubMed] [Google Scholar]
- 87.Amlot PL, Rawlings E, Fernando ON, et al. Prolonged action of a chimeric interleukin-2 receptor (CD25) monoclonal antibody used in cadaveric renal transplantation. Transplantation. 1995;60:748–756. doi: 10.1097/00007890-199510150-00023. [DOI] [PubMed] [Google Scholar]
- 88.Kovarik JM, Rawlings E, Sweny P, et al. Prolonged immunosuppressive effect and minimal immunogenicity from chimeric (CD25) monoclonal antibody SDZ CHI 621 in renal transplantation. Transplant Proc. 1996;28:913–914. [PubMed] [Google Scholar]
- 89.Vincenti F, Kirkman R, Light S, et al. Interleukin-2-receptor blockade with daclizumab to prevent acute rejection in renal transplantation. Daclizumab Triple Therapy Study Group. N Engl J Med. 1998;338:161–165. doi: 10.1056/NEJM199801153380304. [DOI] [PubMed] [Google Scholar]
- 90.Hakimi J, Mould D, Waldeman TA, et al. Antibody therapeutics. Vol. 12. CRC Press; 1997. Development of Zenapax: A humanized anti-tac antibody; pp. 277–300. [Google Scholar]
- 91.Kovarik JM, Rawlings E, Sweny P, et al. Pharmacokinetics and immunodynamics of chimeric IL-2 receptor monoclonal antibody SDZ CHI 621 in renal allograft recipients. Transplant Int. 1996;9(Suppl 1):S32–S33. doi: 10.1007/978-3-662-00818-8_9. [DOI] [PubMed] [Google Scholar]
- 92.Kovarik J, Wolf P, Cisterne JM, et al. Disposition of basiliximab, an interleukin-2 receptor monoclonal antibody, in recipients of mismatched cadaver renal allografts. Transplantation. 1997;64:1701–1705. doi: 10.1097/00007890-199712270-00012. [DOI] [PubMed] [Google Scholar]
- 93.Kovarik J, Breidenbach T, Gerbeau C, Korn A, Schmidt A-G, Nashan B. Disposition and immunodynamics of basiliximab in liver allograft recipients. Clin Pharmacol Ther. 1998;64:66–72. doi: 10.1016/S0009-9236(98)90024-8. [DOI] [PubMed] [Google Scholar]
- 94.Oellerich M, Armstrong VW, Kahan B, et al. Lake Louise Consensus Conference on cyclosporin monitoring in organ transplantation: report of the consensus panel. Ther Drug Monit. 1995;17:642–654. doi: 10.1097/00007691-199512000-00017. [DOI] [PubMed] [Google Scholar]
- 95.Holt DW, Johnston A. Cyclosporin A: analytical methodology and factors affecting therapeutic drug monitoring. Ther Drug Monit. 1995;17:625–630. doi: 10.1097/00007691-199512000-00014. [DOI] [PubMed] [Google Scholar]
- 96.LeGatt DF, Chooi M, Simpson AI, Yatscoff RW. EMIT cyclosporine assay: development of an application protocol for Technicon AXON System. Clin Biochem. 1994;27:387–394. doi: 10.1016/0009-9120(94)90043-4. [DOI] [PubMed] [Google Scholar]
- 97.Perna A, Gotti E, de BE, Perico N, Remuzzi G. A logistic-regression model provides novel guidelines to maximize the anti-acute rejection properties of cyclosporine with a minimum of toxicity. J Am Soc Nephrol. 1996;7:786–791. doi: 10.1681/ASN.V75786. [DOI] [PubMed] [Google Scholar]
- 98.Morris RG. Target concentration strategy for cyclosporin monitoring. Clin Pharmacokin. 1997;32:175–179. doi: 10.2165/00003088-199732030-00001. [DOI] [PubMed] [Google Scholar]
- 99.Schumacher GE, Barr JT. Total testing process applied to therapeutic drug monitoring: impact on patients’ outcomes and economics. Clin Chem. 1998;44:370–374. [PubMed] [Google Scholar]
- 100.Schumacher GE, Barr JT. Bayesian and threshold probabilities in therapeutic drug monitoring: when can serum drug concentrations alter clinical decisions? Am J Hosp Pharm. 1994;51:321–327. [PubMed] [Google Scholar]
- 101.Holt DW, White DJ. How to measure cyclosporin. Lancet. 1984;ii:228. doi: 10.1016/s0140-6736(84)90515-4. [DOI] [PubMed] [Google Scholar]
- 102.Johnston A, Cullen G, Holt DW. Quality assurance for cyclosporin assays in body fluids. Ann Acad Med Singapore. 1991;20:3–8. [PubMed] [Google Scholar]
- 103.Lindholm A, Dahlqvist R, Groth GG, Sjoqvist F. A prospective study of cyclosporine concentration in relation to its therapeutic effect and toxicity after renal transplantation. Br J Clin Pharmacol. 1990;30:443–452. doi: 10.1111/j.1365-2125.1990.tb03796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Johnston A, Holt DW. Calibration of the CYCLO-Trac SP cyclosporine radioimmunoassay. Clin Chem. 1993;39:2532–2533. [PubMed] [Google Scholar]
- 105.McLachlan AJ, Tett SE. Effect of metabolic inhibitors on cyclosporine pharmacokinetics using a population approach. Ther Drug Monit. 1998;20:390–395. doi: 10.1097/00007691-199808000-00007. [DOI] [PubMed] [Google Scholar]
- 106.Schütz E, Svinarov D, Shipkova M, et al. Cyclosporin whole blood immunoassays (AxSYM, CEDIA, and Emit): A critical overview of performance characteristics and comparison with HPLC. Clin Chem. 1998;44:2158–2164. [PubMed] [Google Scholar]
- 107.Taylor PJ, Hogan NS, Lynch SV, Johnson AG, Pond SM. Improved therapeutic drug monitoring of tacrolimus (FK506) by tandem mass spectrometry. Clin Chem. 1997;43:2189–2190. [PubMed] [Google Scholar]
- 108.Schambeck CM, Bedel A, Keller F. Limit of quantitation (Functional Sensitivity) of the new IMx Tacrolimus II microparticulate immunoassay. Clin Chem. 1998;44:2217. ? [PubMed] [Google Scholar]
- 109.Holt DW, Jones K, Johnston A, et al. An immunoassay for the measurement of sirolimus. Clin Chem. 1998;44(6) suppl:A94. doi: 10.1016/s0149-2918(00)89022-0. ? [DOI] [PubMed] [Google Scholar]
- 110.Soldin SJ. Role of immunophilins in therapeutic drug monitoring of immunosuppressive drugs. Clin Biochem. 1998;31:381–384. doi: 10.1016/s0009-9120(98)00046-0. [DOI] [PubMed] [Google Scholar]
- 111.Johnston A, Holt DW. External quality assessment scheme for cyclosporin in body fluids. Scand J Clin Lab Invest Suppl. 1993;212:48–53. doi: 10.3109/00365519309085456. [DOI] [PubMed] [Google Scholar]
- 112.Holt DW, Johnston A. Tacrolimus quality assessment. Ther Drug Monit. 1997;19:243. doi: 10.1097/00007691-199704000-00023. [DOI] [PubMed] [Google Scholar]
- 113.Holt DW, Jones K, Lee T, Stadler P, Johnston A. Quality assessment issues of new immunosuppressive drugs and experimental experience. Ther Drug Monit. 1996;18:362–367. doi: 10.1097/00007691-199608000-00008. [DOI] [PubMed] [Google Scholar]
- 114.Cyclosporin International Proficiency Testing Scheme. URL www.asil.demon.co.uk.
- 115.Holt DW, Johnston A. Monitoring immunosuppressive drugs: The rational for proficiency testing. Ligand Assay. 1998;3:2–6. [Google Scholar]
- 116.Salmaggi A, Corsini E, La ML, et al. Immunological monitoring of azathioprine treatment in multiple sclerosis patients. J Neurol. 1997;244:167–174. doi: 10.1007/s004150050068. [DOI] [PubMed] [Google Scholar]
- 117.Morris R. Modes of action of FK506, cyclosporin A and rapamycin. Transplant Proc. 1994;26:3272–3275. [PubMed] [Google Scholar]
- 118.Schreiber SL, Crabtree GR. The mechanism of action of cyclosporin A and FK506. Immunol Today. 1992;13:136–142. doi: 10.1016/0167-5699(92)90111-J. [DOI] [PubMed] [Google Scholar]
- 119.Clipstone NA, Crabtree GR. Identification of calcineurin as a key signalling enzyme in T-lymphocyte activation. Nature. 1992;357:695–697. doi: 10.1038/357695a0. [DOI] [PubMed] [Google Scholar]
- 120.Batiuk TD, Pazderka F, Halloran PF. Calcineurin activity is only partially inhibited in leukocytes of cyclosporine-treated patients. Transplantation. 1995;59:1400–1404. doi: 10.1097/00007890-199505270-00007. [DOI] [PubMed] [Google Scholar]
- 121.Eugui EM, Almquist SJ, Muller CD, Allison AC. Lymphocyte-selective cytostatic and immunosuppressive effects of mycophenolic acid in vitro: role of deoxyguanosine nucleotide depletion. Scand J Immunol. 1991;33:161–173. doi: 10.1111/j.1365-3083.1991.tb03746.x. [DOI] [PubMed] [Google Scholar]
- 122.Langman LJ, LeGatt DF, Halloran PF, Yatscoff RW. Pharmacodynamic assessment of mycophenolic acid-induced immunosuppression in renal transplant recipients. Transplantation. 1996;62:666–672. doi: 10.1097/00007890-199609150-00022. [DOI] [PubMed] [Google Scholar]
- 123.Yatscoff RW, Aspeslet LJ, Gallant HL. Pharmacodynamic monitoring of immunosuppressive drugs. Clin Chem. 1998;44:428–432. [PubMed] [Google Scholar]
- 124.Chung J, Kuo CJ, Crabtree GR, Blenis J. Rapamycin-FKBP specifically blocks growth-dependent activation of and signaling by the 70 kd S6 protein kinases. Cell. 1992;69:1227–1236. doi: 10.1016/0092-8674(92)90643-q. [DOI] [PubMed] [Google Scholar]
- 125.Gallant HL, Yatscoff RW. P70 S6 kinase assay: a pharmacodynamic monitoring strategy for rapamycin; assay development. Transplant Proc. 1996;28:3058–3061. [PubMed] [Google Scholar]
- 126.Clark K. Monitoring antithymocyte globulin in renal transplantation. Ann Roy Coll Surg Engl. 1996;78:536–540. [PMC free article] [PubMed] [Google Scholar]
- 127.Bogaert M. [Drug policy 1996]. [Dutch] Verh K Acad Geneeskd Belg. 1997;59:227–236. [PubMed] [Google Scholar]
- 128.Ensom MH, Davis GA, Cropp CD, Ensom RJ. Clinical pharmacokinetics in the 21st century. Does the evidence support definitive outcomes? Clin Pharmacokinet. 1998;34:265–279. doi: 10.2165/00003088-199834040-00001. [DOI] [PubMed] [Google Scholar]
- 129.Destache CJ. Use of therapeutic drug monitoring in pharmacoeconomics. Ther Drug Monit. 1993;15:608–610. doi: 10.1097/00007691-199312000-00028. [DOI] [PubMed] [Google Scholar]
- 130.Shaw LM, Kaplan B, Brayman KL. Prospective investigations of concentration-clinical response for immunosuppressive drugs provide the scientific basis for therapeutic drug monitoring. Clin Chem. 1998;44:381–387. [PubMed] [Google Scholar]
- 131.Johnston A, Holt DW. Concentration-controlled trials. What does the future hold? Clin Pharmacokinet. 1995;28:93–99. doi: 10.2165/00003088-199528020-00001. [DOI] [PubMed] [Google Scholar]
- 132.Laskow DA, Vincenti F, Neylan JF, Mendez R, Matas AJ. An open-label, concentration-ranging trial of FK506 in primary kidney transplantation: a report of the United States Multicenter FK506 Kidney Transplant Group. Transplantation. 1996;62:900–905. doi: 10.1097/00007890-199610150-00005. [DOI] [PubMed] [Google Scholar]
- 133.Ponticelli C, Tarantino A. Immunosuppressive protocols for renal transplantation. Nephrol Dial Transplant. 1997;12(Supplement 1):45–50. [PubMed] [Google Scholar]
- 134.Johnston A, Holt DW. Generic substitution for cyclosporine: What should we be looking for in a new formulation. Transplant Proc. 1998;30:1652–1653. doi: 10.1016/s0041-1345(98)00377-7. [DOI] [PubMed] [Google Scholar]
- 135.Al Meshal MA, Khidr SH, Bayomi MA, Al Angary AA. Oral administration of liposomes containing cyclosporine: A pharmacokinetic study. Int J Pharmaceutics. 1998;168:163–168. [Google Scholar]
- 136.Troncoso P, Kahan BD. Preclinical evaluation of a new immunosuppressive agent, FTY720. Clin Biochem. 1998;31:369–373. doi: 10.1016/s0009-9120(98)00043-5. [DOI] [PubMed] [Google Scholar]
- 137.Schuler W, Sedrani R, Cottens S, et al. SDZ RAD, a new rapamycin derivative: pharmacological properties in vitro and in vivo. Transplantation. 1997;64:36–42. doi: 10.1097/00007890-199707150-00008. [DOI] [PubMed] [Google Scholar]
- 138.Bach FH. Xenotransplantation: problems and prospects. Ann Rev Med. 1998;49:301–310. doi: 10.1146/annurev.med.49.1.301. [DOI] [PubMed] [Google Scholar]