

Abstract
Aims
In order to avoid the potential for elevated serum lipid levels as a consequence of long term sedation with propofol, a formulation of propofol 6% in Lipofundin® MCT/LCT 10% (Propofol 6% SAZN) has been developed. The pharmacokinetics, induction of anaesthesia and safety characteristics of this new formulation were investigated after bolus injection and were compared with the commercially available product (propofol 1% in Intralipid® 10%, Diprivan®-10) and propofol 1% in Lipofundin® MCT/LCT 10% (Propofol 1% SAZN).
Methods
In a randomised double-blind study, 24 unpremedicated female patients received an induction dose of propofol of 2.5 mg kg−1 over 60 s which was followed by standardized balanced anaesthesia. The patients were randomized to receive propofol as Propofol 6% SAZN, Propofol 1% SAZN or Diprivan®-10.
Results
For all formulations the pharmacokinetics were adequately described by a tri-exponential equation, as the propofol concentrations collected early after the injection suggested an additional initial more rapid phase. The average values for clearance (CL), volume of distribution at steady-state (Vd,ss), elimination half-life (t1/2,z) and distribution half-life (t1/2,λ2) observed in the three groups were 32±1.5 ml kg−1 min−1, 2.0±0.18 l kg−1, 95±5.6 min and 3.4±0.20 min, respectively (mean±s.e.mean, n = 24) and no significant differences were noted between the three formulations (P > 0.05). The half-life of the additional initial distribution phase (t1/2,λ1) in all subjects ranged from 0.1 to 0.6 min. Anaesthesia was induced successfully and uneventfully in all cases, and the quality of induction was adequate in all 24 patients. The induction time did not vary between the three formulations and the average induction time observed in the three groups was 51±1.3 s which corresponded to an induction dose of propofol of 2.1±0.06 mg kg−1 (mean±s.e.mean, n = 24). The percentage of patients reporting any pain on injection did not vary between the formulations and was 17% for the three groups. No postoperative phlebitis or other venous sequelae of the vein used for injection occurred in any of the patients at recovery of anaesthesia nor after 24 h.
Conclusions
From the above results, we conclude that the alteration of the type of emulsion and the higher concentration of propofol in the new parenteral formulation of propofol does not affect the pharmacokinetics and induction characteristics of propofol, compared with the currently available product. Propofol 6% SAZN can be administered safely and has the advantage of a reduction of the load of fat and emulsifier which may be preferable when long term administration of propofol is required.
Keywords: anaesthetics i.v., propofol, pharmacokinetics, induction characteristics, pharmacology, emulsion formulation
Introduction
Propofol was introduced originally as an intravenous anaesthetic agent for induction and maintenance of anaesthesia and has, until now, extensively been used for this purpose. Propofol is also applied as a sedative agent to accompany either local or regional anaesthesia and is nowadays widely used as an agent to sedate critically ill patients in the Intensive Care Unit (ICU) [1, 2]. Since propofol is formulated in a concentration of 1% in Intralipid® 10% (Diprivan®-10), long term infusions are associated with a progressive increase in serum lipid levels, particularly triglycerides, which is especially of concern in critically ill patients, patients with liver disease or children [2–5]. Infusions for 7 days or more resulted in serum triglyceride levels that were 3 to 4 times normal values and after propofol cessation, serum lipid levels return to normal only after several days [4]. Furthermore, case reports of fatal myocardial failure and metabolic acidosis in children sedated with propofol in the ICU, describe elevated serum lipid levels [6–8]. Additionally, the emulsifier in the lipid emulsion of Diprivan®-10, egg-lecithin, converts to lyso-lecithin which has in vitro haemolytic properties. This compound has been held responsible for the haemolytic effect of Diprivan®-10 in pathologic situations [9].
As the load of lipids and emulsifier can be decreased by using more concentrated formulations of propofol, we developed a new parenteral formulation of propofol 6% in Lipofundin® MCT/LCT 10% (Propofol 6% SAZN). Lipofundin® MCT/LCT 10% is a 10% fat emulsion consisting of medium-chain triglycerides (MCT) and long-chain triglycerides (LCT), whereas Intralipid® 10% contains long-chain triglycerides, exclusively. Changes in the formulation may have an impact on the pharmacokinetics, pharmacodynamics or safety characteristics of a drug, which has previously been demonstrated for propofol [10–13]. As the new formulation Propofol 6% SAZN contains a different type of fat emulsion and has a six times higher concentration of propofol, a preclinical study by Cox et al. [14] investigated the influence of these two alterations in the formulation on the pharmacokinetics and pharmacodynamics of propofol in the rat. In this preclinical study, the difference in the type of fat emulsion and the higher concentration of propofol in the new formulation were not found to affect the pharmacokinetics and pharmacodynamics of propofol [14].
The objectives of the present study are to investigate the pharmacokinetics as well as the induction of anaesthesia and safety characteristics of Propofol 6% SAZN after a single bolus injection in man, and to compare these findings with those of Propofol 1% SAZN (Propofol 1% in Lipofundin® MCT/LCT 10%) and Diprivan®-10 (Propofol 1% in Intralipid® 10%). The formulation Propofol 1% SAZN is added as a third study arm in order to be able to distinguish between the influence of either the concentration of propofol in the fat emulsion or the type of fat emulsion on the pharmacokinetics, induction of anaesthesia and safety characteristics, as Propofol 1% SAZN contains the same concentration of propofol as Diprivan®-10 (1%) and the type of fat emulsion used in Propofol 6% SAZN (Lipofundin® MCT/LCT 10%).
Methods
The investigation was approved by the ethics committee of the St Antonius Hospital, Nieuwegein, The Netherlands. All patients gave their consent in writing after being informed of the aims, nature and procedures of the study.
Patients
Twenty-four women scheduled to undergo a gynaecological operation participated in the randomised double-blind study. Patients were included if they were between 20 and 65 years of age, were classed as American Society of Anesthesiologists (ASA) physical status I or II, weighed between 50 and 85 kg and had normal renal (creatinine concentration < 105 μmol l−1) and hepatic function (liver function tests in normal range) when assessed by routine laboratory testing. Patients were excluded if they were suffering from cardiac disease, impairment of lipid metabolism (triglycerides >1.5 mmol l−1 and/or cholesterol >6.5 mmol l−1) or if they had taken medication with sedative properties.
Study design and procedure
Anaesthesia was induced with 2.5 mg propofol kg−1 bodyweight over 60 s into a fast running saline infusion in an antecubital vein, delivered by a Graseby Medical 3400 infusion pump. No preanaesthetic medication was given. Patients were randomised to receive propofol as a 6% formulation in Lipofundin® MCT/LCT 10% (Propofol 6% SAZN), a 1% formulation in Lipofundin® MCT/LCT 10% (Propofol 1% SAZN) or the commercially available 1% formulation in Intralipid® 10% (Diprivan®-10). Following onset of unconsciousness, fentanyl (0.003-0.005 mg kg−1) and atracurium (0.3-0.6 mg kg−1) were administered, after which the patient was intubated and mechanically ventilated. Anaesthesia was maintained with 0.5-1% isoflurane in a mixture of nitrous oxide (60%) in oxygen (40%). A continuous intravenous infusion of atracurium (0.005 mg kg−1 min−1) was administered after the induction of anaesthesia until 20 min before the expected end of the procedure. During the surgical procedure, fentanyl was given as required in amounts of 50–100 μg. Muscle relaxation was reversed by neostigmine (1.5 mg) and atropine (0.5 mg) before extubation. The duration of anaesthesia was taken as the time interval from the start of the injection of propofol to the time of extubation.
Drugs
Propofol 6% SAZN and Propofol 1% SAZN were prepared in the Department of Clinical Pharmacy of the St Antonius Hospital, Nieuwegein, The Netherlands. Under aseptic conditions 6.7 ml and 1.1 ml of propofol 99.8% (Bufa BV, Uitgeest, The Netherlands) respectively were added to 100 ml of Lipofundin® MCT/LCT 10% through a sterile 0.2 m filter. The vial was shaken for 15 min on a Flask Shaker SF1 (Stuart Scientific, Redhill, Great Britain). Diprivan®-10 was obtained from Zeneca, Ridderkerk, The Netherlands.
Blood sampling
Samples of arterial blood (2 ml) were collected from an indwelling radial arterial cannula prior to the induction of anaesthesia, and at approximately 1, 1.5, 2, 2.5, 3, 4, 5, 8, 11, 15, 20, 30, 45, 60, 90, 120, 150 and 180 min after the start of the propofol injection, the exact time being recorded for each sample. Samples were collected in glass oxalate tubes, mixed thoroughly and were stored at 4° C until analysis.
Induction of anaesthesia
Patients were asked to count from the start of the propofol injection. The induction time was taken as the interval from the start of injection to cessation of counting. During the injection an overall subjective assessment of the quality of induction was made by the anaesthesiologist and rated as adequate or poor. After completion of the propofol injection the presence or absence of the eyelash reflex was noted.
Safety characteristics
All side effects occurring during the induction and maintenance of anaesthesia and recovery from anaesthesia were noted. In particular, any evidence of phlebitis or venous thrombosis at the site of drug administration was sought in the early recovery period and after the 24 h following anaesthesia. On the first postoperative day, the patients were questioned about pain associated with the injection of propofol and were invited to comment on their experience.
Drug assay
Propofol concentrations in whole blood samples were measured using high-performance liquid chromatography with fluorescence detection [15], with the following modifications. Three calibration curves were prepared: 0.05–0.5 mg l−1, 0.5–6 mg l−1 and 6–30 mg l−1. To 200 μl of undiluted blood 20 μl of methanol were added and vortexed. Then 250 μl of internal standard solution were added, vortexed and after centrifugation, 50 μl of the supernatant were analysed. The limit of quantification was 0.05 mg l−1. The coefficients of variation reported for intra- and inter-assay precision were less than 4.1% and 6.0%, respectively over the concentration range studied.
Pharmacokinetic data analysis
Pharmacokinetic analysis of each individual blood concentration versus time profile was performed using the data analysis program WinNonLin (Scientific Consulting, Inc., USA). The following equations were used in order to describe the blood concentration–time profiles after bolus injection:
 |
(1) |
 |
(2) |
where C(t) is the blood concentration of propofol at time t, C1, C2 and C3 are the coefficients and λ1, λ2, λ3 the exponents of the equation. The different models were investigated and tested according to the Akaike Information Criterion [16], to the precision of the parameter estimates obtained, as determined by their standard errors, and by visual assessment of the residuals of the measured concentrations from the lines of best fit. The values of various pharmacokinetic parameters were calculated from the fitted functions [17].
Statistical analysis
Statistical analysis of pharmacokinetic estimates and induction times of the different groups were performed using analysis of variance (ANOVA) by Epistat Statistical Package, version 3.0 (T.L. Gustafson, Wound Rock, TX, USA). A probability level of ≤5% was considered statistically significant.
Results
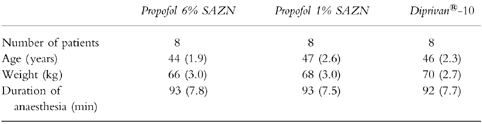
The mean age, weight and the duration of anaesthesia of the patients in the three groups are given in Table 1. Regarding these parameters no differences were noted between the groups studied.
Table 1.
Number of patients and mean (s.e.mean) age, weight and duration of anaesthesia of the patients receiving Propofol 6% SAZN, Propofol 1% SAZN and Diprivan®-10.
Pharmacokinetic analysis
The blood concentration versus time profiles of propofol in representative patients of the three different groups are presented in Figure 1. The data sets were all characterized by high peak concentrations and a very rapid distribution phase followed by the elimination phase. As can be seen in Figure 1, the profiles of the patients who received Propofol 6% SAZN corresponded well with the profiles of those who received Propofol 1% SAZN or Diprivan®-10. Furthermore, it is apparent from this figure that the profiles of all patients could adequately be described using a tri-exponential function. The profiles could not be represented by a two-exponential function as the propofol concentrations collected early after the injection all increased above the projected distribution phase which is suggestive of an additional, initial even more rapid phase. A tri-exponential function was superior over a two-exponential function for all data sets according to the criteria as defined in Methods. Using a tri-exponential function, the complete concentration–time profiles including the early samples, could adequately be represented.
Figure 1.
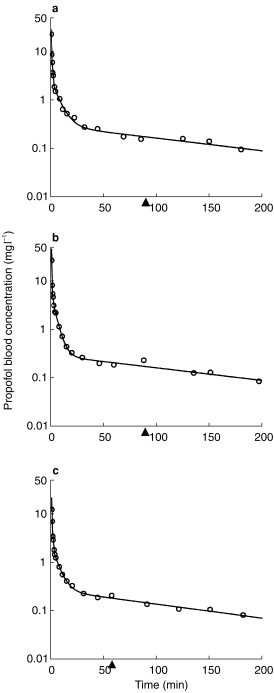
Propofol blood concentrations vs time profiles in three representative patients with the lines of best fit according to a tri-exponential pharmacokinetic model. The patients received 2.5 mg kg−1 propofol formulated as Propofol 6% SAZN (a), Propofol 1% SAZN (b) or Diprivan®-10 (c) over 60 s. The triangles (▴) indicate the moment of extubation.
The pharmacokinetic parameters of propofol were calculated for each patient in the three different groups and are summarized in Table 2 (see also Table 4). No significant differences in pharmacokinetic parameters were noted between the three formulations, except for the half-life of the additional very rapid distribution phase (t1/2,λ1). The average values for clearance (CL), volume of distribution at steady state (Vd,ss), elimination half-life (t1/2,z) and distribution half-life (t1/2,λ2) observed in the three formulations were 32±1.5 ml kg−1 min−1, 2.0±0.18 l kg−1, 95±5.6 min and 3.4±0.20 min, respectively (mean±s.e.mean, n = 24). The half-life of the additional distribution phase (t1/2,λ1) ranged from 0.1 to 0.6 minutes.
Table 2.
Pharmacokinetic parameter estimates (mean (s.e.mean)) of propofol for Propofol 6% SAZN, Propofol 1% SAZN and Diprivan®-10 on the basis of a tri-exponential pharmacokinetic model.
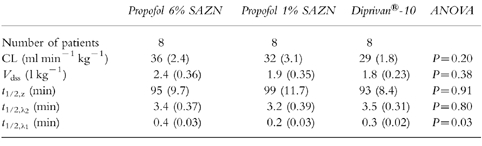
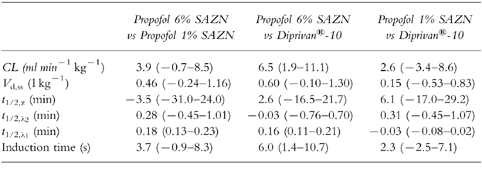
Table 4.
Pairwise comparison between Propofol 6% SAZN and Propofol 1% SAZN, between Propofol 6% SAZN and Diprivan®-10, and between Propofol 1% SAZN and Diprivan®-10 for each pharmacokinetic parameter estimate and the induction time (mean difference (95% confidence interval)).
In 16 of the 24 patients (67%) a rise in the propofol concentration was observed at the very moment of or directly after extubation of the patient. In Figure 1, the profiles in number b and c illustrate this phenomenon, as these profiles show a rise in propofol concentration at the moment of extubation.
Induction of anaesthesia
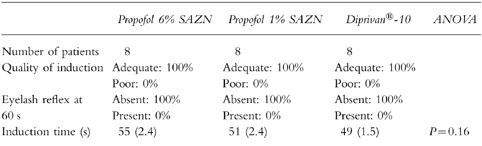
In all patients anaesthesia was induced successfully and uneventfully. The induction characteristics of the three formulations are summarized in Table 3 (see also Table 4). The quality of induction as assessed by the anaesthesiologist was adequate in all cases and the eyelash reflex was abolished after completion of the propofol induction dose in all 24 patients. No significant difference in induction time was observed between the three groups. The average induction time observed in the three groups was 51± 1.3 s which corresponded to a dose of propofol of 2.1±0.06 mg kg−1 (mean±s.e.mean, n = 24).
Table 3.
Induction characteristics of Propofol 6% SAZN, Propofol 1% SAZN and Diprivan®-10: quality of induction rated as adequate or poor (percentage of patients), presence or absence of the eyelash reflex after completion of the induction dose (percentage of patients) and induction time in s (mean (s.e.mean)).
Safety characteristics
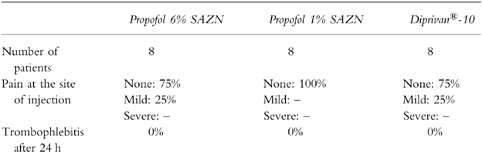
The safety characteristics of the three formulations are shown in Table 5. Mild pain at the site of injection during the bolus injection was reported by two patients who received Propofol 6% SAZN as well as by two patients who received Diprivan®-10. The overall percentage of pain on injection in the three groups was 17%. None of the patients showed evidence of postoperative phlebitis or other venous sequelae of the vein used for injection at recovery of anaesthesia nor after 24 h. No serious adverse events to any of the three propofol formulations were noted. One patient who received Propofol 6% SAZN reported pain in the shoulder/thorax which lasted until a few days after surgery. Lung embolism was excluded and an association with the injection of propofol seemed very unlikely.
Table 5.
Safety characteristics of Propofol 6% SAZN, Propofol 1% SAZN and Diprivan®-10.
Discussion
In order to reduce the lipid as well as the emulsifier load, we developed a new parenteral formulation of propofol with a six times higher concentration compared with the commercially available product. Beside the higher concentration, the new formulation Propofol 6% SAZN also contains a different type of fat emulsion. In this investigation we demonstrate that the pharmacokinetics, induction characteristics and safety characteristics after bolus injection of this new 6% formulation in Lipofundin® MCT/LCT 10% are similar to those of a 1% formulation in Lipofundin® MCT/LCT 10% and of Diprivan®-10. In contrast with our results, other studies have reported that changes in the formulation of propofol may have an impact on the pharmacokinetics, pharmacodynamics or safety characteristics [10–12]. In a comparative study of propofol in the Cremophor EL formulation versus an emulsion formulation, it has been shown that there is a slight loss of potency in the emulsion form when given for induction of anaesthesia. As a consequence the recommended induction dose of propofol has been elevated from 2.0 mg kg−1 to 2.5 mg kg−1 and interestingly less pain on injection has been observed when using emulsion formulation [10, 11, 18–20]. More recently, it has been shown in rats that propofol, when formulated in a lipid free vehicle (2% ethanol), exhibits a slower onset and a prolonged duration of effect. Thus the use of an emulsion formulation accelerates time to onset and maximal effect and enhances the safety of propofol compared to a lipid free vehicle [13]. Doenicke et al. [21] have demonstrated in healthy volunteers that changing the composition of the carrier fat emulsion for propofol to Lipofundin® MCT/LCT 10% does not have an impact on the pharmacokinetics and efficacy of propofol, but exhibits a better patient acceptance by lowering the incidence of pain on injection. The authors have attributed this result to a lower concentration of free propofol in the aqueous phase of the new formulation [21]. Unlike the above reports, our study does not demonstrate differences in pharmacokinetics or pharmacodynamics of propofol as a consequence of alterations in the formulation. The results found in this study in female patients do confirm the results of a preclinical study of Propofol 6% SAZN by Cox et al. [14]. Using the effect on the EEG as pharmacodynamic endpoint, it was found after bolus infusion as well as after a 5 h continuous infusion, that the pharmacokinetics and the pharmacodynamics of propofol were not affected by the type of emulsion or the concentration of propofol in the formulation. The present bolus injection study in female patients confirms these preclinical results in rats, as the pharmacokinetics and the induction characteristics of Propofol 6% SAZN proved to be identical to the commercially available product, containing 1% propofol in a different type of emulsion.
The pharmacokinetic parameter estimates we found in this study in female patients prior to and during gynaecological surgery are in good agreement with those found in other studies [1] except for the additional very rapid distribution phase. We found evidence for the occurrence of this additional early distribution phase for all three formulations. Although the pharmacokinetics of propofol have been extensively studied in man [1, 22–24], only one study reports a similar result [25]. We believe that this difference can be explained by the fact that we sampled arterial blood whereas all others used venous blood. In general, arterial sampling is preferred when pharmacokinetic parameters are studied of drugs which are rapidly distributed, because the steeper arterial concentration profile provides a more sensitive measure for the detection of early distribution phases [26, 27]. For propofol, Major et al. [28] measured significantly higher arterial than venous concentrations directly after bolus injection, which turned into the reverse after 1 min. Wang et al. [29] found similar differences in an infusion setting. They attributed this phenomenon to rapid and extensive uptake of propofol in muscle, fat and skin tissue. The shallower venous concentration profile may have caused the investigators who used venous sampling to fail to detect the early distribution phase. Adam et al. [25] did use venous sampling and found evidence for the early distribution phase in a very limited number of his patients. In many other patients, however, the early phases in his profiles were often uninterpretable, the reason for which, we think, is venous sampling. The half-life of the additional early distribution phase in our study ranged from 0.1 to 0.6 min which is in agreement with the findings of Adam et al. [25], describing in some of the patients a half-life of this additional phase ranging from 0.35 to 0.8 min. In contrast with the results of Adam et al. [25], in our study all arterially collected samples, of which a reasonable amount was taken closely after dosing, could be evaluated, and enabled a description of the additional rapid distribution phase.
Secondary peaks in the drug concentration profile occurring at the time of awakening have been observed previously for propofol [22, 23] and for other lipophilic intravenous anaesthetic agents such as fentanyl [30]. Since the majority of propofol eliminated from the blood during the first 2 h is accounted for by the uptake into peripheral compartments (62%) and not by metabolism [31] we hypothesized that alterations in cardiac output at awakening may have led to the release of propofol from lipid tissues [32].
Regarding the induction characteristics, the present study shows that the hypnotic effect of propofol at the induction of anaesthesia is not affected by the concentration of propofol nor the type of emulsion in the intravenous formulation, as the induction times did not vary significantly between the three different groups. Since it has been demonstrated that the induction time of propofol depends on the dose as well as the rate of injection, our results are in agreement with the observations described previously [11, 20, 33]. The significantly larger time delay between propofol blood concentration and effect (t1/2,keo) observed for Propofol 6% SAZN by Cox et al. [14] in the rat using the effect on the EEG as pharmacodynamic endpoint, was not confirmed in the present study, using the induction time as pharmacodynamic endpoint. This t1/2,keo was one of the eight pharmacodynamic parameters which were derived from the continuous EEG measurements and pharmacokinetic findings, and the only parameter being significantly different for Propofol 6% SAZN. It seems that this larger time delay between concentration and effect in the rat, does not lead to changes in a clinically relevant endpoint in man, such as the time of induction of anaesthesia.
The present study also demonstrates that injection of a higher concentration of propofol (6%) is not associated with a higher incidence of pain at the side of injection when propofol is formulated in Lipofundin® MCT/LCT 10%. Pain on injection is the most common adverse event associated with the use of propofol. Pain of any severity has been reported in 25 to 74% of adult patients, the incidence being higher when propofol was injected into veins on the dorsum of the hand [1, 2]. In this study, the overall incidence of pain on injection is substantially lower (17%) which is probably due to the fact that propofol was infused into a fast running saline infusion in a large antecubital vein. Klement & Arndt [34] and Doenicke et al. [35] have demonstrated that pain on injection is related to the concentration of propofol in the aqueous phase of the emulsion. Babl et al. [36] reported that emulsions with 50% medium chain triglycerides (MCT) in the dispersed oil phase result in significantly lower propofol concentrations in the aqueous phase compared to those containing vegetable oils with long chain triglycerides (LCT) only. Doenicke et al. [21] also showed that after propofol 1% in Lipofundin® MCT/LCT 10%, fewer volunteers reported severe or moderate pain on injection (9%) than after the standard formulation of propofol 1% in Intralipid® 10% (59%) (P < 0.05). Therefore, the presence of 50% MCT in our formulation of propofol 6% may have contributed to the relatively low incidence of pain on injection, in spite of the higher overall concentration of propofol. Another contributing factor may have been the smaller volume of injection of the 6% formulation allowing for a faster dilution of propofol by circulating blood.
In conclusion, the pharmacokinetics, induction of anaesthesia and safety characteristics of Propofol 6% SAZN after bolus injection are in agreement with those of Propofol 1% SAZN and Diprivan®-10. Alteration of the type of emulsion and the higher concentration of propofol in the new parenteral formulation of propofol does not affect the pharmacokinetics, induction characteristics or safety profile of propofol. As a consequence, the higher concentration of propofol in Propofol 6% SAZN will reduce the load of fat and emulsifier, which may be an advantage over the commercially available product when long term administration of propofol is required, especially in critically ill patients, patients with liver disease or children. Further investigations are required to confirm the safety and advantages of Propofol 6% SAZN during long term administration in the Intensive Care Unit.
Acknowledgments
The authors wish to thank the various gynaecologists and their staff for their cooperation and patience; the anaesthesiologists for allowing us to study the patients under their care; all staff in the Operating and Recovery Rooms for their assistance; all staff in the Department of Clinical Pharmacy, in particular L. Lie-A-Huen, for their help and cooperation; the Department of Clinical Chemistry, in particular F. J. L. M. Haas, for their cooperation; V. S. Koster for all her preparatory work; E. H. Cox for all pharmacokinetic discussions; P. M. Berkemeyer for her technical assistance.
References
- 1.Bryson HM, Fulton BR, Faulds D. Propofol. An update of its use in anaesthesia and conscious sedation. Drugs. 1995;50:513–559. doi: 10.2165/00003495-199550030-00008. [DOI] [PubMed] [Google Scholar]
- 2.Fulton B, Sorkin EM. Propofol. An overview of its pharmacology and a review of its clinical efficacy in intensive care sedation. Drugs. 1995;50:636–657. doi: 10.2165/00003495-199550040-00006. [DOI] [PubMed] [Google Scholar]
- 3.Boyle WA, Shear JM, White PF, Schuller D. Tolerance and hyperlipidemia during long-term sedation with propofol [abstract] Anaesthesiology. 1990;73(Suppl. 3A):A245. [Google Scholar]
- 4.Cook S, Palma O. Propofol as a sole agent for prolonged infusion in intensive care. J Drug Dev. 1989;2(Suppl. 2):65–67. [Google Scholar]
- 5.Gottardis M, Khünl-Brady KS, Koller W, Sigl G, Hackl JM. Effect of prolonged sedation with propofol on serum triglyceride and cholesterol concentrations. Br J Anaesth. 1989;62:393–396. doi: 10.1093/bja/62.4.393. [DOI] [PubMed] [Google Scholar]
- 6.Parke TJ, Stevens JE, Rice ASC, et al. Metabolic acidosis and fatal myocardial failure after propofol infusion in children: five case reports. Br Med J. 1992;305:613–616. doi: 10.1136/bmj.305.6854.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Strickland RA, Murray MJ. Fatal metabolic acidosis in a pedriatic patient receiving an infusion of propofol in the intensive care unit: is there a relationship? Crit Care Med. 1995;23:405–409. doi: 10.1097/00003246-199502000-00029. [DOI] [PubMed] [Google Scholar]
- 8.Plötz FB, Waalkens HJ, Verkade HJ, Strengers JLM, Knoester H, Mandema JM. Fatal side-effects of continuous propofol infusion in children may be related to malignant hyperthermia. Anaesth Intensive Care. 1996;24:724–731. doi: 10.1177/0310057X9602400619. [DOI] [PubMed] [Google Scholar]
- 9.Pharma Zeneca., editor. Repertorium. Edition 1998/1999. Utrecht, The Netherlands: Nefarma & Neprofarm; 1998. Product information Diprivan®-10; pp. 916–918. [Google Scholar]
- 10.Glen JB, Hunter SC. Pharmacology of an emulsion formulation of ICI 35868. Br J Anaesth. 1984;56:617–625. doi: 10.1093/bja/56.6.617. [DOI] [PubMed] [Google Scholar]
- 11.Cummings GC, Dixon J, Kay NH, et al. Dose requirements of ICI 35,868 (Propofol, ‘Diprivan’) in a new formulation for induction of anaesthesia. Anaesthesia. 1984;39:1168–1171. doi: 10.1111/j.1365-2044.1984.tb06425.x. [DOI] [PubMed] [Google Scholar]
- 12.Dutta S, Matsumoto Y, Ebling WF. Propofol pharmacokinetics and pharmacodynamics assessed from a cremophor EL formulation. J Pharm Sci. 1997;86:967–969. doi: 10.1021/js970118m. [DOI] [PubMed] [Google Scholar]
- 13.Dutta S, Ebling WF. Emulsion formulation reduces propofol dose requirements and enhances safety. Anesthesiology. 1997;87:1394–1405. doi: 10.1097/00000542-199712000-00019. [DOI] [PubMed] [Google Scholar]
- 14.Cox EH, Knibbe CAJ, Koster VS, et al. Influence of different fat emulsion-based intravenous formulations on the pharmacokinetics and pharmacodynamics of propofol. Pharm Res. 1998;15:442–448. doi: 10.1023/a:1011980432646. [DOI] [PubMed] [Google Scholar]
- 15.Knibbe CAJ, Koster VS, Deneer VHM, Stuurman RM, Kuks PFM, Lange R. Determination of propofol in low volume samples by high-performance liquid chromatography with fluorescence detection. J Chromatogr B Biomed Sci Appl. 1998;706:305–310. doi: 10.1016/s0378-4347(97)00571-9. [DOI] [PubMed] [Google Scholar]
- 16.Akaike H. A new look at the statistical model identification. IEEE Trans Automat Control. 1974;AC-19:716–723. [Google Scholar]
- 17.Gibaldi M, Perrier D. Noncompartmental analysis based on statistical moment theory. In: Gibaldi M, Perrier D, editors. Pharmacokinetics. 2. New York: Marcel Dekker; 1982. pp. 409–424. [Google Scholar]
- 18.Rutter BV, Morgan M, Lumley J, Owen R. ICI 35868 (Diprivan): a new intravenous induction agent. A comparison with methohexitone. Anaesthesia. 1980;35:1188–1192. doi: 10.1111/j.1365-2044.1980.tb05076.x. [DOI] [PubMed] [Google Scholar]
- 19.Briggs LP, Clarke RSJ, Watkins J. An adverse reaction to the administration of disoprofol (Diprivan) Anaesthesia. 1982;37:1099–1101. doi: 10.1111/j.1365-2044.1982.tb01753.x. [DOI] [PubMed] [Google Scholar]
- 20.De Grood PMRM, Coenen LGJ, van Egmond J, Booij LHDJ, Crul JF. Propofol emulsion for induction and maintenance of anaesthesia. A combined technique of general and regional anesthesia. Acta Anaesthesiol Scand. 1987;31:219–223. doi: 10.1111/j.1399-6576.1987.tb02554.x. [DOI] [PubMed] [Google Scholar]
- 21.Doenicke AW, Roizen MF, Rau J, et al. Pharmacokinetics and pharmacodynamics of propofol in a new solvent. Anesth Analg. 1997;85:1399–1403. doi: 10.1097/00000539-199712000-00040. [DOI] [PubMed] [Google Scholar]
- 22.Cockshott ID, Briggs LP, Douglas EJ, White M. Pharmacokinetics of propofol in female patients. Studies using single bolus injections. Br J Anaesth. 1987;59:1103–1110. doi: 10.1093/bja/59.9.1103. [DOI] [PubMed] [Google Scholar]
- 23.Kay NH, Sear JW, Uppington J, Cockshott ID, Douglas EJ. Disposition of propofol in patients undergoing surgery. A comparison in men and women. Br J Anaesth. 1986;58:1075–1079. doi: 10.1093/bja/58.10.1075. [DOI] [PubMed] [Google Scholar]
- 24.Gin T, Gregory MA, Chan K, Buckley Buckley, Oh TE. Pharmacokinetics of propofol in women undergoing elective caesarean section. Br J Anaesth. 1990;64:148–153. doi: 10.1093/bja/64.2.148. [DOI] [PubMed] [Google Scholar]
- 25.Adam HK, Briggs LP, Bahar M, Douglas EJ, Dundee JW. Pharmacokinetic evaluation of ICI 35868 in man. Single induction doses with different rates of injection. Br J Anaesth. 1983;55:97–103. doi: 10.1093/bja/55.2.97. [DOI] [PubMed] [Google Scholar]
- 26.Chiou WL. New physiologically based methods for calculating the apparent steady-state volume of distribution in pharmacokinetic studies. Int J Clin Pharmacol Ther Toxicol. 1982;20:255–258. [PubMed] [Google Scholar]
- 27.Chiou WL. The phenomenon and rationale of marked dependence of drug concentration on blood sampling site: implications in pharmacokinetics, pharmacodynamics, toxicology and therapeutics (Part I) Clin Pharmacokin. 1989;17:175–199. doi: 10.2165/00003088-198917030-00004. [DOI] [PubMed] [Google Scholar]
- 28.Major E, Aun C, Yate PM, et al. Influence of sample site on blood concentrations of ICI 35868. Br J Anaesth. 1983;55:371–375. doi: 10.1093/bja/55.5.371. [DOI] [PubMed] [Google Scholar]
- 29.Wang YP, Cheng YJ, Fan SZ, Liu CC. Arteriovenous concentration differences of propofol during and after a stepdown infusion. Anesth Analg. 1994;79:1148–1150. doi: 10.1213/00000539-199412000-00021. [DOI] [PubMed] [Google Scholar]
- 30.McQuay HJ, Moore RA, Paterson GMC, Adams AP. Plasma fentanyl concentrations and clinical observations during and after operation. Br J Anaesth. 1979;51:543–550. doi: 10.1093/bja/51.6.543. [DOI] [PubMed] [Google Scholar]
- 31.Campbell GA, Morgan DJ, Kumar K, Crankshaw DP. Extended blood collection period required to define distribution and elimination kinetics of propofol. Br J Clin Pharmacol. 1988;26:187–190. doi: 10.1111/j.1365-2125.1988.tb03386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Elfstrom J. Drug pharmacokinetics in the postoperative period. Clin Pharmacokin. 1979;4:16–22. doi: 10.2165/00003088-197904010-00002. [DOI] [PubMed] [Google Scholar]
- 33.Rolly G, Versichelen L, Huyghe L, Mungroop H. Effect of speed of injection on induction of anaesthesia using propofol. Br J Anaesth. 1985;57:743–746. doi: 10.1093/bja/57.8.743. [DOI] [PubMed] [Google Scholar]
- 34.Klement W, Arndt JO. Pain on injection of propofol; effects of concentrations and diluent. Br J Anaesth. 1991;67:281–284. doi: 10.1093/bja/67.3.281. [DOI] [PubMed] [Google Scholar]
- 35.Doenicke AW, Roizen MF, Rau J, Kellermann W, Babl J. Reducing pain during propofol injection: the role of the solvent. Anesth Analg. 1996;82:472–474. doi: 10.1097/00000539-199603000-00007. [DOI] [PubMed] [Google Scholar]
- 36.Babl J, Doenicke A, Mönch V. New formulation of propofol in an LCT/MCT emulsion. EHP. 1995;1:15–21. [Google Scholar]