

Abstract
Aims
The objective of the study was to determine the effect of multiple doses of rifampicin on the steady-state pharmacokinetics of zidovudine and its 5′-glucuronosyl (GZDV) and 3′-amino (AMT) metabolites.
Methods
Eight asymptomatic HIV-infected patients (seven male, one female) participated in this three-period longitudinal study. Each patient received zidovudine (200 mg every 8 h) for 14 days (period 1), followed by rifampicin (600 mg every 24 h) with zidovudine for 14 days (period 2), and then zidovudine alone for a further 14 days (period 3). Blood and urine samples were collected over 6 h on the last day of each period for measurements of zidovudine and GZDV by h.p.l.c.-u.v. and AMT by h.p.l.c.-m.s-m.s.
Results
Compared with zidovudine-alone values in period 1, 14 days of coadministration with rifampicin significantly increased zidovudine oral clearance (89%) and formation clearances to GZDV (100%) and AMT (82%). Correspondingly, there were decreases in maximum plasma concentration (43%), AUC (47%) and urine recovery (37%) of zidovudine. GZDV/zidovudine and AMT/zidovudine AUC ratios increased by 99% and 36%, respectively, despite a significant 29% decrease in AMT AUC. After stopping rifampicin for 14 days, values of these pharmacokinetic parameters returned to within 26% of baseline. Over the three periods AMT plasma levels were < 18 ng ml−1 (n = 6) and < 40 ng ml−1 (n = 2), and molar AMT/zidovudine AUC ratios ranged from 1.7% to 4.5%.
Conclusions
Rifampicin induced zidovudine glucuronidation and amination pathways resulting in decreased plasma and urine exposures to zidovudine. AMT plasma exposure decreased because induction was more pronounced for the major GZDV metabolite. The magnitude of the residual inductive effect was minimal at 14 days after stopping rifampicin.
Keywords: 3′-amino-3′-deoxythymidine, drug interaction, induction, pharmacokinetics, rifampicin, zidovudine
Introduction
Patients infected with human immunodeficiency virus (HIV) are often coinfected with Mycobacterium tuberculosis such that concomitant therapy of M. tuberculosis and HIV is likely [1]. Therefore, the effective management of tuberculosis in HIV disease requires an awareness of potential drug interactions between antituberculosis and antiretroviral agents [2]. Rifampicin is the main drug used in antituberculosis drug regimens. Presently, rifampicin therapy in HIV-associated tuberculosis is limited to coadministration with a single class of HIV antiretroviral agents—the nucleoside reverse transcriptase inhibitors, of which one drug is zidovudine. Rifampicin is contraindicated in patients who are undergoing HIV therapy with combination regimens that include the other two classes of approved antiviral drugs–the protease inhibitors and non-nucleoside reverse transcriptase inhibitors [3]. The induction by rifampicin of the cytochrome P450 (CYP) 3 A subfamily of isoenzymes, which is the major isoenzyme involved in the metabolism of protease inhibitors and non-nucleoside reverse transcriptase inhibitors, causes potentially subtherapeutic levels of these drugs during concurrent therapy [3].
Zidovudine remains an important antiretroviral agent in combination with the other reverse transcriptase inhibitors: didanosine, lamivudine or zalcitabine. Following oral administration, zidovudine is rapidly absorbed and undergoes metabolism by three important pathways: hepatic 5′-glucuronidation to its major metabolite GZDV, intracellular 5′-phosphorylation to mono-, di- and tri-phosphates, and hepatic 3′-reduction of the azide moiety to 3′-amino-3′-deoxythymidine (AMT) [4]. AMT is fivefold to sevenfold more cytotoxic in vitro than zidovudine [5]. A small amount of AMT 5′-glucuronide (GAMT) appears to be formed from GZDV [6]. Uridine diphosphate-glucuronosyl transferase (UGT) in the liver catalyses the glucuronidation of zidovudine [7].
The formation of AMT appears to be mediated by CYP isoenzymes and cytochrome b5 together with their corresponding reductases [8–10]. AMT formation is increased in liver microsomes from rats pretreated with the CYP enzyme inducers phenobarbitone, dexamethasone or clofibrate [9, 10]. The effect of rifampicin, another inducer of CYP, on AMT formation in vivo is unknown.
Rifampicin is a known inducer of specific isoforms of the UGT enzyme system in animals [11–13] and in vitro in human hepatoma cell lines [14, 15]. Clinical evidence that rifampicin induces UGT remains sparse. The inductive effect of rifampicin on the UGT responsible for zidovudine glucuronidation in humans is controversial. Data from a small parallel study suggested that rifampicin induced zidovudine glucuronidation in four HIV-infected individuals receiving coadministration of rifampicin and zidovudine compared with 69 patients receiving zidovudine monotherapy [16]. Another study in 10 healthy volunteers indicated that 10 days of rifampicin pretreatment decreased plasma exposure of a single dose of zidovudine by about 50% [17, 18]. However, the authors speculated that rifampicin did not induce glucuronidation of zidovudine [18].
Induction of zidovudine metabolism will result in lower plasma concentrations of zidovudine, potentially leading to reduced antiviral activity if intracellular triphosphate concentrations are also decreased. Lower zidovudine levels may also decrease drug-induced toxicity depending on the metabolic pathway(s) induced. However, higher AMT levels may occur, possibly with associated increased cytotoxicity, if this pathway is induced (or if GZDV formation is inhibited).
Whether rifampicin induces GZDV, AMT or GAMT formation has not been confirmed. The objectives of the present study were therefore to evaluate the effect of multiple doses of rifampicin on the steady-state pharmacokinetics of zidovudine and its metabolites GZDV and AMT in HIV-infected patients, and to determine the magnitude of the residual inductive effect of rifampicin once rifampicin has been discontinued for 2 weeks.
Methods
Materials
3′-Azido-3′-deoxythymidine (zidovudine), 3′-azido-3′-deoxythymidine β-d-glucuronide (GZDV) sodium salt, 3′-amino-3′-deoxythymidine (3-AMT), and 5′-amino-5′-deoxythymidine (5-AMT) were obtained from Sigma Chemical Co. (St Louis, MO, USA). BW-A22U, a diastereoisomer of zidovudine, was a gift from Wellcome Research Laboratories (Research Triangle Park, NC, USA).
Patient selection
Eight asymptomatic patients (seven male, one female) infected with HIV (CD4 T lymphocytes < 600 cells/mm3) receiving zidovudine (200 mg every 8 h) for >16 weeks participated in the study. At that time, all patients were taking only a combination of zidovudine and zalcitabine (0.75 mg every 8 h) for antiretroviral therapy. Two individuals with CD4 counts < 200 cells/mm3 were concurrently taking cotrimoxazole for prophylaxis of Pneumocystis carinii pneumonia. Two other persons were also receiving acyclovir/trazadone and salbutamol/beclomethasone, respectively. All participants provided written informed consent. The Research Ethics Board of the Ottawa General Hospital approved the study. Exclusion criteria were age younger than 18 years, weight deviating more than 20% from ideal, pregnancy, allergy to rifamycin drugs, results of liver function tests that were more than three times the upper limit of normal, serum creatinine levels >150 μmol l−1, serum albumin levels < 37 g l−1, haemoglobin levels < 100 g l−1, gastrointestinal disease based on physical examination and/or history, active opportunistic disease, or coadministration of drugs known to interfere with the metabolism of zidovudine.
Study design
We conducted a two-treatment, three-period, single sequence, repeated-measures study with a 14 day duration in each period. In period 1, patients received a 200 mg dose of zidovudine (two 100 mg capsules of Retrovir; Burroughs Wellcome, Kirkland, Quebec) every 8 h, followed in period 2 by 600 mg rifampicin (two 300 mg capsules of Rifadin) every 24 h with zidovudine. Rifampicin was ingested simultaneously with the morning dose of zidovudine. In period 3, all patients discontinued rifampicin therapy and maintained zidovudine therapy. On day 14 of each regimen, after fasting for 10 h overnight, patients reported to the Clinical Investigation Unit, Ottawa General Hospital. Participants were studied 8 h after their evening dose of zidovudine. Medications were taken with 150 ml of tap water. Food was allowed 4 h after zidovudine administration. Serial blood and urine samples were collected over 6 h for measurements of zidovudine, GZDV, AMT and rifampicin.
Blood and urine sampling
Blood samples (5 ml) were collected in Vacutainer tubes containing ethylenediamine tetraacetic acid, immediately before drug administration and at 10, 20, 30 and 45 min, and at 1, 1.5, 2, 2.5, 3, 4, 5 and 6 h after dosing. Each sample was allowed to equilibrate for 15 min and then centrifuged (500 g, 24° C, 10 min). A solution of ascorbic acid (0.05 ml of 5 mg ml−1) was added to a separate aliquot of plasma for rifampicin analysis to prevent oxidation of the drug [19]. Immediately after centrifugation, plasma samples for measurement of zidovudine and its metabolites were heated at 58° C for 30 min to inactivate HIV and were stored in polypropylene tubes at −80° C until analysis could be performed. Zidovudine, GZDV and AMT are stable in plasma or serum heated at 58° C for at least 1 h [20, 21]. Patients were requested to completely void their urine before receiving zidovudine, and urine samples were obtained over the intervals of 0–2, 2–4 and 4–6 h after administration. The total volume of urine voided within each interval was recorded, and a 10 ml aliquot of each sample was stored at −80° C until analysis could be conducted.
Zidovudine analysis
A solution of internal standard (A22U) in distilled water (0.5 ml) was added to plasma samples (0.5 ml) and urine samples (0.2 ml). The urine samples were diluted to 1 ml with water. Samples were applied to an Extrelut QE column (1 ml capacity), which was eluted twice with 4 ml aliquots of dichloromethane:isopropanol (9:1, vol:vol). The combined organic extracts were evaporated at 40° C under nitrogen and the residue was reconstituted in mobile phase. An aliquot (0.01 of 1 ml for urine and 0.05 of 0.25 ml for plasma samples) was injected onto two Supelcosil C-18 h.p.l.c. columns coupled in series (150 and 75×4.6 mm; particle size 3 μm). The h.p.l.c. column was maintained at 40° C and was eluted at 1 ml min−1 with a mobile phase of acetonitrile:aqueous (12:88; vol:vol). The aqueous phase contained 0.4 ml concentrated H3PO4 diluted to 1 l with water (pH 2.4). Ultraviolet detection was at 267 nm. Retention times for zidovudine and A22U were 11 and 10 min, respectively. The lower limit of quantification (LOQ) was 2 μg ml−1 (urine) and 20 ng ml−1 (plasma).
GZDV analysis
Urine samples (0.1 ml) were diluted at least 50 times with deionized water or 100-fold diluted control urine to maintain a urine background similar to that of the standards, and analysed for GZDV by direct injection (0.02 ml) into the h.p.l.c. system described above. The retention time for GZDV was 7 min and the LOQ was 200 ng ml−1 for diluted samples.
For plasma samples, an aliquot (0.5 ml) was mixed with A22U in water (0.5 ml) and added to a Varian 500 mg C-18 SPE column, which was previously activated with 2 ml methanol and prewashed twice with 2 ml phosphate-buffered saline (pH 7.2). The column was washed with 1.5 ml of saline and GZDV was eluted with 1.5 ml methanol. The solvent was evaporated at 40° C under nitrogen and the residue was reconstituted in 0.25 ml mobile phase, of which 0.05 ml was injected into the h.p.l.c. system described above. The LOQ was 100 ng ml−1.
3-AMT analysis
Plasma samples (0.5 ml) were mixed with 0.1 m potassium dihydrogen phosphate solution (0.5 ml, adjusted to pH 5 with potassium hydroxide) and internal standard solution containing 300 ng ml−1 5-AMT (0.02 ml). 3-AMT was isolated by solid-phase extraction on 500-mg Bond Elut SCX cation exchange columns according to the procedure of Burger et al. [20], and quantified by h.p.l.c. with tandem mass spectrometric detection (h.p.l.c.-m.s.-m.s.). The two isomers were separated on a Supelcosil LC-ABZ h.p.l.c. column (2.1×250 mm; particle size 5 μm) with an isocratic mobile phase of methanol and water (2:98; vol:vol), each containing 5 mm ammonium acetate and ammonium hydroxide. The pH of the water mobile phase was adjusted to 7.0 with 1 m ammonium hydroxide solution. An identical volume of ammonium hydroxide solution was also added to the methanol mobile phase. Column flow was 0.28 ml min−1. Acetonitrile at a flow rate of 0.12 ml min−1 was added postcolumn to the mobile phase effluent through a t-union resulting in a total solvent flow of 0.4 ml min−1 entering the mass spectrometer. The tandem mass spectrometer operated in an atmospheric pressure chemical-ionization positive-ion mode. M.s.-m.s. data were obtained from multiple-reaction-monitoring of a precusor-to-product ion transition of m/z 242 >116 for 5-AMT and 3-AMT at their respective retention times of 8 and 10 min. The LOQ was 1 ng ml−1.
Urine samples were analysed by diluting 0.005–0.05 ml of each urine sample to 0.5 ml with control plasma and were extracted and chromatographed using the plasma method conditions.
Rifampicin analysis
Random plasma samples from period 2 were measured for rifampicin concentrations by the C-18 reversed-phase h.p.l.c. procedure of Swart & Papgis [19].
Assay data
Pooled within-batch coefficients of variation (CV) were < 12% for duplicate determinations of zidovudine, GZDV and AMT over the concentration ranges of the standard-curve and quality-control samples. The CV for slope of standard curves were 4.7% (n = 3) and 7.0% (n = 8), 4.6% (n = 6) and 8.4% (n = 6), and 12.9% (n = 6) for measurements of zidovudine in urine and plasma, GZDV in urine and plasma, and AMT in plasma or urine samples, and are a reflection of between-batch precision for peak-response data.
Pharmacokinetic analysis
The plasma concentration (C) vs time (t) data were analysed by noncompartmental methods. The highest observed concentration and the corresponding sampling time were defined as Cmax and tmax, respectively. The terminal disposition half-life (t1/2,z) was calculated from the final slope (−λz) of the log-linear concentration-time curve (ln C, t) by least-squares linear regression. The slope was estimated from the data set (n≥3 points) with the smallest 90% confidence interval around the slope. Area under the plasma concentration-time curve (AUC) from time zero to the time of last measurable sample (tz) (AUC(0, tz)) was calculated by the linear trapezoidal method. AUC over the 8-h dosing interval (AUC(0, τ)) was estimated by adding an extrapolated portion from tz to 8 h, which was calculated by adding (C(tz)−C(8 h)) λ−1z to AUC(0, tz), where C(tz) and C(8h) are predicted plasma concentrations that were calculated from the above linear regression equation.
The apparent oral plasma clearance (CLo = CL/F where F is the bioavailability) was calculated by dividing the dose by AUC(0, τ). The urinary recoveries (X) were based on the cumulative milligram equivalents of zidovudine excreted as unchanged drug (ΣXZDV), GZDV (ΣXGZDV), and AMT (ΣXAMT) over the 6 h collection period, and were expressed as a percentage of the administered zidovudine dose. The molar, urinary metabolic ratios were expressed as ΣXGZDV/ΣXZDV and ΣXAMT/ΣXZDV. This ratio is independent of complete urine collections provided that the relative recovery of metabolite to zidovudine is constant in all voids. Renal clearance (CLR) values were calculated as the amount of zidovudine, GZDV or AMT recovered in urine over 6 h divided by the respective AUC(0, 6 h). The values of apparent oral formation clearance to GZDV (CLf(o)GZDV) and to AMT (CLf(o)(AMT) were determined by dividing ΣXGZDV or ΣXAMT by zidovudine AUC(0, 6 h), respectively. This calculation assumes that GZDV and AMT are not metabolized further and that all formed metabolite is recovered in urine, which is reasonable considering that biliary excretion of these metabolites is minor and GAMT is a minor metabolite of GZDV [6].
Statistical analysis
Differences in mean pharmacokinetic parameters of zidovudine, GZDV and AMT between treatments were evaluated by analysis of variance (ANOVA) appropriate for a repeated-measures study, followed by Dunnett's test for multiple comparisons of the treatments in periods 2 and 3 against the reference in period 1. Dunnett's test was used to maintain the experimentwise error rate at 5% (α = 0.05) for the two pairwise comparisons, with a resultant P value of < 0.028 considered statistically significant at the 5% level. All parameters except tmax were logarithmically (ln) transformed before analysis, and ANOVA summary statistics were based on least-squares geometric means. The 90% confidence limits around the ratio of geometric means were calculated with use of the critical t-value from the Dunnett's test. Median tmax values were compared by Friedman's test, followed by the Wilcoxon signed-rank test for pairwise comparisons. The significance level for each comparison was set at 0.025 from the Bonferroni-adjusted P value. For logarithmically transformed data, intraindividual CV (subject-by-treatment interaction) were calculated as 100%×(eMSR−1)1/2 and interindividual CV were estimated as 100%×[(MS−MSR)/3]1/2, where MS is the subject mean square and MSR is the mean-square residual in the anova model. Statistical computations were performed using SAS for Windows version 6.12 (SAS Institute Inc., Cary, NC, USA).
Results
The mean (±s.d.) age and weight of the patients were 34±5 years (range, 26–41) and 72±9 kg (range, 60–82 kg), respectively. The median CD4 T lymphocyte count was 360 cells/mm3 (range, 90–574). All patients completed the study without development of adverse reactions. Pill counts, questioning of participants, and random rifampicin measurements showed that all participants seemed to adhere with their rifampicin regimen. Three individuals had incomplete urine collections, and hence their urine data, including urinary metabolic ratios, were excluded from pharmacokinetic analysis. One patient inadvertently took his zidovudine dose a few hours before his second clinic visit, and except for t1/2,z values his data from period 2 were excluded from pharmacokinetic analysis. AMT concentrations from period 2 for one patient were not determined because of poor chromatography.
Arithmetic mean plasma and urine pharmacokinetic data, differences between geometric treatment means and corresponding intraindividual CV, and median tmax values for zidovudine, GZDV and AMT are summarised in Table 1. Mean plasma zidovudine and AMT concentration-time data are illustrated in Figure 1. Individual and mean changes in zidovudine and AMT AUC, zidovudine formation clearances to GZDV and AMT, molar urinary metabolic ratios, and molar AUC ratio (metabolite/zidovudine) are shown in Figures 2 and 3.
Table 1.
Mean pharmacokinetic parameters of zidovudine, GZDV and AMT for zidovudine administered alone on two occasions (periods 1 and 3) and following coadministration with 14 days rifampin (period 2).
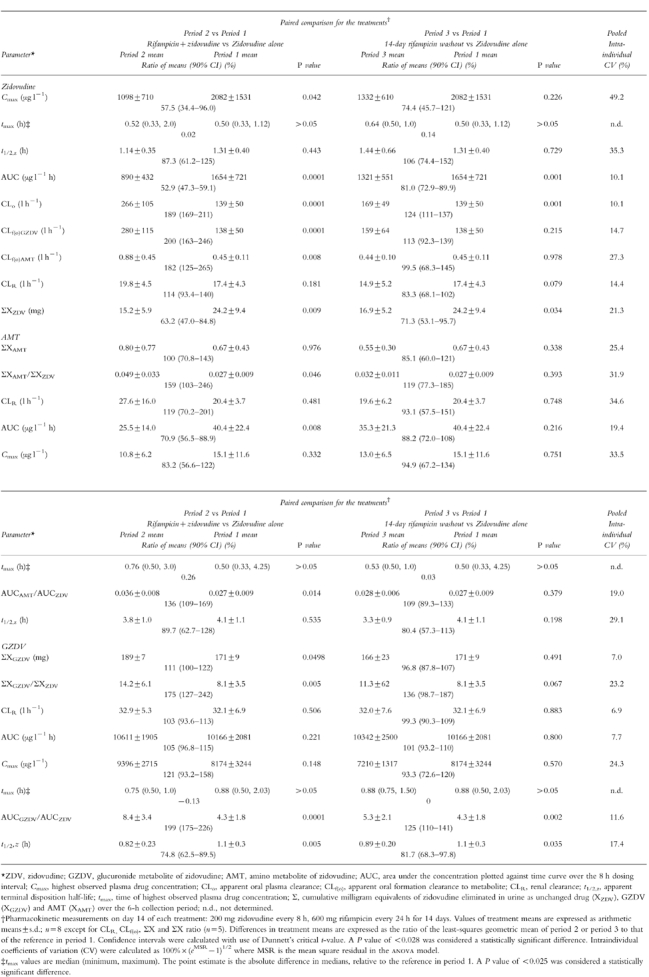
Figure 1.
Mean plasma concentrations of a) zidovudine and b) AMT in eight patients infected with HIV after receiving zidovudine (200 mg every 8 h) alone for 14 days (○), zidovudine concurrently with rifampicin (600 mg every 24 h) for 14 days (▵), or zidovudine (200 mg every 8 h) alone for 14 days after stopping rifampicin (□). Mean concentration data that were less than the lower limit of quantification of assay (zidovudine: 20 ng ml−1, AMT: 1 ng ml−1) are not shown.
Figure 2.
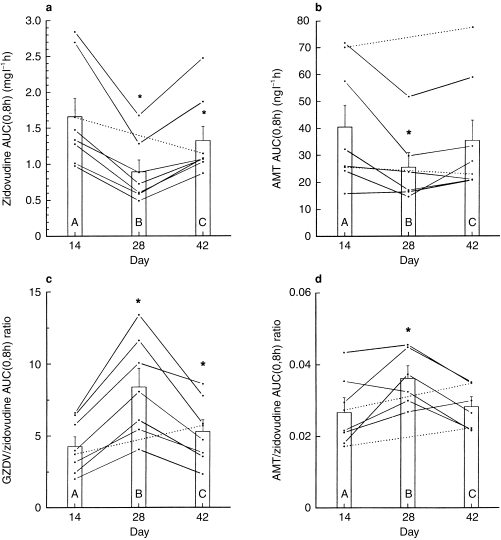
Individual values for a) zidovudine AUC, b) AMT AUC, c) molar GZDV/zidovudine AUC ratio and d) molar AMT/zidovudine AUC ratio on the 14th day of each treatment: (A) zidovudine alone (B) zidovudine plus rifampicin, and (C) zidovudine alone after discontinuing rifampicin. Vertical bars represent mean (±s.e. mean) values for eight participants. Dotted line connects day 14 and day 42 data to indicate missing data on day 28. *P < 0.028 compared with day 14.
Figure 3.
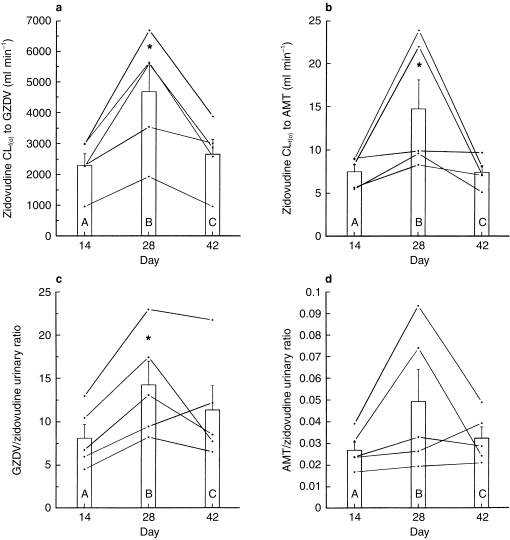
Individual values for a) zidovudine formation clearance to GZDV, b) zidovudine formation clearance to AMT, c) molar urinary GZDV/zidovudine ratio and d) molar urinary AMT/zidovudine ratio on the 14th day of each treatment: (A) zidovudine alone (B) zidovudine plus rifampicin, and (C) zidovudine alone after discontinuing rifampicin. Vertical bars represent mean (±s.e. mean) values for five participants. *P < 0.028 compared with day 14.
Compared with zidovudine-alone values in period 1, 14 days of rifampicin coadministration resulted in significant increases of 89% in CLo (range, 51% to 111%), 100% in CLf(o)GZDV (range, 54% to 146%), and 82% in CLf(o)AMT (range, 9% to 168%). Correspondingly, there were decreases of −43% in Cmax (range, −76% to 139%; P = 0.042) and −47% in AUC (range, −51% to −34%; P < 0.001) with no change in t1/2,z (−13%; range, −50% to 27%; P = 0.443) mean values of zidovudine.
There were small and insignificant increases of 21% in Cmax (range, −31% to 101%) and 5% in AUC (range, −15% to 20%) values of GZDV on the last day of rifampicin therapy. This small increase in AUC together with the large decrease in zidovudine AUC resulted in a significant increase of 99% in the GZDV/ZDV AUC ratio (range, 71% to 152%).
The AMT/ZDV AUC ratio significantly increased by 36% (range, −8% to 106%) despite a significant decrease of −29% (range, −48% to 4%) in AMT AUC. Rifampicin did not significantly alter Cmax of AMT but the trend was towards a decrease (−17%; range, −56% to 56%).
The average amount of zidovudine eliminated in urine over the 6 h collection period as ZDV, GZDV and AMT decreased by −37% (range, −44% to −32%; P = 0.009), increased by 11% (range, 2% to 17%; P = 0.050), and remained unchanged, respectively, during combination therapy. These combined changes in urine excretion resulted in increases of 75% and 59% in the urinary metabolic ratios ΣXGZDV/ΣXZDV (range, 58% to 94%; P = 0.005) and ΣXAMT/ΣXZDV (range, 12% to 139%; P = 0.046). Rifampin did not significantly alter renal clearance values of zidovudine, GZDV or AMT.
After stopping rifampicin for 14 days, the decrease in zidovudine AUC and increase in GZDV/ZDV AUC ratio relative to baseline values in period 1 were still detectable (P < 0.003) but geometric changes were < 20%. The 90% confidence limits for differences in geometric mean AUC (ratios) of zidovudine, GZDV and AMT were between 70% and 143%, indicating that the range of variation in AUC differences for the two zidovudine-alone phases was < 30% for the three compounds. Mean changes for all other pharmacokinetic parameters for the three compounds were insignificant and within 20% of baseline values for most parameters.
Whether administered alone or concurrently with rifampicin, zidovudine was rapidly absorbed with median tmax values of < 1 h and eliminated with average t1/2,zvalues of 1–1.5 h. Formation of GZDV and AMT occurred quickly with median tmax values also of < 1 h. Over the three periods, plasma levels of AMT ranged from 1.7% to 4.5% of ZDV plasma levels on the basis of molar AUC ratio. AMT Cmax ranged from 3 to 39 ng ml−1 and concentrations declined with average elimination half-lives of 3–4 h. Overall, the intraindividual CV for AUC for the three compounds were low at < 20%, but high interindividual variabilities of about 40% were observed in this parameter for zidovudine and AMT.
The mean percentage of administered dose that was recovered up to 6 h in urine as total zidovudine (unchanged plus GZDV and AMT) was 97.7%, 102% and 91.9% for periods 1, 2 and 3, respectively. The latter values were not different (P>0.18) from the control value. AMT accounted for < 0.5% of total urinary recovery on each occasion.
Discussion
Fourteen days of coadministration of rifampicin with zidovudine resulted in increases in oral clearance of zidovudine and formation clearances to GZDV and AMT. These changes led to corresponding decreases in maximum and total systemic exposure of zidovudine with no affect on zidovudine half-life, which is suggestive of a decrease in drug bioavailability rather than an increase in systemic clearance. The increase in formation clearances to the metabolites in the absence of significant changes in renal clearance of zidovudine led to a significant decrease in the amount of zidovudine that was excreted in urine, thereby increasing the urinary metabolic ratios. The higher metabolic AUC ratios of GZDV/zidovudine and AMT/zidovudine in the presence of rifampicin are also reflective of the elevated formation clearances when systemic clearances of the metabolites are unaltered. GZDV and AMT are expected to be mainly cleared by the kidneys, for which renal clearance values were not affected. These changes in plasma and urinary pharmacokinetic variables are consistent with rifampicin inducing the enzymatic conversion of zidovudine to GZDV and AMT. The net effect on AMT was a decrease in exposure because induction was more pronounced for the major metabolite GZDV. The small and insignificant increases in GZDV total exposure in plasma (5%) and amounts recovered in urine (11%) are expected given that this metabolite constituted more than 80% of zidovudine oral clearance and urinary recovery.
Our differential results for urinary recovery of zidovudine and plasma exposures of zidovudine and GZDV during pre and postdosing with rifampin are very similar to those observed by Strolin-Benedetti et al. in 10 healthy volunteers [17] and by Burger et al. in four HIV-infected individuals [16]. However, the quantitative recovery of dose in urine and the additional urine data on GZDV collected in our study allowed us to confirm the induction of glucuronidation by rifampicin. This mechanism was dismissed by Strolin-Benedetti & Dosert [18] mainly because of the lack of change in their subjects' GZDV plasma exposures. Our clinical results are in contrast to in vitro induction data. Li et al. [22] could not confirm the induction of zidovudine glucuronidation by rifampicin in primary human hepatocytes cultured from four individuals, but their experimental conditions for assessing induction potential were optimized for phase I and not phase II metabolism. Also, the inducibility of enyzmes in human hepatocytes varies among liver preparations.
AMT formation is a minor pathway of zidovudine elimination and the proportion of presystemically to systemically formed AMT is unknown. Therefore, the value of the apparent AMT formation clearance, which represents both presystemically and systemically formed AMT, can be artificially affected by systemic changes in non metabolic elimination pathways, such as zidovudine renal clearance. However, zidovudine renal clearance was not significantly altered by rifampicin, indicating that the observed increases in AMT formation clearance are consistent with an induction of zidovudine amination by rifampicin.
The magnitude of the residual inductive effect was minimal at 14 days after discontinuing rifampicin, as most of the pharmacokinetic parameters for zidovudine, GZDV and AMT had returned to within 20% of baseline values. The zidovudine and GZDV plasma and urine data were suggestive of a small residual induction of UGT after 2 weeks, but supportive changes in parameters that depend on urine data (formation clearance and urinary metabolic ratio) were insignificant because only five of eight patients had evaluable urine data and the intraindividual CV for urine data were higher than those for plasma.
The plasma exposures of AMT were much lower and the elimination half-lives of AMT were about three-fold longer than those of zidovudine, and compare favourably with values reported by Hoetelmans et al. [23] and Burger et al. [20] for similar oral doses of zidovudine. Over the three periods, maximum AMT exposures were < 18 ng ml−1 in six patients and < 40 ng ml−1 in the other two, and total exposures were < 4.5% of zidovudine total exposures. The formation of AMT constituted a minor proportion of zidovudine clearance (< 0.5%), and the significantly longer half-life of AMT indicates that its elimination was not limited by its formation, In contrast, GZDV was formation rate-limited. The large interindividual variation in AMT exposure was also observed by Zhou & Sommadossi [24] in six HIV-infected patients receiving a single dose of either 100 or 500 mg zidovudine, and is expected for a metabolite that is a minor pathway of drug elimination. The values of zidovudine and GZDV pharmacokinetic parameters and intraindividual CV are similar to those observed in previous studies [25, 26].
The role of P-glycoprotein in decreasing the bioavailability of zidovudine during rifampicin treatment appears minor. Although the expression of P-glycoprotein is induced by rifampicin in intestinal cells [27], zidovudine is not a substrate of this protein [28]. Moreover, the lack of gut metabolism of zidovudine [29] and our near complete recovery of drug in urine suggest that induction of P-glycoprotein, if present, would contribute minimally to lowering zidovudine levels in the presence of rifampicin.
A number of CYP isoforms (2C9, 2A6, 2E1 and 3A4) with CYP reductase catalyse the reduction of zidovudine to AMT in the presence of either the reduced form of nicotinamide adenine dinucleotide (NADH) or the reduced form of nicotinamide adenine dinucleotide phosphate (NADPH) [8]. In vitro studies in microsomes from humans [30] and from pretreated rats [9, 10] show that activation of AMT can occur with ethacrynic acid, dipyridamole or indomethacin, and the enyzme inducers phenobarbitone, dexamethasone or clofibrate. Rifampicin is an inducer of CYP3A4 and 2C isoforms. That rifampicin induces AMT in the present study implicates the involvement of at least these isoforms with CYP reductase in AMT formation, as proposed by Eagling et al. [10] and Pan-Zhou et al. [8]. Whether other human enzymes involved in AMT formation, such as cytochrome b5, CYP reductase and b5 reductase, are induced by rifampicin is unknown. The specific isoforms of zidovudine UGT that are involved in formation of GZDV and induced by rifampicin are unknown [31].
The clinical significance of this interaction is difficult to establish. Studies have demonstrated that patients receiving 600 mg rifampicin daily as part of an antituberculosis regimen concurrently with zidovudine for more than 8 months tolerated the combination well [16] and showed no additive toxicities compared with those taking zidovudine alone [32]. The lack of significant haematological toxicity reported for these patients is consistent with the decreased plasma levels of zidovudine and overall low plasma exposure of its cytotoxic metabolite AMT observed in our patients during concomitant zidovudine and rifampin therapy. Although the influence of rifampicin on levels of intracellular zidovudine triphosphate, the active form of the drug, was not determined in this study, the approximate 50% decrease in zidovudine plasma exposures by rifampicin is expected to have little effect on the antiviral activity of zidovudine. A pilot study of low-dose zidovudine (300 mg daily) reported clinical and virological effects similar to those of 600 mg daily in HIV-infected patients [33]. Moreover, halving the daily dose of zidovudine from 600 to 300 mg decreased plasma levels accordingly but intracellular triphosphate levels remained similar, though variable [34], suggesting that dose adjustments of zidovudine may not be necessary in rifampicin treatment of tuberculosis in HIV.
Our study demonstrates that therapeutic doses of 600 mg rifampicin daily induced the glucuronidation and amination pathways of zidovudine resulting in decreased plasma and urine exposures to zidovudine, and that the magnitude of the residual inductive effect on both metabolic pathways was minimal at 14 days after stopping rifampicin. Despite the large decreases in zidovudine exposure, the lack of significant haematological toxicity reported for patients taking concurrent zidovudine and rifampicin [16, 32] and the apparent lack of change in intracellular zidovudine triphosphate levels upon decreasing zidovudine exposure [34] suggest that prescribed doses of zidovudine may not need to be modified in the presence of rifampicin.
References
- 1.Drobniewski F. Is death inevitable with multiresistant TB plus HIV infection? Lancet. 1997;349:71–72. doi: 10.1016/S0140-6736(05)60878-1. 10.1016/S0140-6736(05)60878-1. [DOI] [PubMed] [Google Scholar]
- 2.Burman WJ, Gallicano K, Peloquin C. Therapeutic implications of drug interactions in the treatment of HIV-related tuberculosis. Clin Infect Dis. 1999;28:419–430. doi: 10.1086/515174. [DOI] [PubMed] [Google Scholar]
- 3.Centers for Disease Control and Prevention. Prevention and treatment of tuberculosis among patients infected with human immunodeficiency virus: principles of therapy and revised recommendations. MMWR Morb Mortal Wkly Rep. 1998;47:11–15. (no. RR-20) [PubMed] [Google Scholar]
- 4.Veal GJ, Back DJ. Metabolism of zidovudine. General Pharmacol. 1995;26:1469–1475. doi: 10.1016/0306-3623(95)00047-x. 10.1016/0306-3623(95)00047-X. [DOI] [PubMed] [Google Scholar]
- 5.Cretton EM, Xie M, Bevan RJ, Goudgaon NM, Schinazi RF, Sommadossi J-P. Catabolism of 3′-azido-3′-deoxythymidine in hepatocytes and liver enzymes with evidence of formation of 3′-amino-3′-deoxythymidine, a highly toxic catabolic for human bone marrow cells. Mol Pharmacol. 1990;39:258–266. [PubMed] [Google Scholar]
- 6.Stagg MP, Cretton EM, Kidd L, Diasio RB, Sommadossi J-P. Clinical pharmacokinetics of 3′-azido-3′-deoxythymidine (zidovudine) and catabolites with formation of a toxic catabolic, 3′-amino-3′-deoxythymidine. Clin Pharmacol Ther. 1992;51:668–676. doi: 10.1038/clpt.1992.79. [DOI] [PubMed] [Google Scholar]
- 7.Rajaonarison JF, Lacarelle B, De Sousa G, Catalin J, Rahmani R. In vitro glucuronidation of 3′-azido-3′-deoxythymidine by human liver. Role of UDP-glucuronosyltransferase 2 form. Drug Metab Dispos. 1991;19:809–815. [PubMed] [Google Scholar]
- 8.Pan-Zhou XR, Cretton-Scott E, Zhou XJ, Yang MX, Lasker JM, Sommadossi J-P. Role of human liver P450s and cytochrome b5 in the reductive metabolism of 3′-azido-3′-deoxythymidine (AZT) to 3′-amino-3′-deoxythymidine. Biochem Pharmacol. 1998;55:757–766. doi: 10.1016/s0006-2952(97)00538-8. 10.1016/S0006-2952(97)00538-8. [DOI] [PubMed] [Google Scholar]
- 9.Cretton EM, Sommadossi J-P. Reduction of 3′-azido-2′,3′-dideoxynucleosides to their 3′-amino metabolite is mediated by cytochrome P-450 and NADPH-cytochrome P-450 reductase in rat liver microsomes. Drug Metab Dispos. 1993;21:946–950. [PubMed] [Google Scholar]
- 10.Eagling VA, Howe JL, Barry MJ, Back DJ. The metabolism of zidovudine by human liver microsomes in vitro: formation of 3′-amino-3′-deoxythymidine. Biochem Pharmacol. 1994;48:267–276. doi: 10.1016/0006-2952(94)90097-3. 10.1016/0006-2952(94)90097-3. [DOI] [PubMed] [Google Scholar]
- 11.Oesch F, Arand M, Strolin Benedetti MS, Castelli MG, Dostert P. Inducing properties of rifampicin and rifabutin for selected enzyme activities of the cytochrome P-450 and UDP-glucuronosyltransferase superfamilies in female rat liver. J Antimicrob Chemother. 1996;37:1111–1119. doi: 10.1093/jac/37.6.1111. [DOI] [PubMed] [Google Scholar]
- 12.Adachi Y, Nanno T, Yamashita M, Ueshima S, Yamamoto T. Induction of rat liver bilirubin-conjugating enzymes and glutathione S-transferase by rifampicin. Gastroenterol Jpn. 1985;20:104–110. doi: 10.1007/BF02776672. [DOI] [PubMed] [Google Scholar]
- 13.Constantopoulos A, Loupa H, Krikos X. Augmentation of hepatic uridine-diphosphate glucuronyl transferase activity by antituberculosis drugs in hamsters in vivo. Cytobios. 1987;52:185–189. [PubMed] [Google Scholar]
- 14.Doostdar H, Grant MH, Melvin WT, Wolf CR, Burke MD. The effects of inducing agents on cytochrome P 450 and UDP-glucuronyltransferase activities in human hepG2 hepatoma cells. Biochem Pharmacol. 1993;46:629–635. doi: 10.1016/0006-2952(93)90548-b. 10.1016/0006-2952(93)90548-B. [DOI] [PubMed] [Google Scholar]
- 15.Abid A, Sabolovic N, Magdalou J. Inducibility of ethoxyresorufin deethylase and UDP-glucuronosyltransferase activities in two human hepatocarcinoma cell lines KYN-2 and Mz-Hep-1. Cell Biol Toxicol. 1996;12:115–123. doi: 10.1007/BF00143361. 10.1007/BF00143361. [DOI] [PubMed] [Google Scholar]
- 16.Burger DM, Meenhorst PL, Koks CHW, Beijnen JH. Pharmacokinetic interaction between rifampin and zidovudine. Antimicrob Agents Chemother. 1993;37:1426–1431. doi: 10.1128/aac.37.7.1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Strolin Benedetti M, Duchene P, Olliaro P. Proceedings of the Third International Society for the Study of Xenobiotics Meeting; Amsterdam: 1991. Investigation on a possible induction of AZT glucuronidation by rifabutin and rifampicin in humans; p. 295. Book of Abstracts June. [Google Scholar]
- 18.Strolin-Benedetti M, Dostert P. Induction and autoinduction properties of rifamycin derivatives: a review of animal and human studies. Environ Health Perspectives. 1994;102(Suppl 9):101–105. doi: 10.1289/ehp.94102s9101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Swart KJ, Papgis M. Automated high-performance liquid chromatographic method for the determination of rifampicin in plasma. J Chromatogr. 1992;593:21–24. doi: 10.1016/0021-9673(92)80260-2. 10.1016/0021-9673(92)80260-2. [DOI] [PubMed] [Google Scholar]
- 20.Burger DM, Rosing H, Koopman FJ, et al. Determination of 3′-amino-3′-deoxythymidine, a cytotoxic metabolite of 3′-azido-3′-deoxythymidine, in human plasma by ion-pair high-performance liquid chromatography. J Chromatogr. 1993;622:235–242. doi: 10.1016/0378-4347(93)80271-5. [DOI] [PubMed] [Google Scholar]
- 21.Good SS, Reynolds DJ, de Miranda P. Simultaneous quantification of zidovudine and its glucuronide in serum by high-performance liquid chromatography. J Chromatogr. 1988;431:123–133. doi: 10.1016/s0378-4347(00)83075-3. [DOI] [PubMed] [Google Scholar]
- 22.Li AP, Reith MK, Rasmussen A, et al. Primary human hepatocytes as a tool for the evaluation of structure-activity relationship in cytochrome P450 induction potential of xenobiotics: evaluation of rifampin, rifapentine and rifabutin. Chem Biol Interact. 1997;107:17–30. doi: 10.1016/s0009-2797(97)00071-9. 10.1016/S0009-2797(97)00071-9. [DOI] [PubMed] [Google Scholar]
- 23.Hoetelmans RMW, Meenhorst PL, Mulder JW, Burger DM, Koks CHW, Beijnen JH. XI International Conference on AIDS. Vancouver; 1996. Plasma pharmacokinetics of 3′-amino-3′-deoxythymidine, a cytotoxic metabolite of zidovudine. [Abstract Mo. B. 1324] July. [Google Scholar]
- 24.Zhou XJ, Sommadossi J-P. Comparative pharmacokinetics of zidovudine and its toxic catabolic 3′-amino-3′-deoxythymidine in HIV-infected patients. AIDS Res Hum Retroviruses. 1996;12:229–233. doi: 10.1089/aid.1996.12.229. [DOI] [PubMed] [Google Scholar]
- 25.Sahai J, Gallicano K, Garber G, Pakuts A, Cameron W. Pharmacokinetics of simultaneously administered zidovudine and didanosine in HIV-seropositive male patients. J Acquir Immune Defic Syndr Hum Retrovirol. 1995;10:54–60. [PubMed] [Google Scholar]
- 26.Gallicano K, Sahai J, Swick L, Seguin I, Pakuts A, Cameron DW. Effect of rifabutin on the pharmacokinetics of zidovudine in patients infected with human immunodeficiency virus. Clin Infec Dis. 1995;21:1008–1011. doi: 10.1093/clinids/21.4.1008. [DOI] [PubMed] [Google Scholar]
- 27.Schuetz EG, Beck WT, Schuetz JD. Modulators and substrates of P-glycoprotein and cytochrome P 4503A coordinately up-regulate these proteins in human colon carcinoma cells. Mol Pharmacol. 1996;49:311–318. [PubMed] [Google Scholar]
- 28.Glynn SL, Yazdanian M. In vitro blood–brain barrier permeability of nevirapine compared to other antiretroviral agents. J Pharm Sci. 1998;87:306–310. doi: 10.1021/js970291i. 10.1021/js970291i. [DOI] [PubMed] [Google Scholar]
- 29.Howe JL, Back DJ, Colbert J. Extrahepatic metabolism of zidovudine. Br J Clin Pharmacol. 1992;33:190–192. doi: 10.1111/j.1365-2125.1992.tb04024.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fayz S, Inaba T. Zidovudine azido-reductase in human liver microsomes: activation by ethacrynic acid, dipyridamole, and indomethacin and inhibition by human immunodeficiency virus protease inhibitors. Antimicrob Agents Chemother. 1998;42:1654–1658. doi: 10.1128/aac.42.7.1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burchell B, Brierley CH, Rance D. Specificity of human UDP-glucuronosyltransferases and xenobiotic glucuronidation. Life Sci. 1995;57:1819–1831. doi: 10.1016/0024-3205(95)02073-r. [DOI] [PubMed] [Google Scholar]
- 32.Antoniskis D, Easley AC, Espina BM, Davidson PT, Barnes PF. Combined toxicity of zidovudine and antituberculosis chemotherapy. Am Rev Respir Dis. 1992;145:430–434. doi: 10.1164/ajrccm/145.2_Pt_1.430. [DOI] [PubMed] [Google Scholar]
- 33.Collier AC, Bozzette S, Coombs RW, et al. A pilot study of low-dose zidovudine in human immunodeficiency virus infection. N Engl J Med. 1990;323:1015–1021. doi: 10.1056/NEJM199010113231502. [DOI] [PubMed] [Google Scholar]
- 34.Barry MG, Khoo SH, Veal GJ, et al. The effect of zidovudine dose on the formation of intracellular phosphorylated metabolites. AIDS. 1996;10:1361–1367. doi: 10.1097/00002030-199610000-00008. [DOI] [PubMed] [Google Scholar]