

Abstract
Aims
In vitro studies suggest that the oxidation of quinidine to 3-hydroxyquinidine is a specific marker reaction for CYP3A4 activity. To assess the possible use of this reaction as an in vivo marker of CYP3A4 activity, we studied the involvement of cytochromes CYP2C9, CYP2E1 and CYP3A4 in the in vivo oxidative metabolism of quinidine.
Methods
An open study of 30 healthy young male volunteers was performed. The pharmacokinetics of a 200 mg single oral dose of quinidine was studied before and during daily administration of 100 mg diclofenac, a CYP2C9 substrate (n=6); 200 mg disulfiram, an inhibitor of CYP2E1 (n=6); 100 mg itraconazole, an inhibitor of CYP3A4 (n=6); 250 ml single strength grapefruit juice twice daily, an inhibitor of CYP3A4 (n=6); 250 mg of erythromycin 4 times daily, an inhibitor of CYP3A4 (n=6). Probes of other enzyme activities, caffeine (CYP1A2), sparteine (CYP2D6), mephenytoin (CYP2C19), tolbutamide (CYP2C9) and cortisol (CYP3A4) were also studied.
Results
Concomitant administration of diclofenac reduced the partial clearance of quinidine by N-oxidation by 27%, while no effect was found for other pharmacokinetic parameters of quinidine. Concomitant administration of disulfiram did not alter any of the pharmacokinetic parameters of quinidine. Concomitant administration of itraconazole reduced quinidine total clearance, partial clearance by 3-hydroxylation and partial clearance by N-oxidation by 61, 84 and 73%, respectively. The renal clerance was reduced by 60% and the elimination half-life increased by 35%. Concomitant administration of grapefruit juice reduced the total clearance of quinidine and its partial clearance by 3-hydroxylation and N-oxidation by 15, 19 and 27%, respectively. The elimination half-life of quinidine was increased by 19%. The caffeine metabolic index was reduced by 25%. Concomitant administration of erythromycin reduced the total clearance of quinidine and its partial clearance by 3-hydroxylation and N-oxidation by 34, 50 and 33%, respectively. Cmax was increased by 39%.
Conclusions
The results confirm an important role for CYP3A4 in the oxidation of quinidine in vivo, and this applies particularly to the formation of 3-hydroxyquinidine. While a minor contribution of CYP2C9 to the N-oxidation of quinidine is possible, a major involvement of the CYP2C9 or CYP2E1 enzymes in the oxidation of quinidine in vivo is unlikely.
Keywords: CYP2C9, CYP2E1, CYP3A4, diclofenac, disulfiram, erythromycin, grapefruit juice, itraconazole, quinidine
Introduction
The human cytochrome P450 system consists of a number of related enzymes which metabolize many xenobiotics and endogenous compounds. The P4503A4 (CYP3A4) enzyme accounts for approximately 30% of the total human liver cytochrome P450 content, and among many drugs it oxidizes several calcium antagonists, protease-inhibitors, and macrolide antibiotics. Interindividual differences in CYP3A4 activity [1] contribute a significant proportion of the pharmacokinetic variability of many drugs. In order to assess CYP3A4 activity on an individual basis, many attempts have been made to characterize specific in vivo CYP3A4 biomarker reactions, notably the erythromycin breath test [2], the oxidation of midazolam to 1′-hydroxymidazolam [3] or endogenous cortisol metabolism [4] have been suggested among others. However all have proved to be of limited value in one way or another as discussed in detail by Watkins [5] and Kivisto et al. [6], and the quest continues for a readily available and convenient in vivo CYP3A4 assay. Quinidine, a class 1A antiarrythmic, is metabolized primarily by CYP3A4 [7]. Studies of human liver microsomes and yeast recombinant P450 expression systems have shown that the formation of the (3S)-3-hydroxymetabolite is mediated almost exclusively by CYP3A4 [8]. We hypothesize following a low oral single dose that the 3-hydroxylation of quinidine may serve as an in vivo biomarker reaction for CYP3A4 activity.
This study is one of a series of systematic interaction studies, which address the specificity of quinidine for the CYP3A4 enzyme in vivo, as high substrate specificity is obviously an essential feature of any biomarker reaction. Previously a role for CYP2D6 in the oxidation of quinidine has been excluded [9] and a major role for the CYP1A2 and CYP2C19 enzymes seems unlikely [10]. The CYP2C9 enzyme is involved in the oxidation of most nonsteroid anti-inflammatory drugs and tolbutamide. As the CYP2C9 inhibitor of choice, sulphaphenazole, is now only available for in vitro studies, we chose diclofenac as a putative competitive inhibitor of CYP2C9. Diclofenac is a substate with high affinity for this enzyme [11], and has been shown to inhibit the metabolism of other CYP2C9 substrates in vitro [12]. The reductive product of disulfiram, diethylthiocarbamate, is a well documented potent inhibitor of CYP2E1 [13, 14]. Grapefruit juice is an inhibitor of intestinal CYP3A4 [15], itraconazole is a potent inhibitor of CYP3A4 [16, 17], and erythromycin also inhibits CYP3A4 [18]. We hypothesized that concomitant administration of diclofenac or disulfiram would not affect the oxidative metabolism of quinidine but that grapefruit juice, itraconazole and erythromycin indeed would inhibit the oxidative metabolism of quinidine, albeit by different orders of magnitude. The purpose of the present study is to confirm that the 3-hydroxylation of quinidine is mediated by CYP3A4 in vivo.
Methods
Subjects
This was an open study of 30 young healthy male volunteers between 20 and 35 years of age, all phenotyped as extensive metabolizers of sparteine (metabolic ratio <20) and mephenytoin (S/R ratio <0.5). All volunteers gave written informed consent. The study was approved by the Regional Ethics Committee and the Danish National Board of Health. Before inclusion, all volunteers were screened by physical examination, laboratory tests and electrocardiogram.
Study procedure
Subjects were allocated consecutively by the time of inclusion to first a diclofenac group (n=6) and next to a disulfiram group (n=6), then to an itraconazole group (n=6), then to an erythromycin group (n=6) and finally to a grapefruit juice group (n=6). No formal randomization procedure was performed. All volunteers completed two study phases with and without concomitant administration of drug or grapefruit juice according to their allocation. On study day 1 the volunteers took a single oral dose of 250 mg tolbutamide (Tolbutamid ‘Dak’, Nycomed DAK, Denmark), for assessment of CYP2C9 activity, at 08.00 h. Urine was collected for the next 6 h and a sample (10 ml) was frozen at −20° C until analysis. Blood glucose was measured after 6 h. On study day 2 all of the volunteers took 100 mg of sparteine sulphate (Depasan, Giulini Pharma GMBH, Germany), 100 mg of mephenytoin (Mesantoin, Sandoz Pharmaceuticals Corporation, USA) and 200 mg of caffeine (Koffein ‘Dak’, Nycomed DAK, Denmark) orally at 08.00 h, for the assessment of CYP2D6, CYP2C19 and CYP1A2 activities, respectively. It has been demonstrated, that the simultaneous administration of low dose biomarker drugs does not influence the outcome of their respective metabolic indices [19]. Urine was collected for 12 h and three 10 ml samples were frozen at −20° C until analysis. A blood sample (10 ml) was drawn after 6 h and the plasma was separated by centrifugation (10 min, 7000 g) and frozen at −20° C until analysis. On study day 3, following an overnight fast from midnight, the volunteers took a single oral dose of quinidine sulphate (200 mg) (Kinidin ‘Dak’, Nycomed DAK, Denmark), equal to 511 μmol quinidine. Blood samples (10 ml) were collected at t=0, 0.5, 1, 2, 3, 4, 5, 6, 7, 8, 10, 24 and 48 h. Plasma was separated by centrifugation (10 min, 7000 g) and frozen at −20° C until analysis. Volunteers were allowed normal meals after the blood sample at t=1 h was drawn. Urine was collected at intervals of 0–12, 12–24 and 24–48 h. A sample (10 ml) from each period was frozen at −20° C until analysis. Following a washout period of 6–8 weeks, the volunteers according to their allocation group took: 100 mg diclofenac (Voltaren Retard, Ciba-Geigy, Denmark) daily at 08.00 h on study days −1, 0, 1, 2, 3 and 4; 200 mg disulfiram (Antabus, Dumex-Alpharma, Denmark) daily at 08.00 h on study days −2, −1, 0, 1, 2, 3 and 4; 100 mg itraconazole (Sporanox, Janssen-Cilag, Denmark) daily at 08.00 h on study days −2, −1, 0, 1, 2, 3 and 4; 250 mg erythromycin (Erycin, Nycomed Dak, Denmark) four times daily at 08.00, 12.00, 18.00 and 22.00 h, on study days −2, −1, 0, 1, 2, 3, and 4; 250 ml single strength grapefruit juice (Grapefrugt Juice, Rynkeby, Denmark) twice daily at 08.00 and 20.00 h on study days −2, −1, 0, 1, 2, 3 and 4. The initial study procedures with the biomarker reactions of tolbutamide, sparteine, mephenytoin, caffeine and endogenous cortisol followed by the single-dose pharmacokinetics of quinidine were repeated on days 1–4. During the study, the volunteers refrained from any other medication, alcohol or grapefruit juice. Consumption of coffee and xanthine containing foods was not allowed for a period of 48 h before and during the 12 h of urine collection following oral administration of caffeine. The volunteers were asked specifically about adverse events on a daily basis during medication. Following treatment with diclofenac, disulfiram, itraconazole, erythromycin and grapefruit juice control laboratory tests were performed.
Quinidine and metabolites
Quinidine, 3-hydroxyquinidine and quinidine-N-oxide in plasma and urine were analysed by means of h.p.l.c. methods described previously [20]. The limit of quantification was 5 nm for quinidine, 3 nm for 3-hydroxyquinidine and 4 nm for quinidine-N-oxide. The coefficient of variation was in the range of 3–6% at concentrations between 0.25 and 10 μm for all three compounds.
Tolbutamide and metabolites
A method for the quantification of tolbutamide, 4-hydroxytolbutamide and the secondary metabolite carboxytolbutamide in urine was developed. Briefly; urine samples (1 ml) were added 25 μl of 0.01 m internal standard, chlorpropamide, and 30 μl 2 m HCl and 4 ml of tert.-butyl methyl ether. The mixture was shaken for 30 min, and the phases separated by 10 min of centrifugation at 1100 g. The tubes were stored at −30° C until the aqueous phase was frozen. The organic phase was transferred to conical glass tubes and evaporated to dryness under nitrogen at 40° C. The residue was reconstituted in 1 ml of a mobile phase consisting of a 23:77% (v/v) mixture of methanol and 0.01 m sodium acetate buffer adjusted to a pH of 3.0 with 60% perchloric acid. A sample of 100 μl was transferred to h.p.l.c. vials. The injection volume was 20 μl. Chromatography was performed using Merck-Hitachi instruments: an AS-2000 A autosampler with a 100 l injector loop, a L-5025 column thermostat, a L-6200 intelligent Pump and a L-4250 UV-VIS Detector. The system was controlled through a D–6000 interface module h.p.l.c. System Manager (HSM) and an IBM personal computer. Separations were achieved using a Spherisorb S5 Phenyl, 5 μm (250*4.6 mm i.d.) column. The limits of quantification, based on coefficients of variation of less than 20%, were 1.25 μm for tolbutamide, 0.75 μm for 4-hydroxytolbutamide and 2 μm for carboxytolbutamide. The coefficient of variation was in the range of 4.0–4.7% for all three compounds for tolbutamide and hydroxytolbutamide concentrations between 6 and 12 μm and for carboxytolbutamide concentrations between 24 and 48 μm. As the concentration of tolbutamide in urine was mostly below the limits of quantification, a metabolic ratio could not be assessed. To get an index of CYP2C9 activity, we chose to calculate a tolbutamide metabolic index as:
Tolbutamide metabolic index=
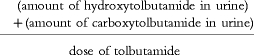 |
As subject compliance was 100% and tolbutamide is almost completely bioavailable, this index, while not validated as the ratio used by Veronese and coworkers [21] (who used a ratio of the sum of the urine concentrations of the two tolbutamide metabolites to the concentration of urine tolbutamide in a 6 h urine sample following an oral single dose of 500 mg tolbutamide), may be useful in assessing effects on CYP2C9 activity.
Sparteine and metabolites
Sparteine and its metabolites, 2,3-and 5,6 dehydrosparteine were analysed by means of gas chromatography [22], and the sparteine metabolic ratio was calculated as:
Sparteine MR=
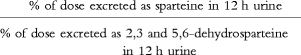 |
Mephenytoin
The chromatographic peak areas of S- and R-mephenytoin in 12 h urine were assayed by means of gas chromatography [23], and the S/R ratio was calculated as:
Mephenytoin S/R=
Caffeine and metabolite
Caffeine and 1.7 dimethylxanthine were analysed by means of h.p.l.c. as described by Rasmussen [24]. The limits of quantification were 1.2 μm and 0.3 μm for caffeine and 1,7 dimethylxanthine, respectively. The coefficients of variation were in the range of 3–8% for both compounds at concentrations between 4 and 30 μm. The caffeine metabolic ratio was calculated according to Fuhr [25] as:
Caffeine metabolic ratio=
Cortisol and metabolite
Cortisol and 6β-hydroxycortisol were analysed by means of h.p.l.c. [26]. The limit of quantification was 14 nm for cortisol and 87 nm for 6β-hydroxycortisol. The coefficient of variation was in the range of 5–10% for both compounds. The metabolic ratio was calculated according to Ged [4] as:
Cortisol metabolic ratio=
Pharmacokinetic analyses
All pharmacokinetic parameters were calculated using noncompartmental methods with the software package Winnonlin Standard Version 1.5 (Scientific Consulting Inc., USA). The area under the plasma concentration-time curves of quinidine (AUC) and its metabolites were calculated using the linear trapezoidal method with extrapolation to infinity. Values of peak concentration (Cmax) and time to reach peak concentration (tmax) were noted directly.
The apparent oral clearance of quinidine was calculated as:
Quinidine renal clearance was calculated as:
Partial clearance by 3-hydroxylation was calculated as:
CL(q⇒3OH−q)=
Partial clearance by N-oxidation was calculated as:
CL(q⇒q−No)=
Residual clearance was calculated as:
The terminal elimination half-life of quinidine was calculated as:
where λz is the terminal slope of the log plasma concentration vs time curve.
Statistical analyses
Data are presented as median and range. Statistical test values are Hodges-Lehmann estimates of median differences with exact 95% confidence intervals. Inter-group comparison was made by the median test. Differences were considered statistically significant when the 95% confidence intervals excluded zero. Statistical analyses were performed using the software packages SPSS 7.5 for Windows (SPSS Inc., USA) and StatXact 3 (Cytel Software Corporation, USA).
Results
All volunteers completed the study. No side-effects were reported during administration of diclofenac. During administration of disulfiram, one volunteer had intermittent diarrhoea, and another volunteer complained of slight abdominal discomfort. Side-effects during administration of itraconazole were nausea (one subject) and intermittent headache (one subject). Side-effects during administration of erythromycin was abdominal discomfort (one subject), while no side-effects were seen during administration of grapefruit juice. All control laboratory tests were within normal values. Six hours after administration of tolbutamide blood glucose concentrations were within the range of 2.6–5.9 mm, without any subjective or objective signs of hypoglycaemia, in all subjects. The only other side-effects noted were slight degrees of headache and irritability due to abstinence from caffeine. The study results with the pharmacokinetic parameters of quinidine and the biomarker reactions are summarized in Tables 1 and 2.
Table 1.
Quinidine (Q) pharmacokinetic parameters in 30 healthy young male volunteers, following a 200 mg single oral dose with and without concomitant administration of diclofenac (Dic, n=6), disulfiram (Dis, n=6), erthyromycin (Ery, n=6) and grapefruit juice (Gra, n=6). For each interaction, the Hodges-Lehmann point estimates of the median differences with the exact 95% confidence intervals are provided.
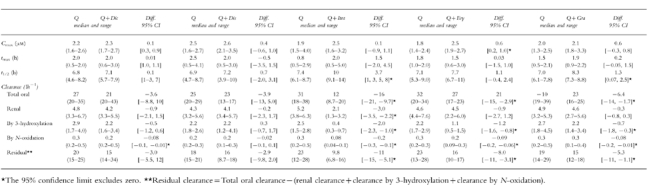
Table 2.
Putative and established metabolic ratios of various CYP marker reactions in 30 healthy young male volunteers, with and without concomitant administration of diclofenac (Dic, n=6), disulfiram (Dis, n=6), itraconazole (Itra, n=6), erthyromycin (Ery, n=6) and grapefruit juice (Gra, n=6). For each interaction, the Hodges-Lehmann point estimates of the median differences with the exact 95% confidence intervals are provided.

Effect of diclofenac
The total apparent oral clearance of quinidine, and clearance by 3-hydroxylation were unchanged. Clearance by N-oxidation was significantly reduced (median difference: −0.08 l h−1; 95% confidence interval of the difference: [−0.1, −0.01 l h−1]). No statistically significant changes were found for the renal clearance of quinidine, Cmax, tmax or elimination half-life. The median sparteine metabolic ratio was significantly reduced (median difference: −0.6; 95% confidence interval of the difference: [−1.6, −0.2]) during administration of diclofenac. No changes were found for the median caffeine metabolic index, mephenytoin S/R ratio, tolbutamide metabolic index or cortisol metabolic ratio.
Effect of disulfiram
The pharmacokinetic parameters of quinidine remained unchanged, and disulfiram did not affect any of the CYP marker reactions.
Effect of itraconazole
The median total oral clearance of quinidine and the partial clearances by 3-hydroxylation and N-oxidation were significantly reduced by 61, 84 and 73%, respectively. The median elimination half life of quinidine increased 35%, while no statistically significant changes was found for Cmax. The median Cmax of 3-hydroxyquinidine decreased from 0.3 to 0.1 μm (median difference: −0.2 μm; 95% confidence intervals of the difference: [−0.6, −0.08 μm]), while the median quinidine N-oxide Cmax decreased from 0.6 to 0.2 μm (median difference: −0.5 μm; 95% confidence intervals of the difference: [−1.1, −0.2 μm]). The renal clearance of quinidine was significantly reduced by a median 60%. No statistically significant effects on the biomarker reactions were found.
Effect of erythromycin
The median total apparent oral clearance of quinidine, clearance by 3-hydroxylation and by N-oxidation were significantly decreased by 34, 50 and 33%, respectively. The median Cmax increased significantly by 39%. The median elimination half-life, the median renal clearance of quinidine and the median quinidine tmax were unchanged. No effects on the marker reactions were seen.
Effect of grapefruit juice: The median total apparent oral clearance of quinidine, clearance by 3-hydroxylation and by N-oxidation were significantly reduced by 15, 19 and 27%, respectively. The median Cmax, tmax and renal clearance of quinidine were unchanged. The median elimination half-life increased significantly by 19%. The median Cmax of quinidine N-oxide decreased significantly from 0.6 to 0.4 μm (median difference: −0.2 μm; 95% confidence intervals of the difference:[−0.5; −0.02 μm]). The caffeine metabolic ratio was significantly reduced by 25%, while there were no effects on the other biomarker reactions.
Intergroup comparisons revealed no statistically significant differences in the pharmacokinetic parameters of quinidine or CYP marker reactions before treatment with disulfiram, diclofenac, itraconazole, erythromycin or grapefruit juice (data not shown).
Discussion
In this study we studied systematically the pharmacokinetics of a single oral dose of 200 mg quinidine before and during concomitant administration of inhibitors of various cytochrome P450 enzymes. Treatment with diclofenac reduced the partial clearance of quinidine by N-oxidation by 33%, while other pharmacokinetic parameters of quinidine were unaffected by concomitant administration of diclofenac. This interaction must have occurred at the metabolic level since the renal clearance was unaffected. As diclofenac is a substrate and hence a putative competitive inhibitor of CYP2C9 [11, 12], these results are consistant with results from in vitro studies, in which up to 23% of the quinidine N-oxidation was accounted for by CYP2C9 [8]. The fact that an effect on the putative marker reaction of CYP2C9 was not seen, could be that the tolbutamide index used here is not a valid marker of this reaction. Alternatively, a type 2 error due to a small sample size and the intraindividual variability of this index is a possibility. As no pharmacokinetic parameters other than the partial clearance by N-oxidation was affected, the results mitigate against an involvement of CYP2C9 in the formation of the 3-hydroxymetabolite of quinidine. The effect on the sparteine: dehydrosparteine metabolic ratio suggesting an induction of CYP2D6 is difficult to explain as anything other than a statistical type 1 error, as CYP2D6 is generally believed to be uninducible. Administration of disulfiram did not alter any of the pharmacokinetic parameters of quinidine. In vitro, less than 2% of the oxidative metabolism of quinidine was accounted for by CYP2E1, and accordingly a very high ki of 248 μm for the reductive product of disulfiram, diethylthiocarbamate, was found [8]. Other studies have concluded that disulfiram is a specific inhibitor of CYP2E1 both in vivo and in vitro [13, 14, 27], a finding supported by our data as disulfiram did not affect any of the other marker reactions. An inhibitory effect of disufiram on the metabolism of caffeine as found by Beach [28], could not be confirmed here.
The effects of itraconazole on the total clearance, renal clearance, partial clearances by 3-hydroxy-lation and N-oxidation and elimination half-life of quinidine were substantial. We found no statistically significant increase in quinidine Cmax, due probably to a large range of plasma concentrations before administration of itraconazole. The magnitude of the effects are similar to those found by Kaukonen et al. though they did not measure the formation clearances of either of the metabolites [16]. Inhibition of intestinal CYP3A4 and P-glycoprotein by itraconazole would only partly account for the magnitude of the effects seen here, and would not explain the elevated elimination half-life. As the bioavialability of quinidine is approximately 70% [29] and its renal clearance is relatively small, a decrease in nonrenal clearance reflected by the increased elimination half-life is obvious. Inhibition of hepatic CYP3A4 by itraconazole readily accounts for the findings though contributions due to some degree of inhibition of P-glycopprotein and intestinal CYP3A4 cannot be excluded. We confirm (Table 1) that the renal clearance of quinidine greatly exceeds glomerular filtration [30], and this is probably due to active secretion by P-glycoprotein in the proximal renal tubules [31–34]. It has previously been shown that itraconazole inhibits the active tubular secretion of digoxin [35], another P-glycoprotein substrate, and we show here that the median renal clearance of quinidine drops from 5.2 to 2.1 l h−1, and this interaction is most likely due to inhibition of P-glycoprotein in the proximal renal tubular cells.
To our knowledge, this is the first formal pharmacokinetic interaction study designed to evaluate the effect of erythromycin on the metabolism of quinidine. Case reports of clinically relevant interactions have previously been described [36–38], as well as pharmacokinetic interactions of erythromycin with other substrates of CYP3A4 [18, 39]. Cmax increased significantly (Table 1), but surprisingly we could not demonstrate a statistically significant effect of erythromycin on the elimination half-life of quinidine. The decrease in total clearance of quinidine and partial clearance by 3-hydroxylation and N-oxidation during administration of erythromycin is somewhat less than the effect found during administration of itraconazole. This is consistant with in vitro CYP3A4 inhibition Ki values of 0.3–2 μm [8, 40] for itraconazole and 37–117 μm [41] for erythromycin. The increase of Cmax and the lack of effect on the elimination half-life seems to indicate that the primary effects of erythromycin on quinidine metabolism are presystemic. Erythromycin has been shown to reverse multidrug resistance in cell lines [42], indicating an inhibitory effect on P-glycoprotein mediated transport. However changes in the clearance of between 33 and 50% are too large to be accounted for solely by inhibition of intestinal CYP3A4 and P-glycoprotein effects as the bioavailability of quinidine is high. Despite the lack of effect on the elimination half-life of quinidine, a result which may represent a type 2 error, inhibition of hepatic CYP3A4 must have occurred, although inhibition of intestinal CYP3A4 and P-glycoprotein may have contributed to the outcome.
Administration of grapefruit juice caused minor, but still clearly significant changes in the metabolism of quinidine. While no effect was found for Cmax, the total and partial clearances of quinidine decreased by 15–27%, and the elimination half-life of quinidine was increased by 19%. These results differ somewhat from those by Min et al. [43], who found an increased tmax and an increase in the AUC of the 3-hydroxy metabolite of quinidine following administration of a single glass of grapefruit juice. No effects on the AUC of quinidine were found, a difference that may be attributable to different schemes of grapefruit juice administration. In a comparative study of the effects of erythromycin and grapefruit juice on the pharmacokinetics of the CYP3A4 substrate felodipine, Bailey et al. [18] found a 93–149% increase in the AUC of felodipine. These changes are much larger than the results found for the present interaction with quinidine and the large difference in bioavailability between felodipine (15%) and quinidine (70%) offers a sufficient explanation. The active inhibitory components in grapefruit juice are believed to be furanocoumarins [44–46], and the effect of grapefruit juice is primarily an irreversible inactivation of intestinal CYP3A4 [15, 45]. Inhibition of intestinal P-glycoprotein has not been found [15], and the lack of effect on the renal clearance of quinidine in this study does not support a hypothesis of inhibition of P-glycoprotein by grapefruit juice components. Keeping in mind the relative high bioavilability of quinidine, only minor effects of grapefruit juice on the metabolism of quinidine were to be expected. The increased elimination half-life found here suggests an inhibitory effect of grapefruit juice components on hepatic CYP3A4, but the magnitude of the effect was marginal with the confidence limits only barely excluding zero. An inhibitory effect on hepatic CYP3A4 is very unlikely as Lown et al. [15] could not demonstrate changes in the result of the erythromycin breath test in healthy volunteers upon prolonged exposure to grapefruit juice.
Except for a 25% reduction in the caffeine metabolic ratio during grapefruit juice administration, no effects of itraconazole, erythromycin or grapefruit juice on the CYP marker reactions were found. The effect of grapefruit juice confirms previous results by Fuhr et al. [47], who found a similar reduction of caffeine clearance during grapefruit juice administration. However, no effects of grapefruit juice were seen on the pharmacokinetics of the CYP1A2 substrate theophylline [48] and the mechanism of this interaction remains unclear, since these findings are unexplained by inhibition of intestinal CYP3A4.
Conclusions
This series of pharmacokinetic interaction studies confirms an important role of CYP3A4 in the oxidative metabolism of quinidine in vivo, as well as a possible role of P-glycoprotein in both intestinal absorbtion and renal excretion of quinidine. CYP2C9 and CYP2E1 appear to make no or only minor contributions to the oxidative metabolism of quinidine in vivo. The formation of the 3-hydroxy metabolite of quinidine seems to depend specifically on the CYP3A4 enzyme, suggesting a possible use of the formation of this metabolite as an in vivo marker reaction of CYP3A4 activity.
Acknowledgments
This study was supported by grants from the Danish Medical Research Council (Reference number 12-9206). The technical assistance of Mrs Birgitte Damby, Mrs Annnelize Casa and Mss Susanne Jørgensen is appreciated.
References
- 1.Shimada T, Yamazaki H, Mimura M, et al. Interindividual variations in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: studies with liver microsomes of 30 Japanese and 30 Caucasians. J Pharmacol Exp Ther. 1994;270:414–423. [PubMed] [Google Scholar]
- 2.Watkins PB, Murray SA, Winkelman LG, et al. Erythromycin breath test as an assay of glucocorticoid-inducible liver cytochromes P-450. Studies in rats and patients. J Clin Invest. 1989;83:688–697. doi: 10.1172/JCI113933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thummel KE, Shen DD, Podoll TD, et al. Use of midazolam as a human cytochrome P450, 3A probe: II. Characterization of inter- and intraindividual hepatic CYP3A variability after liver transplantation. J Pharmacol Exp Ther. 1994;271:557–566. [PubMed] [Google Scholar]
- 4.Ged C, Rouillon JM, Pichard L, et al. The increase in urinary excretion of 6 beta-hydroxycortisol as a marker of human hepatic cytochrome P450IIIA induction. Br J Clin Pharmacol. 1989;28:373–387. doi: 10.1111/j.1365-2125.1989.tb03516.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Watkins PB. Noninvasive tests of CYP3A enzymes. Pharmacogenetics. 1994;4:171–184. doi: 10.1097/00008571-199408000-00001. [DOI] [PubMed] [Google Scholar]
- 6.Kivisto KT, Kroemer HK. Use of probe drugs as predictors of drug metabolism in humans. J Clin Pharmacol. 1997;37:40–48. doi: 10.1177/009127009703700121. [DOI] [PubMed] [Google Scholar]
- 7.Guengerich FP, Muller Enoch D, Blair IA. Oxidation of quinidine by human liver cytochrome P-450. Mol Pharmacol. 1986;30:287–295. [PubMed] [Google Scholar]
- 8.Nielsen TL, Rasmussen BB, Flinois J-P, et al. In vitro metabolism of quinidine: the (3S)-3-hydroxylation is a specific marker reaction for cytochrome P4503A4 activity in human liver microsomes. J Pharmacol Exp Ther. 1999;289:31–37. [PubMed] [Google Scholar]
- 9.Nielsen F, Rosholm JU, Brosen K. Lack of relationship between quinidine pharmacokinetics and the sparteine oxidation polymorphism. Eur J Clin Pharmacol. 1995;48:501–504. doi: 10.1007/BF00194341. [DOI] [PubMed] [Google Scholar]
- 10.Damkier P, Hansen LL, Brosen K. Effect of fluvoxamine on the pharmacokinetics of quinidine. Eur J Clin Pharmacol. 1999;55:451–456. doi: 10.1007/s002280050655. [DOI] [PubMed] [Google Scholar]
- 11.Leemann T, Transon C, Dayer P. Cytochrome P450TB (CYP2C): a major monooxygenase catalyzing diclofenac 4′′-hydroxylation in human liver. Life Sci. 1993;52:29–34. doi: 10.1016/0024-3205(93)90285-b. [DOI] [PubMed] [Google Scholar]
- 12.Kappers WA, Groene EM, Kleij LA, et al. Inhibition of tolbutamide 4-methylhydroxylation by a series of non-steroidal anti-inflammatory drugs in V79-NH cells expressing human cytochrome P4502C10. Xenobiotica. 1996;26:1231–1239. doi: 10.3109/00498259609047227. [DOI] [PubMed] [Google Scholar]
- 13.Kharasch ED, Thummel KE, Mhyre J, et al. Single-dose disulfiram inhibition of chlorzoxazone metabolism: a clinical probe for P450 2E1. Clin Pharmacol Ther. 1993;53:643–650. doi: 10.1038/clpt.1993.85. [DOI] [PubMed] [Google Scholar]
- 14.Brady JF, Xiao F, Wang MH, et al. Effects of disulfiram on hepatic P450IIE1, other microsomal enzymes, and hepatotoxicity in rats. Toxicol Appl Pharmacol. 1991;108:366–373. doi: 10.1016/0041-008x(91)90125-x. [DOI] [PubMed] [Google Scholar]
- 15.Lown KS, Bailey DG, Fontana RJ, et al. Grapefruit juice increases felodipine oral availability in humans by decreasing intestinal CYP3A protein expression [see comments] J Clin Invest. 1997;99:2545–2553. doi: 10.1172/JCI119439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaukonen KM, Olkkola KT, Neuvonen PJ. Itraconazole increases plasma concentrations of quinidine. Clin Pharmacol Ther. 1997;62:510–517. doi: 10.1016/S0009-9236(97)90046-1. [DOI] [PubMed] [Google Scholar]
- 17.Back DJ, Tjia JF. Comparative effects of the antimycotic drugs ketoconazole, fluconazole, itraconazole and terbinafine on the metabolism of cyclosporin by human liver microsomes. Br J Clin Pharmacol. 1991;32:624–626. doi: 10.1111/j.1365-2125.1991.tb03963.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bailey DG, Bend JR, Arnold JM, et al. Erythromycin–felodipine interaction: magnitude, mechanism, and comparison with grapefruit juice. Clin Pharmacol Ther. 1996;60:25–33. doi: 10.1016/S0009-9236(96)90163-0. [DOI] [PubMed] [Google Scholar]
- 19.Frye RF, Matzke GR, Adedoyin A, et al. Validation of the five-drug ‘Pittsburgh cocktail’ approach for assessment of selective regulation of drug-metabolizing enzymes. Clin Pharmacol Ther. 1997;62:365–376. doi: 10.1016/S0009-9236(97)90114-4. [DOI] [PubMed] [Google Scholar]
- 20.Nielsen F, Nielsen KK, Brosen K. Determination of quinidine, dihydroquinidine, (3S)-3-hydroxyquinidine and quinidine N-oxide in plasma and urine by high-performance liquid chromatography. J Chromatogr B Biomed Appl. 1994;660:103–110. doi: 10.1016/0378-4347(94)00259-2. [DOI] [PubMed] [Google Scholar]
- 21.Veronese ME, Miners JO, Randles D, et al. Validation of the tolbutamide metabolic ratio for population screening with use of sulfaphenazole to produce model phenotypic poor metabolizers. Clin Pharmacol Ther. 1990;47:403–411. doi: 10.1038/clpt.1990.46. [DOI] [PubMed] [Google Scholar]
- 22.Vinks A, Inaba T, Otton SV, et al. Sparteine metabolism in Canadian Caucasians. Clin Pharmacol Ther. 1982;31:23–29. doi: 10.1038/clpt.1982.4. [DOI] [PubMed] [Google Scholar]
- 23.Sanz EJ, Villen T, Alm C, et al. S-mephenytoin hydroxylation phenotypes in a Swedish population determined after coadministration with debrisoquin. Clin Pharmacol Ther. 1989;45:495–499. doi: 10.1038/clpt.1989.63. [DOI] [PubMed] [Google Scholar]
- 24.Rasmussen BB, Brosen K. Theophylline has no advantages over caffeine as a putative model drug for assessing CYPIA2 activity in humans. Br J Clin Pharmacol. 1997;43:253–258. doi: 10.1111/j.1365-2125.1997.00546.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fuhr U, Rost KL, Engelhardt R, et al. Evaluation of caffeine as a test drug for CYP1A2, NAT2 and CYP2E1 phenotyping in man by in vivo versus in vitro correlations. Pharmacogenetics. 1996;6:159–176. doi: 10.1097/00008571-199604000-00003. [DOI] [PubMed] [Google Scholar]
- 26.Lykkesfeldt J, Loft S, Poulsen HE. Simultaneous determination of urinary free cortisol and 6 beta-hydroxycortisol by high-performance liquid chromatography to measure human CYP3A activity. J Chromatogr B Biomed Appl. 1994;660:23–29. doi: 10.1016/0378-4347(94)00265-7. [DOI] [PubMed] [Google Scholar]
- 27.Pan J, Hong JY, Li D, et al. Regulation of cytochrome P450 2B1/2 genes by diallyl sulfone, disulfiram, and other organosulfur compounds in primary cultures of rat hepatocytes. Biochem Pharmacol. 1993;45:2323–2329. doi: 10.1016/0006-2952(93)90206-c. [DOI] [PubMed] [Google Scholar]
- 28.Beach CA, Mays DC, Guiler RC, et al. Inhibition of elimination of caffeine by disulfiram in normal subjects and recovering alcoholics. Clin Pharmacol Ther. 1986;39:265–270. doi: 10.1038/clpt.1986.37. [DOI] [PubMed] [Google Scholar]
- 29.Ochs HR, Greenblatt DJ, Woo E. Clinical pharmacokinetics of quinidine. Clin Pharmacokinet. 1980;5:150–168. doi: 10.2165/00003088-198005020-00003. [DOI] [PubMed] [Google Scholar]
- 30.Notterman DA, Drayer DE, Metakis L, et al. Stereoselective renal tubular secretion of quinidine and quinine. Clin Pharmacol Ther. 1986;40:511–517. doi: 10.1038/clpt.1986.216. [DOI] [PubMed] [Google Scholar]
- 31.Wacher VJ, Wu CY, Benet LZ. Overlapping substrate specificities and tissue distribution of cytochrome P450, 3A and P–glycoprotein: implications for drug delivery and activity in cancer chemotherapy. Mol Carcinog. 1995;13:129–134. doi: 10.1002/mc.2940130302. [DOI] [PubMed] [Google Scholar]
- 32.Emi Y, Tsunashima D, Ogawara K, et al. Role of P-glycoprotein as a secretory mechanism in quinidine absorption from rat small intestine. J Pharm Sci. 1998;87:295–299. doi: 10.1021/js970294v. [DOI] [PubMed] [Google Scholar]
- 33.Kimura T, Emi Y, Tsunashima D, et al. Role of P-glycoprotein as a secretory mechanism in quinidine: absorption from rat small intestine. Proc Control Release Soc. 1997;24:371–372. doi: 10.1021/js970294v. [DOI] [PubMed] [Google Scholar]
- 34.Kusuhara H, Suzuki H, Terasaki T, et al. P-Glycoprotein mediates the efflux of quinidine across the blood–brain barrier. J Pharmacol Exp Ther. 1997;283:574–580. [PubMed] [Google Scholar]
- 35.Jalava KM, Partanen J, Neuvonen PJ. Itraconazole decreases renal clearance of digoxin. Ther Drug Monit. 1997;19:609–613. doi: 10.1097/00007691-199712000-00001. [DOI] [PubMed] [Google Scholar]
- 36.Nattel S, Ranger S, Talajic M, et al. Erythromycin-induced long QT syndrome: concordance with quinidine and underlying cellular electrophysiologic mechanism. Am J Med. 1990;89:235–238. doi: 10.1016/0002-9343(90)90305-w. [DOI] [PubMed] [Google Scholar]
- 37.Spinler SA, Cheng JW, Kindwall KE, et al. Possible inhibition of hepatic metabolism of quinidine by erythromycin. Clin Pharmacol Ther. 1995;57:89–94. doi: 10.1016/0009-9236(95)90270-8. [DOI] [PubMed] [Google Scholar]
- 38.Lin JC, Quasny HA. QT prolongation and development of torsades de pointes with the concomitant administration of oral erythromycin base and quinidine. Pharmacotherapy. 1997;17:626–630. [PubMed] [Google Scholar]
- 39.Yasui N, Otani K, Kaneko S, et al. A kinetic and dynamic study of oral alprazolam with and without erythromycin in humans: In vivo evidence for the involvement of CYP3A4 in alprazolam metabolism. Clin Pharmacol Ther. 1996;59:514–519. doi: 10.1016/S0009-9236(96)90179-4. [DOI] [PubMed] [Google Scholar]
- 40.von Moltke LL, Greenblatt DJ, Duan SX, et al. Inhibition of terfenadine metabolism in vitro by azole antifungal agents and by selective serotonin reuptake inhibitor antidepressants: relation to pharmacokinetic interactions in vivo [see comments] J Clin Psychopharmacol. 1996;16:104–112. doi: 10.1097/00004714-199604000-00002. [DOI] [PubMed] [Google Scholar]
- 41.von Moltke LL, Greenblatt DJ, Harmatz JS, et al. Triazolam biotransformation by human liver microsomes in vitro: effects of metabolic inhibitors and clinical confirmation of a predicted interaction with ketoconazole. J Pharmacol Exp Ther. 1996;276:370–379. [PubMed] [Google Scholar]
- 42.Hofsli E, Nissen MJ. Reversal of drug resistance by erythromycin: Erythromycin increases the accumulation of actinomycin D and doxorubicin in multidrug-resistant cells. Int J Cancer. 1989;44:149–154. doi: 10.1002/ijc.2910440126. [DOI] [PubMed] [Google Scholar]
- 43.Min DI, Ku YM, Geraets DR, et al. Effect of grapefruit juice on the pharmacokinetics and pharmacodynamics of quinidine in healthy volunteers. J Clin Pharmacol. 1996;36:469–476. doi: 10.1002/j.1552-4604.1996.tb05034.x. [DOI] [PubMed] [Google Scholar]
- 44.Fukuda K, Ohta T, Yamazoe Y. Grapefruit component interacting with rat and human P450 CYP3A: possible involvement of non-flavonoid components in drug interaction. Biol Pharm Bull. 1997;20:560–564. doi: 10.1248/bpb.20.560. [DOI] [PubMed] [Google Scholar]
- 45.Schmiedlin RP, Edwards DJ, Fitzsimmons ME, et al. Mechanisms of enhanced oral availability of CYP3A4 substrates by grapefruit constituents: Decreased enterocyte CYP3A4 concentration and mechanism-based inactivation by furanocoumarins. Drug Metab Dispos. 1997;25:1228–1233. [PubMed] [Google Scholar]
- 46.Edwards DJ, Bellevue FH, Woster PM. Identification of 6′,7′-dihydroxybergamottin, a cytochrome P450 inhibitor, in grapefruit juice. Drug Metab Dispos. 1996;24:1287–1290. [PubMed] [Google Scholar]
- 47.Fuhr U, Klittich K, Staib AH. Inhibitory effect of grapefruit juice and its bitter principal, naringenin, on CYP1A2 dependent metabolism of caffeine in man. Br J Clin Pharmacol. 1993;35:431–436. doi: 10.1111/j.1365-2125.1993.tb04162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fuhr U, Maier A, Keller A, et al. Lacking effect of grapefruit juice on theophylline pharmacokinetics. Int J Clin Pharmacol Ther. 1995;33:311–314. [PubMed] [Google Scholar]