

Abstract
Aims
The pharmacokinetics of dihydrocodeine (DHC) and its active metabolite dihydromorphine (DHM) were assessed after a single oral dose of DHC and after increasing doses of DHC at steady-state.
Methods
Twelve healthy male volunteers (18–45 years, CYP2D6 extensive metabolizers (EMs), MR<1 took a single oral dose (s.d.) of DHC 60 mg after breakfast. After 60 h DHC 60 mg was administered twice daily for 3 days, the dose was increased to 90 mg twice daily for 3 days, the final dose of 120 mg was administered twice daily for 3 days (multiple dose: m.d.). Blood sampling and urine collection: during 60 h after s.d. and during 12 h after m.d.
Results
No significant differences in the area under the curve (AUC) of both, DHC and DHM could be detected after a single oral dose of 60 mg DHC (AUC (0,∞)) and during steady-state doses of 60 mg DHC (AUC(0,12 h)). During increasing steady-state doses of DHC, the data showed a dose linearity of AUC, maximal serum concentration (Cmax) and minimal steady-state serum levels (Cssmin) of both, DHC and DHM (P<0.0001), point estimates of DHC dose corrected AUCs were well within the bioequivalence range (60 mg: 0.989; 90%CI 0.951–1.028, 90 mg: 0.997; 90%CI 0.959–1.036, 120 mg: 0.977; 90%CI 0.940–1.016). O-demethylation from DHC to DHM remained constant within the increasing steady-state doses of DHC in the 12 extensive metabolizers of CYP2D6.
Conclusions
In the studied dose range (60–120 mg) the pharmacokinetics of DHC and its active metabolite DHM are linear in EMs of CYP2D6.
Keywords: dihydrocodeine, dihydromorphine, multiple dose, pharmacokinetics, single dose
Introduction
Dihydrocodeine (DHC) is a semisynthetic opioid which is frequently used as an analgesic (WHO step 2) and antitussive drug. Furthermore, doses up to 2500 mg day−1 are prescribed in substitution therapy for heroin drug addicted people. Data on the pharmacokinetics and metabolism as well as pharmacodynamics of DHC are very limited [1–4].DHC is structurally related to the natural occurring opioid codeine. In several studies it has been demonstrated that codeine O-demethylation to morphine is mediated by the polymorphically expressed enzyme CYP2D6 [5–8]. Recently it was shown that DHC is also a substrate of the polymorphic enzyme CYP2D6 and the formation of the metabolite dihydromorphine (DHM) is catalysed mainly by this enzyme [3]. Further in vitro studies with human liver microsomes confirm these results; additionally it could be demonstrated that the formation of a second primary metabolite of DHC, nordihydrocodeine, is predominantly catalysed by CYP3 A4 [9].The third primary metabolite is DHC-6-glucuronide [3]. In clinical studies of Eckhardt et al. [10] and Poulsen et al. [11] analgesic effects of codeine could be demonstrated only in extensive and not in poor metabolizers of CYP2D6 using the cold pressure test as a pain model, indicating that the O-demethylated metabolite morphine is responsible for the analgesic effects of codeine in extensive metabolizers of CYP2D6. Similar analgesic mechanisms can be proposed for DHC, although in one recent study analgesic activity was not diminished in quinidine-induced poor metabolizers after a single dose of DHC using different pain models [12].
Regarding the impact of high steady-state doses of DHC in the treatment of chronic pain and in substitution therapy, pharmacokinetic data during higher steady-state doses of DHC and its metabolites are lacking. In addition, there is no knowledge about a possible saturation of the O-demethylating enzyme CYP2D6 during increasing steady state-doses of DHC. Therefore, the aim of our study was to assess the pharmacokinetics of DHC and its probable active metabolite DHM after multiple increasing doses of DHC under steady-state conditions in comparison with a single oral dose of DHC. Because of the genetic polymorphism, only extensive metabolizers of CYP2D6 were included in this study.
Methods
Subjects
Twelve healthy male Caucasian volunteers, extensive metabolizers of the CYP2D6 polymorphism, median age 30 years (23–40 years), median weight 75 kg (67–86 kg), median BMI 23 kg m−2 (21–27 kg m−2), participated in the study.
The study had been approved by the ethics commitee of the local medical board (Landesärztekammer Baden–Württemberg) according to the ethical guidelines of the 1975 Declaration of Helsinki. All volunteers gave their written informed consent prior to inclusion.
The subjects were healthy according to history, physical examination and laboratory tests, had no history of drug abuse and did not take any medication. The volunteers had been previously phenotyped with respect to sparteine oxidation with a single dose of 100 mg sparteine sulphate and subjects were classified according to the urinary metabolic ratio (MR) of sparteine and its 2- and 5-dehydrometabolite [13]. In order to minimize the variability of CYP2D6, only subjects with a MR<1 were included in this study. With regard to genotyping, using the methods of Stüven et al. [14] and Griese et al. [15], there was no genotype specific for poor metabolizers of CYP 2D6.
Study design
The study was performed as an open study with each subject receiving first a single oral dose of dihydrocodeine 60 mg after breakfast with 100 ml water. After 60 h 60 mg dihydrocodeine was administered twice daily for 3 days, thereafter the dose was increased to 90 mg twice daily for 3 days, thereafter the highest dose of 120 mg was administered twice daily for 3 days.
After single dose (60 mg) blood samples were taken before and 0.5, 1, 2, 3, 4, 5, 6, 8,10, 12, 24, 25, 35, 36, 48 and 49 h after administration, after multiple dosing before and 1, 2, 3, 4, 5, 6, 8, 10, 12 h after the sixth (last) administration of each dihydrocodeine dose regimen. Blood samples were centrifuged after 30 min and serum was stored at –20° C until analysed.
Urine was collected before drug administration, at 0–12, 12–24, 24–48 and 48–60 h after administration of single dose (60 mg) and during the last dosing interval (12 h) of each of the three dihydrocodeine dose regimens. A 10 ml aliquot of each urine sample was stored at −20° C until analysed. Vital signs (heart rate, blood pressure, respiration) and adverse events were assessed frequently before each drug administration and until 8 h post administration.
Study medication
Codicontin®; active compound: Dihydrocodeine tartrate (MWt: 451.47).
Formulation, strength: sustained release tablet with 60, 90, 120 mg dihydrocodeine tartrate Manufacturer: Mundipharma GmbH, Basel, Switzerland.
Analytical measurements
Determination of dihydrocodeine and dihydrocodeine metabolites in serum and urine using a modified GC–MS–MS method of Hofmann et al. [16]. 1 pmol [2H3]-dihydromorphine and 25 pmol [2H3]-dihydrocodeine were added as internal standards to 1 ml serum. Extraction was performed as described, and the pentafluoropropionyl derivatives prepared by treatment with 30 μl pentafluoropropionic anhydride, 10 μl pentafluoropropanol and 20 μl acetonitrile for 30 min at 60° C. The limits of quantification were 1 pmol ml−1 serum for dihydrocodeine and 50 fmol ml−1 serum for dihydromorphine. Quality control samples of dihydrocodeine (1.0, 100, 500 and 1000 pmol ml−1) and dihydromorphine (0.1, 2.0, 5.0 and 15.0 pmol ml−1) were routinely assayed with an intra-assay coefficient of variation of 10.2% or better and an interassay coefficient of variation less than 14%. Dihydromorphine and dihydrocodeine in urine (50 μl) were determined after 1:20 dilution with water as described above. The total amounts of dihydrocodeine and dihydromorphine (free and conjugated) were measured in a 1:20 dilution of 50 μl urine in water after acid hydrolysis (330 μl 37% hydrochloric acid) at 100° C for 45 min. Dihydrocodeine bitartrate was obtained from Merck (Darmstadt, Germany), and dihydromorphine hydrochloride was a gift from Mundipharma (Limburg, Germany).
Pharmacokinetic evaluation
Standard noncompartmental analysis for calculations using serum concentration-time data was performed with TOPFIT 2.0 (Gustav Fischer Verlag 1993).
The following pharmacokinetic parameters of dihydrocodeine (DHC) and dihydromorphine (DHM) were determined from the serum concentration-time data und the urine concentration data:
Cmax peak serum concentration (pmol ml−1): obtained from visual inspection of the serum concentration-time curves of DHC and DHM
tmax time to attain peak serum concentration (h): obtained from visual inspection of the serum concentration time curves of DHC and DHM
t1/2 terminal phase half-life calculated according to ln(2)/λz (h)
AUC area under the serum concentration time curve (nmol l−1 h): calculated by the trapezoidal rule, the segment to infinity was determined from the last concentration measured divided by the elimination rate constant (single dose); AUC after multiple doses was calculated by the trapezoidal rule using the serum concentration time data during the dosing interval (0,12 h).
ratio AUC AUC DHC: AUC DHM
Ae cumulative amount of drug or metabolite excreted in urine [% of dose]
CLo apparent oral clearance of DHC determined by dosep.o./AUCp.o. (ml min−1)
CLmetDHC→DHM metabolic clearance of DHC to DHM calculated as Ae total DHM
(free+glucuronidated)/AUC DHC (ml min−1). The true CLmet is probably higher due to the formation of nordihydromorphine and its glucuronides.
CLmet DHC→DHC-G metabolic clearance of DHC to DHC-glucuronide calculated as Ae DHC-glucuronide/AUC DHC (ml min−1)
CLmet DHM→DHM-G metabolic clearance of DHM to DHM-glucuronide calculated as Ae DHM-glucuronide/AUC DHM (ml min−1)
CLren DHC renal clearance of DHC calculated as Ae free DHC/AUC DHC (ml min−1)
CLren DHM renal clearance of DHM calculated as Ae free DHM/AUC DHM (ml min−1)
Cssmin minimal steady-state-concentration of DHC: average of values of 0 h and 12 h of the dosing interval (pmol ml−1) during multiple dose.
Statistical evaluation
The data are presented as mean±95% CI.
Statistical analysis of the obtained pharmacokinetic parameters within the different DHC doses were performed with a repeated measures anova analysis using the statistical program Instat 3.0 (Graphpad Software, 1990). If anova supported a statistically significant difference (P<0.05), Bonferroni multiple comparison test was performed as post test. All statistical tests were performed as two-tailed test with a level of significance of 0.05.
Additional assessment of bioequivalence under single and multiple dose conditions: bioequivalence testing was performed by calculating the individual ratios
test/reference (test=dose–corrected AUC multiple dose, normalized to 60 mg DHC dose; reference=AUC 60 mg single dose).
Point estimates (geometric means) and shortest 90% CI after logarithmic transformation and re-transformation were given for the ratios. Test was considered bioequivalent to reference if the 90% CI of the AUCs(0,∞) (single dose) and AUC(0,12 h)(multiple dose) ratios were within 0.80–1.25. (Statgraphics Plus 2.0)
In order to verifiy if steady-state was reached, a Wilcoxon matched pairs signed rank test was applied to the DHC serum concentrations before the last dose of each dose level and the concentration measured 12 h thereafter.
Results
The serum time-concentrations of dihydrocodeine (DHC) and dihydromorphine (DHM) after single and multiple oral dosages of DHC are presented in Figures 1, 2a and 2b.
Figure 1.

Serum—time concentrations of DHC (○) and DHM (▵) after 60 mg DHC single dose; blood sampling: 0–49 h; n =12; mean±s.d.
Figure 2.

a) Serum—time concentrations of DHC after 60 mg (○), 90 mg (▵), 120 mg (◊)DHC multiple dose; blood sampling: 0–12 h; n =12; mean±s.d. and b) Serum—time concentrations of DHM after 60 mg (▵), 90 mg (□), 120 mg (○) DHC multiple dose; blood sampling: 0–12 h; n =12; mean±s.d.
No significant differences in the area under the curve (AUC) of both, DHC and DHM could be detected after a single oral dose of 60 mg DHC and during steady-state doses of 60 mg DHC. The mean percentage extrapolation of AUC to infinity after single dose was 0.3% (95% CI 0.2–0.4) for DHC and 6.2% (95% CI 3.6–8.8) for DHM. Concerning DHC, there were no significant changes in apparant oral clearance (CLo), glucuronidation (Cmet DHC→DHC-G) and renal clearance (CLren). In addition, no significant difference in the urinary excretion of total DHC+total DHM (free+glucuronidated drug) was observed (see Table 1, 2). After single dose administration glucuronidated DHC was the main metabolite (49.3% of dose), free DHC represented 23.5% of dose, total DHM represented 5.4% of dose in urinary recovery.
Table 1.
Pharmacokinetic data of DHC after single dose (s.d.) and multiple doses (m.d.) (n =12; mean, 95% CI).
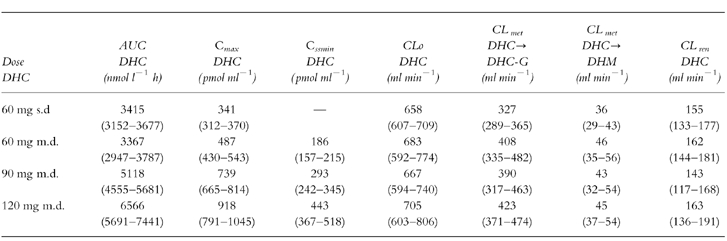
Table 2.
Pharmacokinetic data of DHM and urinary excretion of total DHC and DHM (free+glucuronidated drug) after single dose (s.d.) and multiple doses (m.d.) (n =12; mean, 95% CI).
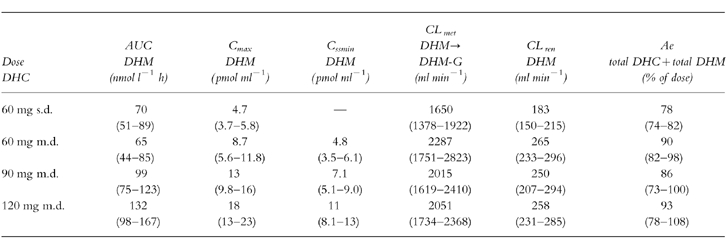
The pharmacokinetic data show an increase of AUC, maximal serum concentration (Cmax) and minimal steady-state serum levels (Cssmin) of both, DHC and DHM after increasing steady-state doses of DHC (P<0.0001) (Tables 1 and 2). In contrast, no significant dose-dependent differences were observed concerning apparant oral clearance of DHC, renal clearance of DHC and partial metabolic clearance from DHC to DHC-glucuronide and to the O-demethylated metabolite DHM (Tables 1 and 2). DHC O-demethylation to DHM remained constant within the increasing steady-state doses of DHC in the 12 extensive metabolizers of CYP2D6: Regression analysis of the ratios AUC DHC/AUC DHM demonstrated no significant changes within the three steady-state doses (Figure 3). In addition, there was no significant difference in the urinary excretion of DHC and DHM (Table 2).
Figure 3.

Regression analysis: Ratio AUC DHC/AUC DHM and the corresponding DHC doses (mg) after multiple doses of DHC; n =12; slope: −0.09±0.12; 95%CI of slope; −0.33–0,15; P: NS.
Additional assessment of bioequivalence of DHC after single and multiple doses of DHC
Dose–corrected AUCs (AUC per mg dose, normalized to 60 mg dose) of DHC and DHM after the three multiple doses (DHC 60 mg, 90 mg, 120 mg) were tested in reference to corresponding AUC obtained after 60 mg DHC single dose administration. Point estimates and shortest 90% CI were all inbetween the bioequivalence ranges from 0.80 to 1.25:0.989 (0.951–1.028), 0.997 (0.959–1.036), 0.977 (0.940–1.016) for DHC, 0.949 (0.905–0.995), 0.972 (0.927–1.020), 0.974 (0.928–1.021) for DHM.
Steady-state
For each of the three different doses (60, 90, 120 mg) steady-state was achieved as no differences in DHC serum concentrations were observed between Cssmin before the last dose of each dose interval and 12 h after the last dose.
Side-effects
Opiate related side-effects occurred especially during higher steady state doses of DHC: headache, nausea, vomiting, dizziness, constipation, pain of stomach, mild urinary retention, dry mouth, and mild euphoria. The side-effects were mild and reversible and none of the subject had to discontinue the study.
Discussion
Although DHC is included in the treatment of chronic pain (WHO step 2), there is a lack of pharmacokinetic data especially during increasing steady-state doses of DHC. Our data show that in the dose range studied (60–120 mg twice daily) the pharmacokinetics of DHC and its active metabolite DHM are linear in extensive metabolizers of CYP2D6; there was a linear, dose dependent increase of AUC, Cmax and Cssmin of both, DHC and DHM, for the three doses of DHC used. Clearance of DHC and DHM (CLo, partial metabolic clearance from DHC to DHC-glucuronides, DHC to DHM, DHM to DHM-glucuronides, CLren of DHC and DHM and renal excretion were not affected during the three steady-state doses of DHC, indicating that metabolism and excretion of DHC and its metabolites are not dose-dependent.
There is further evidence from in vitro data with human liver microsomes that formation of DHC-metabolites is linear because of high Km values for O-demethylation (981 μm), N-demethylation (9541 μm) and glucuronidation (1566 μm) of DHC [9, 17]; these Kmvalues are more than 1000-fold higher than the maximal serum concentrations of DHC after 120 mg multiple dosing in this study.
Bioequivalence assessment of dose–corrected AUCs after single and multiple doses demonstrated point estimators and 90% CI well within the range of 0.8–1.25, therefore the dose–corrected AUCs in the dose range studied can be regarded as bioequivalent with the single dose AUC.
The AUC ratios DHC/DHM did not differ between the three steady-state doses, indicating that there is no saturation of CYP2D6-depending O-demethylation of DHC in extensive metabolizers. These data are in agreement with Mikus et al. [18], who could show that the AUC ratios DHC/DHM in EM subjects undergoing DHC replacement therapy for opioid dependence using daily doses of 435–1350 mg DHC (actual given single dose before blood sampling 120–390 mg) were in the same range.
The pharmacokinetic parameters for DHC after single dose are similar to those reported by Fromm et al. [3] using the same amount of DHC, confirming the results of that study.
Dihydrocodeine is structurally related to codeine. Codeine O-demethylation is also mediated by the polymorphic enzyme CYP2D6 [5–7]. The pharmacokinetics of codeine appears to be linear [19]. The major metabolic pathway of codeine is the formation of codeine-6-glucuronide. O-demethylation of codeine to morphine constitutes a minor pathway accounting for about 7% of the dose administered [19, 20]. In our study, the Ae of total DHM was 6.7%, 6.5% and 6.7% for the three steady-state doses of DHC.
Comparing the pharmacokinetics of DHC after 60 mg single dose and 60 mg multiple dose, our data show no differences in the AUC of DHC,CLo, clearance to DHC-glucuronides, renal clearance of DHC and the urinary recovery of total DHC and DHM; partial metabolic clearance from DHC to DHM increased slightly, just reaching statistical significance, but it did not differ between the three steady-state doses.
In conclusion, in the dose range studied (60–120 mg) the pharmacokinetics of DHC and its active metabolite DHM were linear in extensive metabolizers of CYP2D6. There was a linear, dose dependent increase of AUC, Cmax and Cssmin of both, DHC and DHM, within the three steady-state doses of DHC. Metabolic ratio, clearance and renal excretion were not affected, indicating that metabolism and excretion of DHC and its metabolites is not dose dependent.
Acknowledgments
The study was supported by the Robert Bosch–Foundation, Stuttgart, Germany and by Mundipharma Pharmaceutical Company, Basel, Switzerland. We appreciate the excellent technical assistance of Mrs Erika Schneider and Mrs Anja Ziegler.
References
- 1.Rowell FJ, Seymour RA, Rawlins MD. Pharmacokinetics of intravenous and oral dihydrocodeine and its acid metabolites. Eur J Clin Pharmacol. 1983;25:419–424. doi: 10.1007/BF01037958. [DOI] [PubMed] [Google Scholar]
- 2.Davies KN, Castleden CM, McBurney A, Jagger C. The effect of ageing on the pharmacokinetics of dihydrocodeine. Eur J Clin Pharmacol. 1989;37:375–379. doi: 10.1007/BF00558503. [DOI] [PubMed] [Google Scholar]
- 3.Fromm MF, Hofman U, Griese E-U, Mikus G. Dihydrocodeine: a new opioid substrate for the polymorphic CYP2D6 in humans. Clin Pharmacol Ther. 1995;58:374–382. doi: 10.1016/0009-9236(95)90049-7. [DOI] [PubMed] [Google Scholar]
- 4.Wotherspoon HA, Kenny GNC, McArdle CS. Analgesic efficacy of controlled release dihydrocodeine. Anaesthesia. 1991;46:915–917. doi: 10.1111/j.1365-2044.1991.tb09845.x. [DOI] [PubMed] [Google Scholar]
- 5.Chen ZR, Somogyi AA, Bochner F. Polymorphic O-demethylation of codeine. Lancet. 1988;ii:914–915. doi: 10.1016/s0140-6736(88)92529-9. [DOI] [PubMed] [Google Scholar]
- 6.Dayer P, Desmeules J, Leeman T, Striberni R. Bioactivation of the narcotic drug codeine in humans is medicated by the polymorphic monooxygenase catalysing debrisoquin 4-hydroxylation. Biochem Biophys Res Commun. 1988;152:411–416. doi: 10.1016/s0006-291x(88)80729-0. [DOI] [PubMed] [Google Scholar]
- 7.Yue QJ, Svennson JO, Alm C, Sjöquist F, Säwe J. Codeine O-demethylation co-seggregates with polymorphic debrisoquine hydroxylation. Br J Clin Pharmacol. 1989;28:639–645. doi: 10.1111/j.1365-2125.1989.tb03556.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mikus G, Trausch B, Rodewald C, et al. Effects of codeine on gastrointestinal motility in relation to CYP2D6 phenotype. Clin Pharmacol Ther. 1997;61:459–465. doi: 10.1016/S0009-9236(97)90196-X. [DOI] [PubMed] [Google Scholar]
- 9.Kirkwood LC, Nation RL, Somogyi AA. Characterisation of the human cytochrome P450 enzymes involved in the metabolism of dihydrocodeine. Br J Clin Pharmacol. 1997;44:549–555. doi: 10.1046/j.1365-2125.1997.t01-1-00626.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eckhardt K, Li S, Ammon S, Schänzle G, Mikus G, Eichelbaum M. Same incidence of adverse drug events after codeine administration irrespective of the genetically determined differences in morphine formation. Pain. 1998;76:27–33. doi: 10.1016/s0304-3959(98)00021-9. [DOI] [PubMed] [Google Scholar]
- 11.Poulsen L, Brosen K, Arendt-Nielsen L, Gram LF, Elbaek K, Sindrup SH. Codeine and morphine in extensive and poor metabolizers of sparteine: pharmacokinetics, analgesic effect and side effects. Eur J Clin Pharmacol. 1996;51:289–295. doi: 10.1007/s002280050200. [DOI] [PubMed] [Google Scholar]
- 12.Wilder-Smith CH, Hufschmid E, Thormann W. The visceral and somatic antinociceptive effects of dihydrocodeine and ist metabolite, dihydromorphine. A cross-over study with extensive and quinidine-induced poor metabolizers. Br J Clin Pharmacol. 1998;45:575–581. doi: 10.1046/j.1365-2125.1998.00727.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eichelbaum M, Bertilsson L, Säwe J, Zekorn C. Polymorphic oxidation of sparteine and debrisoquine: related pharmacogenetic entities. Clin Pharmacol Ther. 1982;31:184–186. doi: 10.1038/clpt.1982.29. [DOI] [PubMed] [Google Scholar]
- 14.Stüven T, Griese E-U, Kroemer HK, Eichelbaum M, Zanger U. Rapid detection of CYP2D6 null alleles by long distance—and multiplex—polymerase chain reaction. Pharmacogenetics. 1996;6:417–421. doi: 10.1097/00008571-199610000-00005. [DOI] [PubMed] [Google Scholar]
- 15.Griese E-U, Zanger U, Brudermanns U, et al. Assessment of the predictive power of genotypes for the in-vivo catalytic function of CYP2D6 in a German population. Pharmacogenetics. 1998;8:15–26. doi: 10.1097/00008571-199802000-00003. [DOI] [PubMed] [Google Scholar]
- 16.Hofmann U, Fromm MF, Johnson S, Mikus G. Simultaneous determination of dihydrocodeine and dihydromorphine in serum by gas chromatography-tandem mass spectrometry. J Chromatogr B Biomed Appl. 1995;663:59–65. doi: 10.1016/0378-4347(94)00423-3. [DOI] [PubMed] [Google Scholar]
- 17.Kirkwood L, Nation RL, Somogyi AA. Glucuronidation of dihydrocodeine by human liver microsomes and the effect of inhibitors. Clin Exp Pharmacol Physiol. 1998;25:266–270. doi: 10.1111/j.1440-1681.1998.t01-19-.x. [DOI] [PubMed] [Google Scholar]
- 18.Mikus G, Ulmer A, Mörike K, Hofmann U. Heroin substitution therapy: Pharmacokinetics of dihydrocodeine. Naunyn-Schmiedeberg’s Arch Pharmacol. 1998;358(Suppl 2):47–28. [Google Scholar]
- 19.Chen ZR, Somogyi AA, Reynolds G, Bochner F. Disposition and metabolism of codeine after single and chronic doses in one poor and seven extensive metabolizers. Br J Clin Pharmacol. 1991;31:381–390. doi: 10.1111/j.1365-2125.1991.tb05550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yue QY, Hasselström J, Svensson JO, Säwe J. Pharmacokinetics of codeine and ist metabolites in Caucasian healthy volunteers: comparisons between extensive and poor hydroxilators of debrisoquine. Br J Clin Pharmacol. 1991;31:635–642. doi: 10.1111/j.1365-2125.1991.tb05585.x. [DOI] [PMC free article] [PubMed] [Google Scholar]