

Abstract
Aims
The frequency of CYP2C19 poor metabolizers (PMs) in populations of African descent has been reported to range from 1.0% to 35.4%. In order to determine with greater certainty the frequency of CYP2C19 PMs in such black populations we have performed a meta-analysis of the studies.
Methods
Relevant data on the frequency of both the PM phenotype of probe drugs (mephenytoin, omeprazole, and proguanil), and the distribution frequencies of CYP2C19 alleles and genotypes in black populations were summarized and reanalysed using a meta-analytical approach.
Results
Of nine reported studies two were excluded because of significant heterogeneity (χ2 = 115, P < 0.0001). The combined data from the remaining seven studies showed that the frequency of the PM phenotype in 922 healthy unrelated black Africans and black Americans ranged from 1.0% to 7.5% (n = 7 for combined data) with an overall frequency being 3.9% (36 of 922; 95%CI: 2.7%–5.2%). The frequency of the PM genotypes in blacks was 3.7% (36 of 966; 95%CI: 2.5%–4.9%), in agreement with the frequency of the PM phenotype. In the extensive metabolizers (EMs) 29% (271 of 930) were heterozygotes (wt/m). The observed frequencies of the three Mendelian genotypes were 0.68 for wt/wt, 0.28 for wt/m, and 0.04 for m/m. The allelic distribution was appropriate at 82.3% (95%CI: 80.5%-83.9%) for CYP2C19*1, 17.3% (95%CI:15.7%–19.0%) for CYP2C19*2 (m1), and 0.4% (95%CI: 0.1%–0.7%) for CYP2C19*3 (m2) in these populations.
Conclusions
We conclude that subjects of African ancestry have a low frequency of the CYP2C19 PM phenotype and genotype; that the defective CYP2C19 alleles are uncommon, and that a small proportion of heterozygotes exists in the EM subpopulation.
Keywords: (S)-mephenytoin hydroxylation, black subjects, CYP2C19, database, genetic polymorphism, meta-analysis
Introduction
Genetic polymorphism of (S)-mephenytoin 4′-hydroxylation is a well-documented polymorphically distributed pharmacogenetic trait in humans [1, 2]. Recently, human (S)-mephenytoin 4′-hydroxylase has been identified as cytochrome P450 (CYP) 2C19 [3, 4] and shown to be genetically polymorphic [5–8], with individuals being phenotypically characterized as either extensive (EMs) or poor metabolizers (PMs) according to their ability to oxidize CYP2C19 substrates. This polymorphism is relevant to the oxidative metabolism of a number of clinically important drugs [1, 2], such as diazepam, certain barbiturates, tricyclic antidepressants, omeprazole and proguanil. As a result, pronounced genetically determined differences in the disposition of these drugs may affect their efficacy and toxicity. Also, CYP2C19 polymorphism may contribute to a metabolic predisposition to certain diseases including non-aggressive bladder cancer [9], lung cancer (squamous cell carcinoma) [10], scleroderma or systemic sclerosis [11] and the eosinophilia–myalgia syndrome [12]. Thus defining the frequency of this polymorphism in different populations has considerable epidemiologic importance. Several polymorphisms in the CYP2C19 gene have been identified and shown to cause phenotypic variability. To date, nine alleles have been described which include the active CYP2C19*1A (wt1) and 2C19*1B (wt2) alleles and seven defective alleles CYP2C19*2A (m1A), 2C19*2B (m1B), 2C19*3 (m2), 2C19*4 (m3), 2C19*5A (m4 or TRP433), 2C19*5B, and 2C19*6 (m5) (nomenclature from Ibeanu et al. 1998) [8]. There is an excellent correlation between genotype and phenotype, with defective CYP2C19 forms resulting in the PM phenotype as determined using a range of substrates [1, 2].
The frequency of the CYP2C19 PM phenotype is 4–5 times more common in Asian than in Caucasian populations, for example, 14.3% (95% CI:12.3%–16.4%) of 1117 Chinese subjects were CYP2C19 PMs [13] as compared with 3.2%–3.4% in Caucasian populations [1, 14, 15]. However, a wide variation has been reported in the frequency of the PM phenotype in populations of African ancestry (1.0%–35.4%), depending on the populations tested and the probe drugs used for phenotyping [16–24]. To synthesize these data and determine an overall population frequency of the PM phenotype and/or genotype in black subjects of African ancestry, all relevant eligible data were reanalysed.
Methods
All available information was retrieved by electronic searching of the English-language publications which appeared in the MEDLINE database (1980– October 1998), hand-searching abstracts of major related conferences since 1980 and checking relevant references cited in other publications. Any publications describing nonrelated and randomly selected healthy black subjects phenotyped as EMs or PMs, or genotyped as carriers of defective and/or active CYP2C19 alleles were included. As described elsewhere [13, 15, 25, 26], the text of each report was carefully reviewed using a meta-analytical approach, and the total sample size and numbers of phenotypes and/or genotypes of CYP2C19 were extracted. Because only the final data are eligible for meta-analysis, some reports in abstract form were not included in this meta-analysis.
Allocation of an individual to the PM phenotype is based on the subject’s ability to oxidize CYP2C19 substrates, such as (S)-mephenytoin [1, 5, 6, 14, 16, 18, 24], omeprazole [19, 24], and proguanil [17, 22]. All genotyping analysis was based on the recently developed method by de Morais et al. [5, 27, 28]. The principal genetic defect found in PMs of (S)-mephenytoin is a single G→A mutation in exon 5 of CYP2C19 gene (CYP2C19*2), creating an aberrant splice site [27]. A second defective allele (CYP2C19*3) is a single G→A mutation at position 636 of exon 4, creating a premature stop codon [28]. These single-point mutations remove, respectively, restriction endonucleases BamHI and SmaI sites [5, 6, 10, 18–21, 27–31].
The geographical distributions of the CYP2C19 phenotypes and genotypes reported in black populations are summarized in Table 1 and 2, respectively. To determine whether available data from each individual study could legitimately be pooled with the other studies, the homogeneity among the independent reports was first estimated on the basis of the overlap of the 95% confidence interval (CI) of the PM incidence in each study, since the intersection of plots of 95%CI of the actual incidence of PMs in each study may be used to visually judge the homogeneity of the studies [31]. Homogeneity across the studies was finally assessed with a standard χ2 test on 2×n– contingency tables as described elsewhere [13, 15, 25, 26]. Lack of a significant χ2 test indicated the homogeneity of eligible data for pooled analysis [31].
Table 1.
Distribution of S-mephenytoin 4′-hydroxylation phenotypes in various populations of African ancestry.
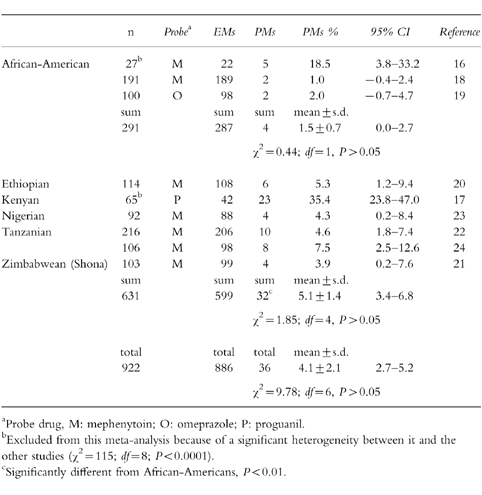
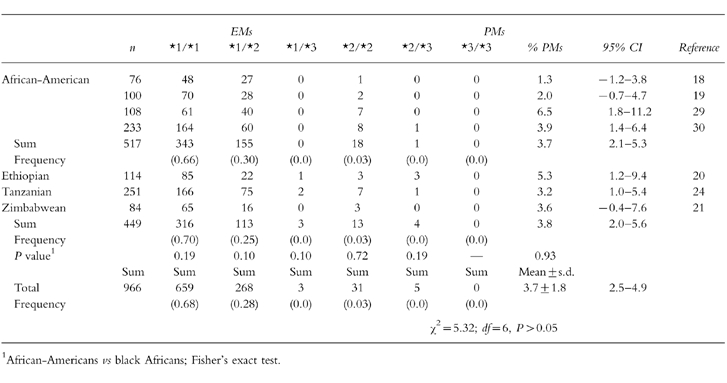
Table 2.
Distribution of the CYP2C19 genotypes in various populations of African ancestry.
It has been shown that the PM phenotype of CYP2C19 is inherited as an autosomal recessive trait [32–34]. According to the Hardy–Weinberg theory, the square root of the observed incidence of the PMs in a certain population tested should equal the total gene frequency (q) of the recessive alleles controlling the PM phenotype; the total gene frequency (p) of the dominant alleles controlling the expression of the EM phenotypes is equal to 1-q; the expected frequencies for the homozygous (wt/wt) and heterozygous (wt/m) high activity phenotypes are estimated to be p2 and 2pq, respectively [35]. Thus, the actual and predicted frequencies of the PM phenotype and/or genotype were compared.
Results
CYP2C19 phenotype
A statistically significant inconsistency existed across the three studies in African-Americans (χ2 = 26.62; d.f. = 2, P < 0.0001), and across the six studies in black Africans (χ2 = 75.63; d.f. = 5, P < 0.0001). When the two small studies, showing a PM frequency of 18.5% [16] and 35.4% [17] in an African-American and Kenyan population, respectively, were excluded from the meta-analysis, an excellent consistency was found in African-Americans (χ2 = 0.44; d.f. = 1, P = 0.51), and in black Africans (χ2 = 1.85; d.f. = 4, P = 0.76). This was due to significant heterogeneity between these and the other studies (χ2 = 114.96; d.f. = 8, P < 0.0001). A significant overlap of the 95% CIs of the PM phenotype frequencies was found between the separate observations (χ2 = 9.78; d.f. = 6, P = 0.13) in the remaining seven studies (Table 1). Because all the data with homogeneity may be used for pooled analysis, the overall estimate of the PM phenotype was 3.9% (36 of 922; 95%CI: 2.7%-5.2%) of all the blackpopulations tested, with a mean value of 4.1±2.1%. To further assess the potential population-based differences in this polymorphism, we compared the frequencies of CYP2C19 PM phenotype and found that there was no intersection of 95%CIs of both subtotal PM frequencies between African-Americans (0%-2.7%) and black Africans (3.4%-6.8%), and that black Africans have a higher frequency of CYP2C19 PM phenotype than African-Americans (5.1%vs 1.4%; P = 0.0056) as shown in Table 1.
CYP2C19 genotype
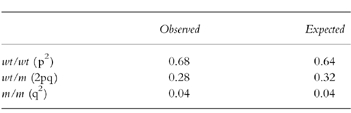
In the African-Americans studied, a good consistency existed in the distributions of various genotypes (χ2 = 12.24; d.f. = 9, P = 0.20), and in the frequencies of EMs and PMs genotypically identified (χ2 = 4.41; d.f. = 3, P = 0.22) as presented in Table 2. Similarly, a significant homogeneity occurred in the black African populations for the distributions of the genotypes (χ2 = 12.71; d.f. = 8, P = 0.12), and for the frequencies of genotyped EMs and PMs (χ2 = 0.94; d.f. = 2, P = 0.62). By comparison, there were no population-based differences in the prevalence of genotyped PMs (African-Americans vs Africans; 19 of 517 vs 17 of 449; χ2 = 0.0083; d.f. = 1, P = 0.93), or in the distributions of both major genotypes 2C19*1/*1 (343 of 517 vs 316 of 449; P = 0.19) and 2C19*1/*2 (155 of 517 vs 113 of 449; P = 0.10) between African-Americans and black Africans. Furthermore, systematic analysis of the genotypic data on PMs in all 7 studies indicated that there was no significant heterogeneity between the different studies (Table 2) (χ2 = 5.34; d.f. = 6, P = 0.50). The frequency of the PM genotype was 3.7% (36 of 966; 95%CI:2.5%–4.9%), with a mean value of 3.7±1.8%. Based on the estimate of the PM phenotype of 3.9%, the expected frequencies of the three Mendelian genotypes were calculated to be 0.64 for wt/wt, 0.32 for wt/m, and 0.04 for m/m in black populations (Table 3). In contrast, the observed frequencies of the three Mendelian genotypes were 0.68 (659 of 966) for wt/wt, 0.28 (271 of 966) for wt/m, and 0.04 (36 of 966) for m/m (Table 2 and Table 3), very close to expected.
Table 3.
Comparison of both observed and expected frequencies of the three Mendelian genotypes of CYP2C19 in various populations of African ancestry.
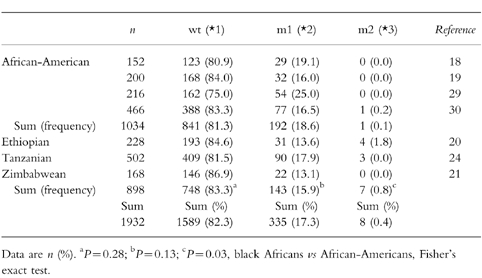
As shown in Table 4, an excellent consistency was found in the distributions of various alleles in the African-Americans (χ2 = 9.27; d.f. = 6, P = 0.16) or black Africans (χ2 = 7.70; d.f. = 4, P = 0.10), and also found in the distributions of active (wt) and inactive (m1 plus m2) alleles across the seven studies (χ2 = 12.30; d.f. = 6, P = 0.06) but not in the distributions of three major alleles (wt, m1, and m2) (χ2 = 27.63; d.f. = 12, P = 0.006), showing that black Africans have a relatively higher prevalence of the allele m2 than African-Americans (0.8%vs 0.1%; P = 0.03). In both populations, the observed allelic frequencies were as follows: CYP2C19*1 (82.3%; 95%CI: 80.5%-83.9%), 2C19*2 (17.3%; 95%CI: 15.7%-19.0%), and 2C19*3 (0.4%; 95%CI: 0.1%-0.7%).
Table 4.
The distribution frequency of the CYP2C19 alleles in various populations of African ancestry.
Discussion
The interethnic differences in the frequency of defective CYP2C19 alleles and/or diminished CYP2C19 catalytic activity are a subject of active research [1, 2]. This study has provided a systematic overview of the population distribution of the CYP2C19 PM phenotype and CYP2C19 genotypes and alleles in black subjects in different geographical areas. From Table 1, a precise estimate of the PM phenotype frequency based on the overview of the data Table 1) was 3.9% (95%CI: 2.7%-5.2%) in 922 healthy black subjects, in accordance with actual PM incidence (range:1.0%–7.5%; mean: 4.1%) in each study. As shown in Table 2, the frequency of PM genotypes was 3.7% (95%CI: 2.5%-4.9%) in 966 healthy black individuals. The PM frequencies by both phenotyping and genotyping are in complete agreement. The observed frequencies of the three Mendelian genotypes are in good agreement with the expected (Table 3), indicating that in this study the distribution of the defective or inactive alleles is in Hardy–Weinberg genetic equilibrium and the total sample size is adequate. Thus the combination of genotyping and phenotyping tests gives a ‘true value’ of the deficient PM frequency of CYP2C19 in black populations (~4%).
Greater variability in the frequency of CYP2C19 PM phenotype (1.0%-35.4%) was reported among different black populations (Table 1). The apparent discrepancy between the two excluded studies and the remaining seven studies may be due to their inadequate sample size, differences in ethnic origin or genetic background, or the different test drugs used for the phenotyping studies [16–24]. In the first study, a small study of elderly African-Americans (n = 27), 18.5% were found to be PMs [16], a finding perhaps influenced by the small sample size. In the second study, 35.4% of healthy Kenyan adults (n = 65) were identified as PMs using proguanil as the test drug [17]. Recent evidence has shown that the oxidative cyclization of proguanil (PG) to cycloguanil (CG) is catalysed by both CYP2C19 and CYP3A4 [36, 37] although the latter makes only a minor contribution in vivo [37]. The larger interindividual variability in CYP3A in vivo [38] and hence variability in its contribution to CG formation may mask a clear distinction between the ratios of PG to CG (PG/CG ratio) of the EMs and PMs, resulting in the frequency distribution pattern of the PG/CG ratio being highly skewed and continuous rather than bimodal in Caucasian [39] or black African [17] populations, and further making it difficult to assign an antimode. Also, the secondary parallel metabolic pathway of PG produces 4-chlorophenylbiguanide (4-CPBG) under of the control of CYP2C19 [40]. In theory, the ratio of PG to both metabolites (CG+4-CPBG), but not the PG/CG ratio, should reflect the true status of CYP2C19 activity in any individual. However, the two CYP2C19-mediated metabolites (CG and 4-CPBG) may not be the terminal metabolites, and hence variability in their subsequent metabolism will also affect the ratios, PG/CG, or PG/(CG+4-CPBG).
Potential widespread use of antimalarial drug(s) (e.g. chloroquine, and/or proguanil) may have occurred in the black African subjects who reside in a malaria-endemic area. Although chloroquine is not likely to interact with drug metabolism catalysed by CYP2C19 [41], the use of proguanil as a malaria chemoprophylactic drug might contribute to the relatively higher frequency of the CYP2C19 PM phenotype in black Africans in contrast to the African-Americans who are not exposed to antimalarials (5.1%vs 1.4%; P = 0.0056) (Table 1). The genotyping studies provide a direct marker for CYP2C19 status, and showed that the overall frequency of the CYP2C19 PM phenotype in black Africans, while not different from their genotypic frequency (phenotype vs genotype; 5.1%vs 3.8%, P = 0.21), was much more similar to that in African-Americans (CYP2C19 PM genotype: 3.8%vs 3.7%; P = 0.93) as were the three major genotypes, wt/wt, wt/m1, and m1/m1 (all P > 0.10) (Table 2).
Black populations are evolutionarily distinct from Asian and Caucasian populations [2]. The ancestors of African-Americans originated in Africa but significant genetic admixture has subsequently occurred. Furthermore, within Africa genetic and cultural diversity is substantial, and may thus affect the distribution of CYP2C19 polymorphic drug oxidation in these populations. As shown in Table 1–Table 3, there is some racial microheterogeneity in CYP2C19 activity between African-American and black African populations. Historically, it was from west Africa, particularly the coastal regions, that Africans were transported to North and South America as slaves. Thus the origin of many African-Americans is thought to have been west Africa. The African populations studied in Africa have been from east and central Africa. In fact, there were some distinct differences among the African populations living in ethnically diverse regions because of evolution in isolation and separation by race. The interaction with different genetic and environmental factors may result in the microheterogeneity of phenotypes and genotypes of CYP2C19 among the populations of African ancestry. In addition, it is possible that the estimated 20%–25% admixture of Caucasian alleles in the African-Americans’ gene pool [42] has diluted some alleles (e.g. m2) in black Americans.
The antimalarial agent proguanil is widely used in Africa because of endemic malaria. This has potential clinical implications for black PMs in endemic malarial areas. Proguanil is an inactive pro-drug that requires biotransformation to its therapeutically active metabolite cycloguanil, which is predominantly catalysed by CYP2C19 and to a minor extent by CYP3A4 [36, 37]. Therefore, decreased activation of proguanil to cycloguanil in CYP2C19 PMs may result in failure of malaria chemoprophylaxis. However, the low prevalence of the PM trait (~4%) in black Africans suggests that this will not be a major problem. The results from this study show that the relative proportions of the heterozygotes and homozygotes in the EM group in blacks was 29% (271 of 930) and 71% (659 of 930), respectively, which are very close to those reported in white EMs [26] but different from those in Chinese subjects [26]. Based on the relevant evidence from omeprazole clinical trials [43, 44], it is expected that gene dosage of CYP2C19 will affect the antimalarial action of proguanil when it is used in patients with malaria.
In summary, the present study has defined the prevalence of the genetic deficiency of CYP2C19 at 4% in the black populations of African ancestry. This estimate has utility in designing large scale epidemiological investigations and clinical trials in the population.
Acknowledgments
This study was supported by USPHS grants HL56251, GM-RR00095, and GM 31304. Dr Xie is a Merck International Fellow in Clinical Pharmacology. Dr Stein is a recipient of a Pharmaceutical Research and Manufacturers Association Foundation Career Development Award.
References
- 1.Wilkinson GR, Guengerich FP, Branch RA. Genetic polymorphism of S-mephenytoin hydroxylation. Pharmacol Ther. 1989;43:53–76. doi: 10.1016/0163-7258(89)90047-8. [DOI] [PubMed] [Google Scholar]
- 2.Bertilsson L. Geographic/interracial differences in polymorphic drug oxidation: Current state of the knowledge of cytochrome P450 (CYP) 2D6 and 2C19. Clin Pharmacokinet. 1995;29:192–209. doi: 10.2165/00003088-199529030-00005. [DOI] [PubMed] [Google Scholar]
- 3.Wrighton SA, Stevens JC, Becker GW, VandenBranden M. Isolation and characterization of human liver cytochrome P450 2C19: Correlation between 2C19 and S-mephenytoin 4′-hydroxylation. Arch Biochem Biophys. 1993;306:240–245. doi: 10.1006/abbi.1993.1506. [DOI] [PubMed] [Google Scholar]
- 4.Goldstein JA, Faletto MB, Romkes-Sparks M, et al. Evidence that CYP2C19 is the major (S) -mephenytoin 4′-hydroxylase in humans. Biochemistry. 1994;33:1743–1752. doi: 10.1021/bi00173a017. [DOI] [PubMed] [Google Scholar]
- 5.de Morais SMF, Goldstein JA, Xie HG, et al. Genetic analysis of the S-mephenytoin polymorphism in a Chinese population. Clin Pharmacol Ther. 1995;58:404–411. doi: 10.1016/0009-9236(95)90053-5. [DOI] [PubMed] [Google Scholar]
- 6.Xiao ZS, Goldstein JA, Xie HG, et al. Differences in the incidence of the CYP2C19 polymorphism affecting the S-mephenytoin phenotype in Chinese Han and Bai populations and identification of a new rare CYP2C19 mutant alleles. J Pharmacol Exp Ther. 1997;281:604–609. [PubMed] [Google Scholar]
- 7.Ferguson RJ, de Morais SMF, Benhamou S, et al. A new genetic defect in human CYP2C19: Mutation of the initiation codon is responsible for poor metabolism of S-mephenytoin. J Pharmacol Exp Ther. 1998;284:356–361. [PubMed] [Google Scholar]
- 8.Ibeanu GC, Goldstein JA, Meyer U, et al. Identification of new human CYP2C19 alleles (CYP2C19*6 and CYP2C19*2B) in a Caucasian poor metabolizer of mephenytoin. J Pharmacol Exp Ther. 1998;286:1490–1495. [PubMed] [Google Scholar]
- 9.Kaisary A, Smith P, Jacqz E, et al. Genetic predisposition to bladder cancer: ability to hydroxylate debrisoquine and mephenytoin as risk factors. Cancer Res. 1987;47:5488–5493. [PubMed] [Google Scholar]
- 10.Tsuneoka Y, Fukushima K, Matsuo Y, Ichikawa Y, Watanabe Y. Genetic analysis of the CYP2C19 gene in the Japanese population. Life Sci. 1996;59:1711–1715. doi: 10.1016/s0024-3205(96)00507-3. [DOI] [PubMed] [Google Scholar]
- 11.May DG, Black CM, Olsen NJ, et al. Scleroderma is associated with differences in individual routes of drug metabolism: a study with dapsone, debrisoquine, and mephenytoin. Clin Pharmacol Ther. 1990;48:286–295. doi: 10.1038/clpt.1990.151. [DOI] [PubMed] [Google Scholar]
- 12.Flockhart DA, Clauw DJ, Sale EB, Hewett J, Woosley RL. Pharmacogenetic characteristics of the eosinophilia–myalgia syndrome. Clin Pharmacol Ther. 1994;56:398–405. doi: 10.1038/clpt.1994.154. [DOI] [PubMed] [Google Scholar]
- 13.Xie HG, Xu ZH, Luo X, Huang SL, Zeng FD, Zhou HH. Genetic polymorphisms of debrisoquine and S-mephenytoin oxidative metabolism in Chinese populations: a meta-analysis. Pharmacogenetics. 1996;6:235–238. doi: 10.1097/00008571-199606000-00005. [DOI] [PubMed] [Google Scholar]
- 14.Reviriego J, Bertilsson L, Carrillo JA, Llerena A, Valdivielso MJ, Benítez J. Frequency of S-mephenytoin hydroxylation deficiency in 373 Spanish subjects compared to other Caucasian populations. Eur J Clin Pharmacol. 1993;44:593–595. doi: 10.1007/BF02440867. [DOI] [PubMed] [Google Scholar]
- 15.Alván G, Bechtel P, Iselius L, Gunder-Remy U. Hydroxylation polymorphisms of debrisoquine and mephenytoin in European populations. Eur J Clin Pharmacol. 1990;39:533–537. doi: 10.1007/BF00316090. [DOI] [PubMed] [Google Scholar]
- 16.Pollock BG, Perel JM, Kirshner M, Altieri LP, Yeager AL, Reynolds Cf., III S-mephenytoin 4-hydroxylation in older Americans. Eur J Clin Pharmacol. 1991;40:609–611. doi: 10.1007/BF00279979. [DOI] [PubMed] [Google Scholar]
- 17.Watkins WM, Mberu E, Nevill CG, Ward SA, Breckenridge AM, Koech DK. Variability in the metabolism of proguanil to the active metabolite cycloguanil in healthy Kenyan adults. Trans Roy Soc Trop Med Hyg. 1990;84:492–495. doi: 10.1016/0035-9203(90)90010-c. [DOI] [PubMed] [Google Scholar]
- 18.Edeki TI, Goldstein JA, de Morais SMF, et al. Genetic polymorphism of S-mephenytoin 4′-hydroxylation in African-Americans. Pharmacogenetics. 1996;6:357–360. doi: 10.1097/00008571-199608000-00009. [DOI] [PubMed] [Google Scholar]
- 19.Marinac JS, Balian JD, Foxworth JW, et al. Determination of CYP2C19 phenotype in black Americans with omeprazole: correlation with genotype. Clin Pharmacol Ther. 1996;60:138–144. doi: 10.1016/S0009-9236(96)90129-0. [DOI] [PubMed] [Google Scholar]
- 20.Persson I, Aklillu E, Rodrigues F, Bertilsson L, Ingelman-Sundberg M. S-mephenytoin hydroxylation phenotype and CYP2C19 genotype among Ethiopians. Pharmacogenetics. 1996;6:521–526. doi: 10.1097/00008571-199612000-00005. [DOI] [PubMed] [Google Scholar]
- 21.Masimirembwa C, Bertilsson L, Johansson I, Hasler JA, Ingelman-Sundberg M. Phenotyping and genotyping of S-mephenytoin hydroxylase (cytochrome P450 2C19) in a Shona population in Zimbabwe. Clin Pharmacol Ther. 1995;57:656–661. doi: 10.1016/0009-9236(95)90228-7. [DOI] [PubMed] [Google Scholar]
- 22.Skjelbo E, Mutabingwa TK, Bygbjerg IB, Nielson KK, Gram LF, Brøsen K. Chloroguanide metabolism in relation to the efficiency in malaria prophylaxis and the S-mephenytoin oxidation in Tanzanians. Clin Pharmacol Ther. 1996;59:304–311. doi: 10.1016/S0009-9236(96)80008-7. [DOI] [PubMed] [Google Scholar]
- 23.Iyun AO, Tucker GT, Woods HF, Lennard MS. The 4-hydroxylation of (S) -mephenytoin in Nigerians: a population study. Br J Clin Pharmacol. 1990;30:312P. [Abstract] [Google Scholar]
- 24.Herrlin K, Massele AY, Jande M, et al. Bantu Tanzanians have a decreased capacity to metabolize omeprazole and mephenytoin in relation to their CYP2C19 genotype. Clin Pharmacol Ther. 1998;64:391–401. doi: 10.1016/S0009-9236(98)90070-4. [DOI] [PubMed] [Google Scholar]
- 25.Xie HG, Xu ZH, Ou-Yang DS, et al. Meta-analysis of phenotype and genotype of NAT2 deficiency in Chinese populations. Pharmacogenetics. 1997;7:503–514. doi: 10.1097/00008571-199712000-00009. [DOI] [PubMed] [Google Scholar]
- 26.Xie HG. Direct evidence for the higher frequency of CYP2C19 allelic heterozygotes in Chinese subjects than in white subjects. Clin Pharmacol Ther. 1997;62:691–692. doi: 10.1016/S0009-9236(97)90088-6. [Letter] [DOI] [PubMed] [Google Scholar]
- 27.de Morais SMF, Wilkinson GR, Blaisdell J, Nakamura K, Meyer UA, Goldstein JA. The major genetic defect responsible for the polymorphism of S-mephenytoin in humans. J Biol Chem. 1994;269:15419–15422. [PubMed] [Google Scholar]
- 28.de Morais SMF, Wilkinson GR, Blaisdell J, Nakamura K, Meyer UA, Goldstein JA. Identification of a new genetic defect responsible for the polymorphism of S-mephenytoin metabolism in Japanese. Mol Pharmacol. 1994;46:594–598. [PubMed] [Google Scholar]
- 29.Goldstein JA, Ishizaki T, Chiba K, et al. Frequencies of the defective CYP2C19 alleles responsible for the mephenytoin poor metabolizer phenotype in various Oriental, Caucasian, Saudi Arabian, and American black populations. Pharmacogenetics. 1997;7:59–64. doi: 10.1097/00008571-199702000-00008. [DOI] [PubMed] [Google Scholar]
- 30.Martin DE, Tran JQ, Flockhart DA, Jorkasky DK. Analysis of CYP2D6 and CYP2C19 genotypes in large African-American and Caucasian populations. Clin Pharmacol Ther. 1998;63:206. [Abstract] [Google Scholar]
- 31.D’Agostino RB, Weintraub M. Meta-analysis: a method for synthesizing research. Clin Pharmacol Ther. 1995;58:605–616. doi: 10.1016/0009-9236(95)90016-0. [DOI] [PubMed] [Google Scholar]
- 32.Inaba T, Jurima M, Kalow W. Family studies of mephenytoin hydroxylation deficiency. Am J Hum Genet. 1986;38:768–772. [PMC free article] [PubMed] [Google Scholar]
- 33.Ward SA, Goto F, Nakamura K, Jacqz E, Wilkinson GR, Branch RA. S-mephenytoin 4-hydroxylase is inherited as an autosomal recessive trait in Japanese families. Clin Pharmacol Ther. 1987;42:96–99. doi: 10.1038/clpt.1987.114. [DOI] [PubMed] [Google Scholar]
- 34.Brøsen K, de Morais SMF, Meyer UA, Goldstein JA. Multifamily study on the relationship between CYP2C19 genotype and S-mephenytoin oxidation phenotype. Pharmacogenetics. 1995;5:312–317. doi: 10.1097/00008571-199510000-00007. [DOI] [PubMed] [Google Scholar]
- 35.Xie HG. How to correctly calculate the proportions of the heterozygotes and homozygotes in the extensive metabolizers of a certain drug-metabolizing enzyme in an ethnic or racial population? Clin Pharmacol Ther. 1998;64:575. doi: 10.1016/S0009-9236(98)90142-4. [Letter] [DOI] [PubMed] [Google Scholar]
- 36.Birkett DJ, Rees D, Andersson T, Gonzalez FJ, Miners JO, Veronese ME. In vitro proguanil activation to cycloguanil by human liver microsomes is mediated by CYP3A4 isoforms as well as by S-mephenytoin hydroxylase. Br J Clin Pharmacol. 1994;37:413–420. doi: 10.1111/j.1365-2125.1994.tb05707.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Funck-Brentano C, Becquemont L, Leneveu A, Roux A, Jaillon P, Beaune P. Inhibition by omeprazole of proguanil metabolism: Mechanism of the interaction in vitro and prediction of in vivo results from the in vitro experiments. J Pharmacol Exp Ther. 1997;280:730–738. [PubMed] [Google Scholar]
- 38.Thummel KE, Wilkinson GR. In vitro and in vivo interactions involving human CYP3A. Ann Rev Pharmacol Toxicol. 1998;38:389–430. doi: 10.1146/annurev.pharmtox.38.1.389. [DOI] [PubMed] [Google Scholar]
- 39.Ward SA, Watkins WM, Mberu E, et al. Intersubject variability in the metabolism of progaunil to the active metabolite cycloguanil in man. Br J Clin Pharmacol. 1989;17:781–787. doi: 10.1111/j.1365-2125.1989.tb03440.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jeppesen U, Rasmussen BB, Brøsen K. Fluvoxamine inhibits the CYP2C19-catalyzed bioactivation of chloroguanide. Clin Pharmacol Ther. 1997;62:279–286. doi: 10.1016/S0009-9236(97)90030-8. [DOI] [PubMed] [Google Scholar]
- 41.Masimirembwa CM, Gustafsson LL, Dahl ML, Abdi YA, Hasler JA. Lack of effect of chloroquine on debrisoquine (CYP2D6) and S-mephenytoin (CYP2C19) hydroxylation phenotype. Br J Clin Pharmacol. 1996;41:344–346. doi: 10.1046/j.1365-2125.1996.30713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reed TE. Caucasian genes in American Negroes. Science. 1969;165:762–768. doi: 10.1126/science.165.3895.762. [DOI] [PubMed] [Google Scholar]
- 43.Ohashi K, Furuta T, Kosuge K, et al. Disposition and effects of omeprazole are related to CYP2C19 genotype. Clin Pharmacol Ther. 1998;63:152. abstract. [Google Scholar]
- 44.Furuta T, Ohashi K, Kamata T, et al. Effect of genetic differences in omeprazole metabolism on cure rates for Helicobacter pylori infection and peptic ulcer. Ann Intern Med. 1998;129:1027–1030. doi: 10.7326/0003-4819-129-12-199812150-00006. [DOI] [PubMed] [Google Scholar]