

Abstract
Colorectal carcinoma is an important cause of cancer morbidity and mortality. 5-fluorouracil has been the major chemotherapeutic agent for the treatment of colorectal carcinoma for the past four decades. This regimen is noncurative, and its impact on survival is unclear. Attempts at identifying more effective chemotherapeutic agents for colorectal cancer have yielded oral formulations and prodrugs of 5-fluorouracil with apparently equivalent efficacy. Specific thymidylate synthase inhibitors are now available. Platinum analogues with activity in colorectal carcinoma, and no cross-resistance to the antimetabolites have also been developed. The topoisomerase I inhibitors represent a new class of agents with a novel mechanism of action. These agents are in phase II and Phase III clinical trials, others have been approved for clinical use within the last 3 years.
Keywords: chemotherapy, colorectal cancer, new agents
Introduction
Colorectal cancer is the third leading cause of cancer deaths for men and women in the United States. More than 130 000 new cases will be diagnosed in 1999 with more than 56 000 fatalities [1]. Approximately 60% of patients with colorectal cancer will require systemic therapy for metastatic disease, either at diagnosis or for disease recurrence. Most patients with metastases die from their disease, with less than 12% of patients alive at 2 years. Standard therapy remains 5-fluorouracil (5-FU) modulated with folinic acid. This regimen achieves response rates in only the 20–30% range, and its impact on survival is unclear [2, 3]. Infusional 5-FU regimens have yielded higher responses in patients, and confer a modest survival advantage over bolus schedules [4]. Until recently, patients failing optimally modulated 5-FU therapy had no other therapeutic options. However, new approaches to 5-FU modulation, and the discovery of new classes of cytotoxic agents with activity in colorectal cancer, promise a new era in the systemic treatment of this fatal disease. This review outlines the pharmacology and early clinical results of agents in phase II/III trials, and recently approved agents for the treatment of colorectal cancer.
Fluoropyrimidine modulation
Attempts at improving the efficacy of 5-FU have focused on the enhancement of the intratumoral concentrations of critical cytotoxic 5-FU anabolites. This has been achieved through combination with modulatory agents, usually with minimal intrinsic cytotoxic activity themselves. These agents, such as leucovorin, N-(phosphonacetyl)-l-aspartate (PALA), and interferon, interact with enzymes involved in the anabolic pathway [5–7]. In contrast to these earlier approaches, current modulatory efforts have focused on the catabolism of 5-FU. Administered 5-FU is almost entirely eliminated through catabolism (Figure 1). The initial rate-limiting enzyme in this pathway is dihydropyrimidine dehydrogenase (DPD), which is abundant in the GI tract, liver and peripheral blood mononuclear cells [8]. An inherited variability in the activity of this enzyme has been described, and is thought to account for the wide interpatient variability in 5-FU pharmacokinetics and oral bioavailability [9, 10]. Inhibitors of DPD, by blocking the degradation of orally administered 5-FU in gastrointestinal mucosa and other tissues will be expected to make the oral formulation of 5-FU more bioavailable, and increase the systemic exposure to the drug.
Figure 1.

The metabolic pathway of 5-fluorouracil. DPD, dihydropyrimidine dehydrogenase; EU, ethynyluracil; FdUMP, 5-fluorodeoxyuridine monophosphate; FUMP, 5-fluorouridine monophosphate; 5-FUTP, 5-fluorouridine triphosphate; 5-FUR, 5-fluorouridine; 5-dUdR, 5-deoxyuridine; H25-FU, dihydro-5-fluorouracil; FBal, fluoro-beta-alanine; PPRT, pyrimidine phosphoribosyl transferase; TK, thymidine kinase; ThdPh, thymidine phosphorylase; Urd Ph, uridine phosphorylase. Inactive metabolites (5-FUR, FBal, Urea, CO2) are in bold italics.
Eniluracil (776C85)
Eniluracil (ethynyluracil, EU) is an analogue of uracil which is a potent, irreversible inactivator of DPD in vivo [11]. It lacks antitumour activity but improves the therapeutic activity of 5-FU when compared with 5-FU alone. In rats with advanced colorectal cancer, eniluracil increased the therapeutic index of 5-FU six-fold compared with a two-fold increase with leucovorin and PALA. Cures were only achieved with the eniluracil/5-FU combination [12]. In dogs, dose-limiting neurotoxicity of 5-FU at high doses was abolished with the coadministration of eniluracil [13]. This observation has been explained by the prevention of formation of the potentially neurotoxic metabolite of 5-FU, alpha-fluoro-beta-alanine, after the inhibition of DPD [14]. In initial human bioavailability studies, eniluracil made oral 5-FU completely bioavailable (80–120%), increased its plasma half-life from 12 min to 4 h and made chronic oral dosing, which mimics prolonged infusional schedules, possible [15]. Phase I studies utilized two schedules of administration. When 5-FU was administered intravenously (i.v.) on days 1–5 in combination with oral eniluracil, the recommended phase II dose was 20 mg of eniluracil and 25 mg m−2 of 5-FU. Therapeutic activity was noted in 5-FU refractory colon cancer [16], and the dose-limiting toxicity was neutropaenia. Eniluracil and 5-FU have also been studied on a 28 day chronic oral dosing schedule. The recommended doses for further studies on this schedule are 1 mg m−2 5-FU twice a day, 12 h apart, and 10 mg eniluracil twice a day. Dose-limiting toxicity was diarrhoea [17]. Phase II trials utilising this dose and schedule in colorectal cancer have been completed. The North Central Cancer Treatment Group is conducting another phase II trial in colorectal cancer on a 5-day schedule of oral 5-FU 20 mg m−2 a day, and eniluracil 50 mg a day, starting a day before and ending a day after the administration of 5-FU [18]. A pivotal phase III trial in advanced colorectal cancer is currently ongoing. Patients are randomised to receive either standard 5-FU (425 mg m−2) and leucovorin (20 mg m−2) daily for 5 days every 4 weeks; or eniluracil 11.5 mg m−2 plus 5-FU 1.15 mg m−2 (a 10:1 ratio of eniluracil:5-FU) twice daily for 28 days, separated by a 7-day rest period.
UFT (Uracil-Ftorafur)
Ftorafur (Ft, 1-(2-tetrahydrofuranyl)-5-fluorouracil), is an oral pro-drug of 5-FU. It is completely absorbed through the gastrointestinal mucosa and metabolised through two major pathways to 5-FU. The first mechanism involves hydrolysis via the action of thymidine phosphorylase, and the second mechanism is oxidation through the action of cytochrome P450 (Figure 2a). Metabolic conversion occurs mainly in the liver and tumour tissue [19]. This drug was synthesised and introduced into clinical practice in Russia and Japan in 1967 [20]. Initial development in North America was abandoned because of lack of efficacy [21]. Review of the early Russian experience showed ftorafur to have only modest clinical activity [22]. In 1978, Fujii and coworkers demonstrated that 5-FU levels in tumour tissue were disproportionately increased compared with plasma levels when uracil was coadministered with ftorafur, leading to an increase in antitumour activity [23]. UFT is a combination of uracil and ftorafur in the optimal molar ratio of 4:1. Uracil is a natural substrate for DPD, and in this combination acts as a reversible, competitive inhibitor of DPD. UFT has been studied extensively in Japan on a chronic daily oral dosing schedule. Response rates of approximately 25% have been documented in stomach, colorectal, breast and pancreatico-biliary cancers [24]. In an attempt to improve its therapeutic activity, UFT has been combined with oral leucovorin. A phase II study of this combination (UFT 300–350 mg m−2 with leucovorin 150 mg day−1, orally for 28 days followed by a 7 day rest period) has been performed. An objective response rate of 42% (95% confidence interval 28% to 58%) was found in patients with metastatic colon cancer. The regimen was well-tolerated with diarrhoea, abdominal cramping and rash being the common toxicities. Neutropaenia and mucositis were uncommon [25]. Objective response rates of 25–43% in colorectal cancer patients have been reported in three other trials which employed different doses and schedules of oral leucovorin and UFT [26–28]. This combination is currently undergoing broad worldwide prospective randomised studies, in comparison with intravenous 5-FU plus leucovorin evaluating response, survival, quality of life and pharmacoeconomics.
Figure 2.

The activation and metabolic pathway of a) S-1 and b) UFT. CDHP, chlorodihydropyrimidine; FT, ftorafur; Oxo, oxonic acid; PPRT, pyrimidine phosphoribosyl transferase; P-450, cytochrome P450; U, uracil. — — → inhibition, ⇝ activation.
S-1 (FT-CDHP-Oxonic acid)
S-1 is an oral formulation of ftorafur and its modulators in a molar ratio of 1.0:1.0:0.4. Oxonic acid is a potent inhibitor of gastrointestinal pyrimidine phosphoribosyl transferase (PPRT) and 5-chlorodihydropyrimidine (CDHP), is a DPD inhibitor (Figure 2b). Diarrhoea is the commonest dose-limiting toxicity when 5-FU is administered as a prolonged infusion. This has been documented in studies with chronic dosing schedules of 5-FU plus eniluracil [17] and UFT plus leucovorin [25]. PPRT activates 5-FU to 5-FU monophosphate and consequently 5-FU triphosphate which is incorporated into RNA. This incorporation of 5-FUTP into RNA is thought to primarily mediate the diarrhoea from 5-FU [29]. Theoretically, S-1 should have similar antitumour activity but less gastrointestinal toxicity compared with 5-FU plus eniluracil, or UFT. In rats bearing implanted Yoshida sarcoma, continuous venous infusion (CVI) of 5-FU was compared with oral S-1. CVI 5-FU produced a greater than 50% weight loss and severe diarrhoea in order to achieve a 100% tumour growth inhibition. The dose of S-1 required for a similar effect led to a 10% weight loss and no diarrhoea. In Japanese phase II trials, S-1 (80 mg m−2 day−1 FT for 28 days followed by a 14-day rest period) yielded a response rate of 17% with no cases of grade 3 gastrointestinal toxicity [30]. This drug is now undergoing trials in North America and Europe. The structures of 5-FU, eniluracil, ftorafur and S-1 are depicted in Figure 3.
Figure 3.

The structures of 5-FU, ethynyluracil, ftorafur and S-1.
Capecitabine (Xeloda)
Pyrimidine nucleoside phosphorylase (thymidine phosphorylase) catalyses the formation of pyrimidine bases from nucleosides. Levels of this enzyme have been shown to be significantly higher in tumour tissue compared to normal tissue. Recently, pyrimidine nucleoside phosphorylase has been shown to be a tumour-associated angiogenic factor, identified as platelet derived endothelial cell growth factor (PDEGF), whose expression in tumours has correlated with promotion of tumour growth [31]. 5′-deoxyfluorouridine, which is a substrate for this enzyme, was synthesised in an attempt to increase the efficacy of 5-FU by increasing intratumoral delivery of drug [32]. In subsequent human testing, response rates similar to those achieved by 5-FU in colorectal carcinoma (30–35%) could be obtained only after protracted continuous venous infusion, up to 3 months in one study [33]. Unfortunately, a trial of oral administration was limited by diarrhoea, due presumably to the release of 5-FU in the gastrointestinal tract through the activity of intestinal pyrimidine nucleoside phosphorylase [34]. Capecitabine (N4-pentyloxycarbonyl-5′-deoxy-5-fluorocytidine) has been developed to circumvent the problem of gastrointestinal toxicity from oral 5′-deoxyfluorouridine. This oral prodrug of 5-FU is absorbed through the gastrointestinal mucosa as an intact molecule. It is sequentially activated by carboxylesterase, cytidine deaminase and pyrimidine nucleoside phosphorylase (Figure 4). This cascade results in the formation of 5′-deoxy-5-fluorocytidine (5′-DFCR), 5′-deoxy-5-fluorouridine (5′-DFUR) and finally the intratumoral release of 5-FU [35]. In human tumour xenograft models, capecitabine yields substantially higher concentrations of 5-FU within tumours than in plasma or normal tissue. In addition, capecitabine yields higher intratumoral concentrations of 5-FU than equitoxic doses of 5-FU. In phase I studies on a twice daily oral schedule for 6 weeks, the maximal tolerated dose was 1657 mg m−2 day−1 [36]. On a twice daily dosing schedule for 14 days with 1 week off, the recommended phase II dose was 2510 mg m−2. This latter dose had the best toxicity profile, and has been selected for subsequent studies. Pharmacokinetic data revealed a rapid, near complete absorption with rapid conversion to metabolites and low systemic exposure to 5-FU. Diarrhoea was dose-limiting with other toxicities such as palmar-plantar erythrodysesthesia (hand–foot syndrome) and stomatitis being typical of toxicities of protracted infusion 5-FU [36]. The antitumour activity of capecitabine has been demonstrated in metastatic breast cancer. In a phase II study of patients with metastatic breast cancer who had previously received paclitaxel and doxorubicin, an overall response rate of 20% with a median survival of 384 days was achieved. Toxicities were mild. Based on these data, this drug has been approved for use in refractory breast cancer in the United States. Phase III trials in colorectal cancer have been completed, and results are awaited with interest.
Figure 4.

Intracellular activation of capecitabine. 5-DFCR, 5-deoxy-5-fluorocytidine: 5-DFUR, 5-deoxy-5-fluorouridine.
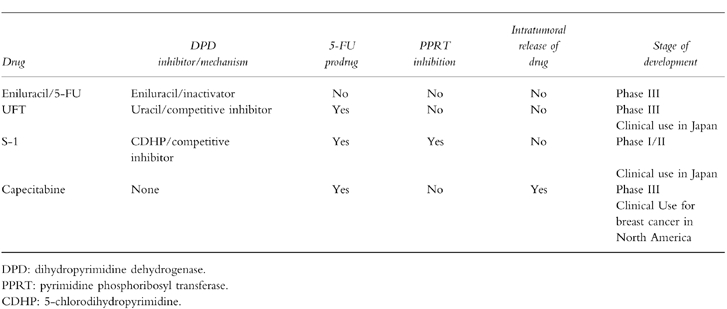
The major impetus behind the development of all these oral formulations of 5-fluorouracil is the realization that continuous infusion 5-FU may have a better therapeutic index than bolus schedules. Oral administration may simulate continuous infusion schedules without the cost, inconvenience and morbidity associated with central venous catheters and infusion pumps. The major pharmacologic properties of these agents are outlined in Table 1.
Table 1.
Pharmacologic properties of oral fluoropyrimidines.
Specific thymidylate synthase inhibitors
Thymidine monophosphate (TMP) is anabolized in cells to the triphosphate, which is essential for DNA synthesis and repair. An enzyme critical to the de novo synthesis of TMP is thymidylate synthase (TS). The substrate for TS is deoxyuridine monophosphate (dUMP), which is converted to TMP. The carbon donor for this reaction is the folate cofactor, 5,10-methylenetetrahydrofolate (CH2FH4). After intracellular anabolism to FdUMP, the fluoropyrimidine 5-FU inhibits TS by forming a ternary complex with TS and CH2FH4. Fluoropyrimidine resistance in several tumours, including colorectal cancer has been shown to be mediated through increased TS protein and mRNA levels [37]. In addition, high levels of dUMP have been found in colorectal tumour samples of patients not responding to 5-FU−1 leucovorin [38]. Pure, specific inhibitors of TS may overcome these resistance mechanisms and lead to greater therapeutic activity, compared to the indirect inhibition of modulated 5-FU. Various heterocyclic folate analogues have been found to be potent TS inhibitors. The first such inhibitor to be evaluated clinically was CB3717. This agent was active in several tumour types in vitro and in vivo but exhibited life-threatening nephrotoxicity thought to be related to aqueous insolubility in phase I/II testing [39]. Several second generation agents are in different stages of clinical development.
Raltitrexed (Tomudex®)
Raltitrexed (N-[5-(N-[3, 4-dihydro-2-methyl-4-oxoquinazolin-6-ylmethyl]-N-methylamino)-2-th enoyl]-l- glutamic acid) is the first specific TS inhibitor to be approved for clinical use. This is a water soluble second-generation quinazoline analogue of folic acid which is transported into cells by the reduced folate carrier and is extensively anabolized to the more active (600-fold more cytotoxic) polyglutamated forms. Cellular retention is increased because of the extensive polyglutamation. Preclinical activity was documented in a wide variety of human tumour xenografts in nude mice [40]. A 26% response rate was achieved in a phase II trial of raltitrexed in colorectal cancer [41]. Based on this study, two international phase III studies comparing raltitrexed 3 mg m−2 as a single i.v. infusion every 3 weeks with 5-FU plus low-dose leucovorin (Mayo regimen) or high-dose leucovorin (Machover regimen) have been completed [42, 43]. A North American study was set up to compare two raltitrexed doses (3.0 and 4.0 mg m−2) with 5-FU and low-dose leucovorin, but the 4.0 mg m−2 arm was discontinued because of excessive toxicity. Total objective response rates were similar for raltitrexed and 5-FU+LV, and palliative benefits were similar in extent in all three studies [44]. After a follow-up of 26, 12, and 17 months, respectively, median survival in months was statistically identical for the first two studies (10.1/10.2, 10.9/12.3 for raltitrexed and 5-FU, respectively) and inferior for raltitrexed (9.7 vs 12.7 for 5-FU) in the North American study. The tolerability profile of raltitrexed on these studies appeared to be slightly superior. Severe leucopaenia (WHO grades 3 and 4) occurred in 6–18% of patients compared with 13–41% in the 5-FU/LV group. Grade 3 and 4 mucositis occurred in 2–3% of patients on raltitrexed compared with 10–22% of patients in the 5-FU/LV group. The incidence of diarrhoea was equivalent for both agents. However raltitrexed caused more severe anaemia (5–9%vs 2–4%) and elevation of hepatic transaminases (9–10% vs 0–1%) compared with 5-FU/LV. Based on these data, raltitrexed was approved in the UK and several European countries as well as Canada for the treatment of metastatic colorectal cancer. Results of the North American phase III study has raised doubts about the approval of raltitrexed for the treatment of colorectal cancer in the United States. Another classical folate analogue, BW1843 U89 is in clinical development. BW1843 U89 is a 3-methyl-benzoquinazoline analogue which is a very potent TS inhibitor, with a Ki of 0.09 nm. It is an excellent substrate for the human reduced folate carrier and folylpolyglutamate synthase (FPGS), but polyglutamylation proceeds to the diglutamate only. The diglutamate form does not possess any increased TS inhibitory properties over the parent compound. Clinical activity in colon carcinoma has been noted in phase I-testing [45].
LY231514 (MTA)
MTA (LY231514, N-[4-[2-(2-amino-3, 4-dihydro-4- oxo-7H-pyrrolo[2, 3-d]pyrimidin-5-yl)ethyl]-benzoyl]- l-glutamic acid), is a novel multitargeted antifolate which inhibits TS, dihydrofolate reductase (DHFR), and glycinamide ribonucleotide formyl transferase (GARFT) [Figure 5] [46] [47], [48]). GARFT is a folate-dependent enzyme that is involved in purine synthesis. The Ki values for these enzymes are 1.3, 7.1 and 65 nm, respectively. The implication of two and possibly all of these three targets in the cytotoxicity of MTA is supported since both thymidine and hypoxanthine are required to circumvent cellular death caused by MTA [49]. This drug gains entry into cells via the reduced folate carrier, and has a high affinity for FPGS. The predominant pentaglutamate form in cells has a greater than 60 fold potency in its inhibition of TS, compared with the monoglutamate form [50]. In initial phase I studies, two partial responses were seen in colorectal cancer patients, lasting 7 and 11 months, respectively. The recommended dose for further testing was 600 mg m−2 administered every 3 weeks. Dose-limiting toxicity on this schedule was neutropaenia. Other toxicities were rash, mucositis, nausea, vomiting, fatigue, anorexia and elevation of hepatic transaminases [51]. Cumulative results from several clinical trials indicate that the folate status of patients is a sensitive predictor of toxicity from MTA. The most sensitive indicator of folate status appears to be serum homocysteine. Patients with serum homocysteine levels above a threshold concentration of 10 μm are at significant risk of developing severe myelosuppression, mucositis or diarrhoea [52]. The dose of MTA has been successfully escalated up to 1000 mg m−2 every 3 weeks with folate supplementation, which may not adversely affect the antitumour activity of MTA [53]. MTA is undergoing broad phase II studies and has shown activity in colon, breast, lung, and bladder cancers, as well as in mesothelioma [54–56].
Figure 5.

a) Structures of folic acid, tomudex and MTA (LY231514) b) Inhibition of multiple folate enzymes (TS, thymidylate synthase: DHFR, dihydrofolate reductase: GARFT, glycinamide ribonucleotide formyltransferase) by MTA and its polyglutamated metabolites.
Platinum compounds
Cytotoxic platinum compounds are activated by an aquation reaction in which a leaving group is replaced by water forming a positively charged species which cross-links DNA leading to cytotoxicity [57]. The common platinum agents, cisplatin and carboplatin are ineffective in colorectal cancer. However, second and third generation platinum agents differing in the carrier ligand have been synthesized in an attempt to improve the clinical activity and decrease the toxicity of these compounds.
Oxaliplatin (L -OHP, Eloxatin®)
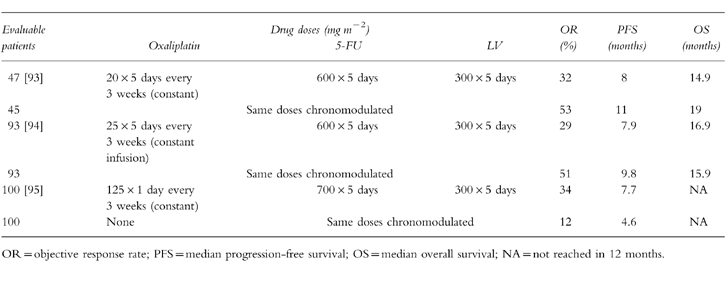
Oxaliplatin (oxalato-trans-l-1, 2 diaminocyclohexane platinum, Figure 6) is a third generation platinum compound synthesized in Japan in 1969 [58]. This agent produces inter and intrastrand DNA cross-links which are qualitatively and quantitatively similar to those produced by cisplatin [59]. However, the reaction kinetics are more rapid, and DNA-platinum adducts produced by oxaliplatin appear to be more resistant to repair by cellular mechanisms, and more cytotoxic than those produced by cisplatin [60]. This has been explained by the retention of the bulky diaminocyclohexane ring by activated oxaliplatin (Figure 6). Oxaliplatin exhibits markedly diminished cross-resistance with cisplatin both in vitro and in vivo. Deficiencies in mismatch repair (MMR) and increases in replicative bypass (the ability of the replication complex to synthesize DNA past the site of DNA damage), which contribute to resistance to cisplatin, have not been shown to induce resistance to oxaliplatin [61]. Deficiency in MMR occurs in patients with familial nonpolyposis colon cancer. Oxaliplatin may be a good therapeutic choice in these patients. This drug has a different toxicity profile from cisplatin [62, 63]. Its dose-limiting toxicity is peripheral neuropathy. It is non-nephrotoxic, and has minimal haematological, auditory, or cardiac toxicity. Oxaliplatin has exhibited in vitro and in vivo activity in 5-FU sensitive and 5-FU resistant colon cancer cells. In phase I studies, a dose of 130 mg m−2 given as a 2 h infusion every 3 weeks was identified for phase II evaluation. This schedule has been tested in 63 previously untreated and 101 patients with disease refractory to 5-FU in phase II trials. In first-line therapy, the response rate was 18%, while the response was 10% in patients previously treated with 5-FU, illustrating the modest activity of single-agent oxaliplatin in colorectal cancer. However, oxaliplatin has a greater than additive effect with 5-FU/leucovorin in vitro, and its activity in vivo is significantly enhanced by combination with 5-FU. Unfortunately, the development of oxaliplatin in combination with 5-FU/leucovorin has been complicated by the utilisation of a variety of doses and schedules.
Figure 6.

Biotransformation of cisplatin, carboplatin and oxaliplatin.
The concept of adapting the delivery of chemotherapeutic agents to circadian rhythms, chronotherapy, has been utilised in the European development of this drug. In one phase II study of a combination of oxaliplatin and 5-FU/leucovorin in colorectal cancer, a 5-day infusion of chronomodulated drugs were administered with a maximum rate of drug delivery at 16.00 h and a minimum at 04.00 h [64, 65]. A 58% response rate was seen in 93 patients, 47 of whom had received prior therapy. To dispel the notion that the promising results observed with oxaliplatin/5-FU+LV combinations were due to the chronomodulated regimen used, oxaliplatin has been tested in constant-rate infusion schedules and in regimens using bolus administration followed by 5-FU/LV infusion. Current data comparing chronomodulated and constant-rate infusions demonstrate a lower response rate for the latter regimen, but comparable progression-free and median survival, respectively.
Tables 2–4 summarize certain key studies testing oxaliplatin as a single agent and in combination with 5-FU+LV in the first-line and second-line setting. A broad multicentre trial of oxaliplatin and 5-FU in patients with 5-FU refractory colon cancer is near completion in North America. In an elegant French study, 53 patients out of a total of 330 with unresectable hepatic metastases from colorectal carcinoma had their liver lesions resected after down-staging following systemic chemotherapy with chronomodulated oxaliplatin and 5-FU/LV. In this group of 53 patients, cumulative 3-and 5-year survival rates were 54 and 40%, respectively [66]. A similar trial utilizing bolus regimens of oxaliplatin and 5-FU/LV will be started by the North Central Cancer Treatment Group in the United States. Oxaliplatin was approved in the spring of 1996 in France for use as a single agent or in combination with 5-FU, in patients with colorectal cancer which is refractory to fluoropyrimidine-based therapy.
Table 2.
Clinical activity of single-agent oxaliplatin in colorectal cancer.
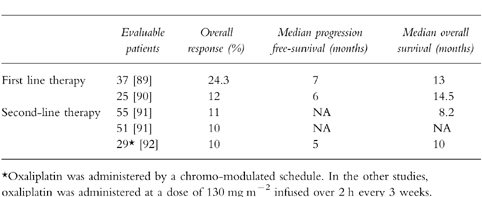
Table 4.
Selected studies of Oxaliplatin plus 5-fluorouracil and folinic acid in previously treated metastatic colorectal cancer.
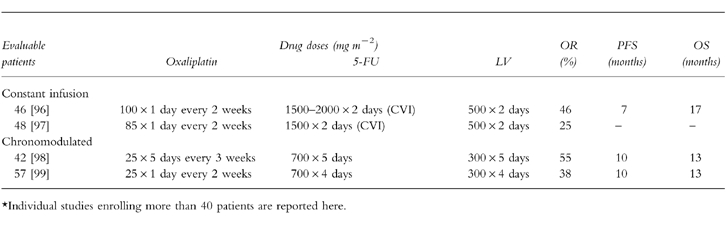
Table 3.
Phase III trials of oxaliplatin plus 5-fluorouracil and folinic acid in previously untreated metastatic colorectal cancer.
Topoisomerase I inhibitors
Topoisomerase I is a 100-kDa nuclear enzyme which is critical for DNA replication and transcription. It causes transient breaks in a single strand of DNA, by forming a transient DNA-enzyme ‘cleavable complex’. These breaks release the torsional strain caused by synthesis of a new strand of DNA or RNA around a double helix, thus relaxing supercoiled DNA [67]. The camptothecins target the DNA-topoisomerase 1 complex, preventing the reannealing of the nicked DNA strand. This inhibition results in intracellular accumulation of drug-stabilized topo I-DNA cleavable complexes, arrest of DNA replication and subsequent cell death [68]. Camptothecin is a plant alkaloid obtained from the Camptotheca acuminata tree. This drug was tested for antitumour activity and abandoned in the 1960s because of severe and unpredictable haemorrhagic cystitis, myelosuppression, nausea and vomiting. Three derivatives of camptothecin, have been introduced into clinical trials in recent years. Topotecan has been approved for the treatment of refractory ovarian cancer, and in the United States approval has been recommended for extensive-stage small cell lung cancer. This agent is however, inactive in colorectal cancer. 9-aminocamptothecin has been evaluated on different schedules for the treatment of colorectal cancer but does not appear to have significant enough activity in this disease to warrant further development.
Irinotecan (CPT-11, Camptosar®)
Irinotecan (7-ethyl-10-[4-(1-piperidino)-1-piperidino] carbonyloxycamptothecin) is a semisynthetic, water-soluble analogue of camptothecin, with greater in vivo and in vitro activity and less severe and more predictable toxicity (Figure 7a). It is the most active agent in this class in the therapy of colorectal cancer, and is approved for the treatment of 5-FU refractory colon cancer. Irinotecan is a pro-drug which is converted in vivo primarily by hepatic microsomal carboxylesterases to an active metabolite, SN-38. The topoisomerase 1 inhibition of irinotecan is accounted for by the intracellular concentrations of SN-38, which is about 250–1000 times more potent than the parent drug. Two human carboxylesterase isoforms responsible for SN-38 formation; a high-affinity, low Km and a low-affinity, high-Km isoform, have been described [69]. Intestinal carboxylesterases can also generate SN-38, followed by subsequent absorption. SN-38 is mainly eliminated through conjugation by the 1 A1 isoform of uridine diphosphate glucuronosyltransferase (UGT1 A1), the same isoenzyme responsible for glucuronidation of bilirubin [70]. Patients with Gilbert’s syndrome are deficient in UGT1 A1 activity. Severe irinotecan-related toxicity (neutropaenia and diarrhoea) has been described in these patients. Patients with elevated bilirubin or partial UGT1 A1 deficiency (Crigler–Najjar syndrome type II), as well as patients receiving valproic acid (an inhibitor of UGT1 A1) may also be at risk for increased irinotecan toxicity [71]. A second major metabolite of irinotecan, aminopentanecarboxylic acid (APC), is a product of CYP3 A4-mediated oxidation (Figure 7b). APC is a relatively weak inhibitor of acetylcholinesterase and two orders of magnitude less potent than SN-38 as a topoisomerase 1 inhibitor.
Figure 7.

a) Structure of camptothecin and its analogues (left panel). b) Metabolism of irinotecan (right panel). APC, aminopentanecarboxylic acid; UGT1A1, uridine diphosphate glucuronosyltransferase (isozyme 1A1).
The presence of an intact lactone ring in camptothecin and related compounds, including irinotecan, enhances antineoplastic activity. The lactone functional group undergoes a pH-dependent hydrolytic ring opening to the relatively inactive hydroxyacid form, with the closed ring form predominating at low pH [72]. In earlier Japanese and US phase 1 studies of irinotecan, the maximum tolerated dose was defined as 240–250 mg m−2 using a once every 3 week schedule, and 100 and 150 mg m−2 using a weekly schedule, respectively [73]. In these studies, the weekly intermittent schedule was associated with greater dose intensity and was therefore chosen for further studies.
The dose-limiting toxiciiesy in all these studies were diarrhoea and neutropaenia. European studies with a single infusion every 3 weeks showed diarrhoea to be dose-limiting at 350 mg m−2 but with concomitant administration of high dose loperamide, CPT-11 doses of up to 600 mg m−2 were administered [73]. This once every 3 week schedule was felt to be better tolerated with superior dose intensity compared to the weekly schedule, and was chosen for further studies in Europe. In a confirmatory phase I trial at Mayo Clinic [74], the recommended phase II dose of CPT-11 on a 3-weekly schedule was found to be 320 mg m−2. Alternate dosage schedules studied are continuous infusion for 5 days and daily for 3 days [75]. These studies have demonstrated that the dose-limiting toxicities of CPT-11 are similar regardless of the administration schedule. Early onset diarrhoea, emesis, diaphoresis, abdominal cramping and less commonly, hyperlacrimation and rhinorrhoea occurring during or within 24 h of CPT-11 infusion has been identified [76]. These symptoms are consistent with cholinergic hyperstimulation and are easily controlled with anticholinergic therapy such as atropine 0.5–1.0 mg intravenously. Irinotecan has been shown to inhibit acetylcholinesterase, and also binds to and stimulates muscarinic receptors [77]. Late-onset diarrhoea appearing 5–10 days after drug administration, is difficult to treat and appears to be related to SN-38-induced GI mucosal toxicity. Early recognition and prolonged administration of loperamide is effective for the late-onset diarrhoea and has decreased the incidence of grade 4 diarrhoea from 20 to 30% to 5–10% in different studies [78]. Leucopaenia, primarily neutropaenia was the other dose-limiting toxicity in several phase I trials. The incidence of grade 4 neutropaenia is approximately 6% and reversible, lasts mostly for less than 5 days and is usually asymptomatic. The appearance of concomitant severe neutropenia and diarrhoea has been fatal in a few cases [79]. Other toxicities have included nausea, vomiting, malaise and alopecia [80–82]. Two recently reported European trials have provided some insight into the benefit of irinotecan in 5-FU refractory colorectal cancer. In a randomised trial comparing irinotecan with best supportive care, the group receiving irinotecan had a survival rate of 9.2 months compared to 6.5 months for best supportive care (P = 0.0001, log-rank test), after a median follow-up of 13 months. The irinotecan group also had an improved quality of life and better control of disease-related symptoms [83]. The second trial compared irinotecan in second-line therapy with one of three infusional 5-FU schedules. With a median follow-up of 15 months, the overall survival of irinotecan-treated patients was 10.8 months as compared with 8.5 months for the 5-FU-treated patients (P = 0.04) [84]. Combinations of CPT-11 and 5-FU/LV have been tested on different schedules in phase I trials [85–87]. Future development of CPT-11 involves studies in untreated metastatic colorectal cancer, and in the adjuvant setting. An oral formulation of CPT-11 is undergoing phase I-testing. This route is pharmacologically suited to the highly schedule-dependent activity of irinotecan [88]. The high concentration of tissue carboxylesterases in the GI tract could promote the presystemic conversion of irinotecan to SN-38. In addition the low gastric and upper jejunal pH would favour the retention of irinotecan and SN-38 in the active lactone ring form.
Conclusions
For the first time since 5-FU was discovered four decades ago, there are new antimetabolites and promising modulatory approaches to therapy with 5-FU. There are also new classes of agents which are noncross resistant with 5-FU. These agents achieve response rates similar to 5-FU but hold the promise of more convenient dosing and some may possess milder toxicity profiles. Appropriate combinations of these agents and selection of therapy based on disease characteristics may improve the therapeutic outcome of colorectal cancer. For example, one may avoid the fluoropyrimidines and TS inhibitors in patients with tumours expressing high levels of TS; capecitabine could be used in tumours with high levels of thymidine phosphorylase; and eniluracil/5-FU could be used in tumours with high levels of DPD.
This work was supported in part by Grant CA77112 from the National Cancer Institute. The author wishes to thank Richard M. Goldberg, M.D. for reviewing the manuscript, and Ms. Gail L. Prechel for expert secretarial assistance.
References
- 1.Landis SH. Taylor M. Bolden S. Wingo PA. Cancer Statistics, 1997. CA-Acancer J for Clinicians. 1998;48:6–28. doi: 10.3322/canjclin.48.1.6. [DOI] [PubMed] [Google Scholar]
- 2.Advanced Colorectal Cancer Meta-analysis Project. Modulation of fluorouracil by leucovorin in patients with advanced colorectal cancer: evidence in terms of response rate. J Clin Oncol. 1992;10:896–903. doi: 10.1200/JCO.1992.10.6.896. [DOI] [PubMed] [Google Scholar]
- 3.Arbuck SG. Overview of clinical trials using 5-fluorouracil and leucovorin for the treatment of colorectal cancer. Cancer. 1989;6:1036–1047. doi: 10.1002/1097-0142(19890315)63:6+<1036::aid-cncr2820631309>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 4.Meta-analysis Group in Cancer. Efficacy of intravenous continuous infusion of fluorouracil compared with bolus administration in advanced colorectal cancer. J Clin Oncol. 1998;16:301–308. doi: 10.1200/JCO.1998.16.1.301. [DOI] [PubMed] [Google Scholar]
- 5.O’Dwyer PJ, Paul AR, Walczak J, et al. Phase II study of biochemical modulation of fluorouracil by low-dose PALA in patients with colorectal cancer. J Clin Oncol. 1990;88:1497–1503. doi: 10.1200/JCO.1990.8.9.1497. [DOI] [PubMed] [Google Scholar]
- 6.Wadler S, Lembersky B, Atkins M, et al. Phase II trial of fluorouracil and recombinant interferon alpha-2a in patients with advanced colorectal cancer: an Eastern Cooperative Oncology Group study. J Clin Oncol. 1991;9:1806–1810. doi: 10.1200/JCO.1991.9.10.1806. [DOI] [PubMed] [Google Scholar]
- 7.Poon MA, O’Connell MJ, Moertel CG, et al. Biochemical modulation of fluorouracil: Evidence of significant improvement of survival and quality of life in patients with advanced colorectal carcinoma. J Clin Oncol. 1989;10:1407–1418. doi: 10.1200/JCO.1989.7.10.1407. [DOI] [PubMed] [Google Scholar]
- 8.Diasio RB, Harris BE. Clinical pharmacology of 5-fluorouracil. Clin Pharmacokinet. 1989;16:215–237. doi: 10.2165/00003088-198916040-00002. [DOI] [PubMed] [Google Scholar]
- 9.Fleming RA, Milano G, Thyss A. Correlation between dihydropyrimidine dehydrogenase activity in peripheral mononuclear cells and systemic clearance of fluorouracil in cancer patients. Cancer Res. 1992;52:2899–2902. [PubMed] [Google Scholar]
- 10.Spector T, Harrington JA, Porter DJT. 5-ethynyluracil (776C85): inactivation of dihydropyrimidine dehydrogenase in vivo. Biochem Pharmacol. 1993;46:2243–2248. doi: 10.1016/0006-2952(93)90615-4. [DOI] [PubMed] [Google Scholar]
- 11.Porter DJT, Chestnut WG, Merrill BM, et al. Mechanism-based inactivation of dihydropyrimidine dehydrogenase by 5-ethynyluracil. J Biol Chem. 1992;267:5236–5242. [PubMed] [Google Scholar]
- 12.Cao S, Rustum YM, Spector T. 5-ethynyluracil (776C85): Modulation of 5-fluorouracil efficacy and therapeutic index in rats bearing advanced colorectal carcinoma. Cancer Res. 1994;54:1507–1510. [PubMed] [Google Scholar]
- 13.Baccanari DP, Davis ST, Knick VC, et al. 5-ethynyluracil (776C85): a potent modulator of the pharmacokinetics and anti-tumor efficacy of 5-fluorouracil. Proc Natl Acad Sci. 1993;90:11064–11068. doi: 10.1073/pnas.90.23.11064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Okeda R, Shibutani M, Matsuo T, et al. Experimental neurotoxicity of 5-fluorouracil and its derivatives is due to poisoning by the monofluorinated organic metabolites, monofluoroacatic acid and alpha-fluoro-beta-alanine. Acta Neuropathol. 1990;81:66–73. doi: 10.1007/BF00662639. [DOI] [PubMed] [Google Scholar]
- 15.Adjei AA, Doucette M, Spector T, et al. 5-ethynyluracil (776C85) an inhibitor of dihydropyrimidine dehydrogenase, permits reliable oral dosing of 5-fluorouracil and prolongs its half-life. Proc Am Soc Clin Oncol. 1995;14:459. [Google Scholar]
- 16.Schilsky RL, Burris H, Ratain M, et al. Phase I clinical and pharmacologic study of 776C85 plus 5-fluorouracil (5-FU) in patients with advanced cancer. Proc Am Soc Clin Oncol. 1996;15:485. doi: 10.1200/JCO.1998.16.4.1450. [DOI] [PubMed] [Google Scholar]
- 17.Baker SD, Diasio R, Lucas VS, et al. Phase I and pharmacologic study of oral 5-fluorouracil (5-FU) on a chronic 28-day schedule in combination with the dihydropyrimidine dehydrogenase (DPD) inactivator 776C85. Proc Am Soc Clin Oncol. 1996;15:486. doi: 10.1200/JCO.2000.18.4.915. [DOI] [PubMed] [Google Scholar]
- 18.Goldberg RM, Kugler J, Sargent DJ, et al. A Phase II trial of a seven day regimen of oral 776C85 plus a five day regimen of oral 5-fluorouracil (5-FU) in untreated patients (pts) with metastatic colorectal cancer: a North Central Cancer Treatment Group Study. J Clin Oncol. 1998;17:1084. [Google Scholar]
- 19.Creaven PJ, Rustum YM, Petrelli N, Gorbunova VA. Clinical studies of the modulation of ftorafur. In: Rustum YM, editor. Novel Approaches to Selective Treatments of Human Solid Tumors: Laboratory and Clinical Correlation. New York: Plenum Press; 1993. pp. 253–262. [Google Scholar]
- 20.Hillers S, Zhuk RA, Lidaks M. Analogs of pyrimidine nucleosides I. N-(alpha-furanidyl) derivatives of natural pyrimidine bases and their antimetabolites. Dokl Akad Nauk SSSR. 1967;176:332–335. [PubMed] [Google Scholar]
- 21.Friedman MA, Ignoffo RJ. A review of the United States clinical experience of the fluoropyrimidine, ftorafur (NAC-148958) Cancer Treatment Reviews. 1980;7:205–213. doi: 10.1016/s0305-7372(80)80037-5. [DOI] [PubMed] [Google Scholar]
- 22.Blokhina NG, Vozny EK, Garin AM. Results of treatment of malignant tumors with ftorafur. Cancer. 1972;30:390–392. doi: 10.1002/1097-0142(197208)30:2<390::aid-cncr2820300214>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 23.Fujii S, Ikrenaka K, Fukushima M, Shirasaka T. Effect of uracil and its derivatives on antitumor activity of 5-fluorouracil and 1-(2-tetrahydrofuryl) -5-fluorouracil. Gann. 1978;69:763–772. [PubMed] [Google Scholar]
- 24.Ota K, Taguchi T, Kimura K. Report on nationwide pooled data and cohort investigation in UFT phase II study. Cancer Chemother Pharmacol. 1988;22:333–338. [PubMed] [Google Scholar]
- 25.Pazdur R, Lassere Y, Rhodes V, et al. Phase II trial of uracil and tegafur plus oral leucovorin: an effective oral regimen in the treatment of metastatic colorectal carcinoma. J Clin Oncol. 1994;12:2296–2300. doi: 10.1200/JCO.1994.12.11.2296. [DOI] [PubMed] [Google Scholar]
- 26.Sanchez F, Milla A. Tegafur. Uracil (UFT) plus folinic acid in advanced rectal cancer. Jpn J Clin Oncol. 1994;24:322–326. [PubMed] [Google Scholar]
- 27.Saltz LB, Leichman CG, Young CW, et al. A fixed-ratio combination of uracil and Ftorafur (UFT) with low dose leucovorin. An active regimen for advanced colorectal cancer. Cancer. 1995;75:782–785. doi: 10.1002/1097-0142(19950201)75:3<782::aid-cncr2820750306>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 28.Nogue M, Segui M, Batiste E, et al. Phase II study of oral tegafur (TF) and low-dose oral leucovorin (LV) in advanced colorectal cancer (ACC) Proc Am Soc Clin Oncol. 1996;15:200. [Google Scholar]
- 29.Mandel HG. The incorporation of 5-FU into RNA and its molecular consequences. Prog Mol Subcellular Biol. 1969;1:82–135. [Google Scholar]
- 30.Taguchi T, Shirasaka T. New oral anticancer agent. S-1 Ann Oncol. 1996;7(Suppl 1):223. [Google Scholar]
- 31.Moghaddam A, Zhang H-T, Fan T, et al. Thymidine phosphorylase is angiogenic and promotes tumor growth. Proc Natl Acad Sci USA. 1995;92:998–1002. doi: 10.1073/pnas.92.4.998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meropol NJ, Creaven PJ, Petrelli NJ. Metastatic colorectal cancer: advances in biochemical modulation and new drug development. Sem Oncol. 1995;22:509–524. [PubMed] [Google Scholar]
- 33.Schuster D, Heim ME, Decoster G, et al. Phase I-II trial of doxifluridine (5′DFUR) administered as long-term continuous infusion using a portable infusion pump for advanced colorectal cancer. Eur J Cancer Clin Oncol. 1989;25:1543–1548. doi: 10.1016/0277-5379(89)90295-2. [DOI] [PubMed] [Google Scholar]
- 34.Bajetta E, Colleoni M, DiBartolomeo M, et al. Doxifluridine: an active agent in advanced colorectal cancer (CRC) Proc Am Soc Clin Oncol. 1994;13:192. [Google Scholar]
- 35.Ishitsuka H, Miwa M, Ishikawa T, et al. Capecitabine: an orally available fluoropyrimidine with tumor selective activity. Proc Am Ass Cancer Res. 1995;36:2426. [Google Scholar]
- 36.Twelves C, Budman DR, Creaven PJ, et al. Pharmacokinetics (PK) and pharmacodynamics (PD) of capecitabine in two phase I studies. Proc Am Ass Clin Oncol. 1996;15:1509. [Google Scholar]
- 37.Berger SH, Chung-Her J, Johnson LF, et al. Thymydylate synthase overproduction and gene amplification in fluorodeoxyuridine-resistant human cells. Mol Pharmacol. 1985;28:461–467. [PubMed] [Google Scholar]
- 38.Ardalan B, Chua L, Tian E, et al. A phase II study of weekly 24-hour infusion with high-dose fluorouracil with leucovorin in colorectal carcinoma. J Clin Oncol. 1991;9:625–630. doi: 10.1200/JCO.1991.9.4.625. [DOI] [PubMed] [Google Scholar]
- 39.Cantwell BMJ, Mccaulay V, Harris AL, et al. Phase II study of the antifolate N10-propargyl-5,8-dideazafolic acid (CB3717) in advanced breast cancer. Eur J Cancer Clin Oncol. 1988;24:769–775. [Google Scholar]
- 40.Stephens TC, Valaccia BE, Sheader ML, et al. The thymidylate synthase inhibitor ICI D1694, is superior to CB3717, 5-fluorouracil and methotrexate against a panel of human tumour xenografts. Proc Am Assoc Cancer Res. 1991;32:328. [Google Scholar]
- 41.Cunningham D, Francois E, Van Cutsem E, et al. Tomudex (ZD1694) a new thymidylate synthase inhibitor with good antitumor activity in advanced colorectal cancer (ACC) Proc Am Assoc Clin Oncol. 1994;13:199. [Google Scholar]
- 42.Cunningham D, Zalcberg JR, Rath U, et al. Final results of a randomised trial comparing Tomudex (raltitrexed) with 5-fluorouracil plus leucovorin in advanced colorectal cancer. Ann Oncol. 1996;7:961–965. doi: 10.1093/oxfordjournals.annonc.a010800. [DOI] [PubMed] [Google Scholar]
- 43.Harper P Study Group. Advanced colorectal cancer (ACC): results from the latest (raltitrexed) Tomudex comparative study. Proc Am Soc Clin Oncol. 1997;16:802. [Google Scholar]
- 44.Pazdur R, Vincent M. Raltitrexed (Tomudex) vs 5-fluorouracil+leucovorin (5-FU+LV) in patients with advanced colorectal cancer (ACC): results of a randomised multicenter North American trial. Proc Am Soc Clin Oncol. 1997;16:801. [Google Scholar]
- 45.Burris HA, Smetzer LA, Eckardt GI, et al. A phase I trial of the novel thymidylate synthase inhibitor 1843U89 with and without high dose oral folate. Proc Am Soc Clin Oncol. 1996;15:490. [Google Scholar]
- 46.Grindley GB, Shih C, Barnett CJ, et al. LY231514, a novel pyrrolopyrimidine antifolate that inhibits thymidylate synthase (TS) Proc Am Assoc Cancer Res. 1992;33:411. [Google Scholar]
- 47.Schilsky RL. Antimetabolites. In: Perry MC, editor. The Chemotherapy Source Book. Baltimore, MD: Williams & Wilkins; 1992. pp. 301–315. [Google Scholar]
- 48.Shih C, Gosset L, Gates S, et al. LY231514 and its polyglutamates exhibit potent inhibition against both human dihydrofolate reductase (DHFR) and thymidylate synthase (TS): multiple folate enzyme inhibition. Ann Oncol. 1996;7(Suppl 1):85. [Google Scholar]
- 49.Schultz R, Andis S, Chen V, et al. Comparative antitumor activity of the multitargeted antifolate LY231514 and the thymidylate synthase inhibitor ZD1694. Ann Oncol. 1996;7(Suppl 1):85. [Google Scholar]
- 50.Chen VJ, Bawley JR, Gossett L, et al. Activity of LY231514 against several enzymes in the folate-dependent pathways. Proc Am Assoc Cancer Res. 1996;37:2598. [Google Scholar]
- 51.Rinaldi DA, Burris HA, Dorr FA, et al. A phase I evaluation of LY231514, a novel multitargeted antifolate, administered every 21 days. Proc Am Soc Clin Oncol. 1996;15:489. [Google Scholar]
- 52.Niyikiza C, Walling J, Thornton D, et al. LY231514 (MTA): Relationship of vitamin metabolite profile to toxicity. Proc Am Soc Clin Oncol. 1997;17:2139. [Google Scholar]
- 53.Hammond L, Villalona-Calero M, Eckhardt SG, et al. A phase I and pharmacokinetic (PK) study of the multitargeted antifol (MTA) LY231514 with folic acid. Proc Am Soc Clin Oncol. 1997;17:866. [Google Scholar]
- 54.Lind MJ, Smith IE, Coleman RE, et al. Phase II study of MTA in patients with locally recurrent or metastatic breast cancer. Proc Am Soc Clin Oncol. 1997;17:433. [Google Scholar]
- 55.John W, Picus J, Blanke C, et al. Activity of MTA in patients with advanced colorectal cancer—results from a phase II study. Proc Am Soc Clin Oncol. 1997;17:1067. [Google Scholar]
- 56.Paz-Ares L, Tarbernero J, Moyano A, et al. A phase II study of the multi-targeted antifolate, LY231514 in patients with advanced transitional cell carcinoma of the bladder. Proc Am Soc Clin Oncol. 1997;17:1307. [Google Scholar]
- 57.Lyss AP. Enzymes and random synthetics. In: Perry MC, editor. The Chemotherapy Source Book. Baltimore: Williams & Wilkins; 1991. pp. 398–412. [Google Scholar]
- 58.Kidani Y, Inagaki K, Saito R, et al. Synthesis and anti-tumor activities of platinum (II) complexes of 1,2-diaminocyclohexane isomers and their related derivatives. J Clin Hematol Oncol. 1977;7:197. [Google Scholar]
- 59.Boudny V, Vrana O, Gaucheron F, et al. Biophysical analysis of DNA modified by 1,2-diaminocyclohexane Platinum (II) complexes. Nucl Acid Res. 1992;20:267–272. doi: 10.1093/nar/20.2.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jennerwein MM, Eastman A, Kokhar AR. The role of DNA repair in resistance of L1210 cells to isomeric 1,2-diaminocyclohexane Platinum (II) complexes. Chem–Biol Interacts. 1989;70:39–49. [Google Scholar]
- 61.Raymond E, Faivre S, Woynarowski JM, et al. Oxaliplatin: Mechanism of action and antineoplastic activity. Sem Oncol. 1998;25(Suppl 5):4–12. [PubMed] [Google Scholar]
- 62.Kidani Y. A coordination chemical approach to prepare organ-specific antitumor platinum complexes in cancer chemotherapy. In: Nicolini M, editor. Platinum and Other Metal Coordination Compounds in Chemotherapy. Boston, MA: Martinus Nijhoff; 1988. pp. 555–562. [Google Scholar]
- 63.Extra JM, de Gramont A, Gamelin E, et al. Oxaliplatin (L-OHP) synergistic activity with 5-fluorouracil (5-FU) in 5-FU resistant colorectal cancer patients is independent of the 5-FU administration modality and/or modulation by folinic acid. Ann Oncol. 1996;7(Suppl 1):236. [Google Scholar]
- 64.Levi F, Misset JL, Brienza S, et al. A chronopharmacologic phase II clinical trial with 5-fluorouracil, folinic acid and oxaliplatin using an ambulatory multichannel programmable pump. High antitumor effectiveness against metastatic colorectal cancer. Cancer. 1992;69:893–900. doi: 10.1002/1097-0142(19920215)69:4<893::aid-cncr2820690410>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 65.Levi FA, Zidani R, Vannetzel J-M, et al. Chronomodulated versus fixed-infusion rate delivery of ambulatory chemotherapy with oxaliplatin, fluorouracil, and folinic acid (leucovorin) in patients with colorectal cancer metastases: a randomized multi-institutional trial. J Natl Cancer Inst. 1994;86:608–1617. doi: 10.1093/jnci/86.21.1608. [DOI] [PubMed] [Google Scholar]
- 66.Bismuth H, Adam R, Levi F, et al. Resection of nonresectable liver metastases from colorectal cancer after neoadjuvant chemotherapy. Ann Surg. 1996;224:509–522. doi: 10.1097/00000658-199610000-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Slichenmeyer WR, Rowinsky EK, Donehower RC, et al. The current status of camptothecin analogues as antitumor agents. J Natl Cancer Inst. 1993;85:271–291. doi: 10.1093/jnci/85.4.271. [DOI] [PubMed] [Google Scholar]
- 68.Pommier Y. Eukaryotic DNA, topoisomerase I. Genome gatekeeper and its intruders, camptothecins. Sem Oncol. 1996;23(1) Suppl 3:3–10. [PubMed] [Google Scholar]
- 69.Slatter JG, Siu P, Sams JP, et al. Bioactivation of the anticancer agent CPT-11 to SN-38 by human hepatic microsomal carboxylesterases and the in vitro assessment of potential drug interactions. Drug Metab Dispos. 1997;25:1157–1164. [PubMed] [Google Scholar]
- 70.Gupta E, Wang X, Ramirez J, et al. Modulation of glucoronidation of SN-38, the active metabolite of irinotecan, by valproic acid and phenobarbital. Cancer Chemother Pharmacol. 1997;39:440–444. doi: 10.1007/s002800050595. [DOI] [PubMed] [Google Scholar]
- 71.Wasserman E, Myara A, Lokiec F, et al. Severe CPT-11 toxicity in patients with Gilbert’s syndrome: two case reports. Ann Oncol. 1997;8:1049–1052. doi: 10.1023/a:1008261821434. [DOI] [PubMed] [Google Scholar]
- 72.Fassberg J, Stella VJ. A kinetic and mechanistic study of the hydrolysis of camptothecin and some analogues. J Pharm Sci. 1992;81:676–684. doi: 10.1002/jps.2600810718. [DOI] [PubMed] [Google Scholar]
- 73.Armand JP. CPT-11: Clinical experience in phase I studies. Sem Oncol. 1996;23(1) Suppl 3:27–33. [PubMed] [Google Scholar]
- 74.Pitot HC, Erlichman C, Goldberg RM, et al. Phase I trial of irinotecan (CPT-11) given once every three weeks to patients with advanced solid tumors. Proc Am Soc Clin Oncol. 1996;15:494. [Google Scholar]
- 75.Rothenberg ML. CPT-11: an original spectrum of clinical activity. Sem Oncol. 1996;23(1) Suppl 3:21–26. [PubMed] [Google Scholar]
- 76.Bleiberg H, Cvitkovic E. Characterization and clinical management of CPT-11 (irinotecan)-induced adverse events. The European perspective. Eur J Cancer. 1996;32A(Suppl 3):S18–S23. doi: 10.1016/0959-8049(96)00293-6. [DOI] [PubMed] [Google Scholar]
- 77.Kawato Y, Sakiguchi M, Akahane K, et al. Inhibitory activity of camptothecin derivatives against acetylcholinesterase in dogs and their binding activity to acetylcholine receptors in rats. J Pharm Pharmacol. 1993;45:444–448. doi: 10.1111/j.2042-7158.1993.tb05573.x. [DOI] [PubMed] [Google Scholar]
- 78.Abigerges D, Armand J-P, Chabot GG, et al. Irinotecan (CPT-11) high-dose escalation using intensive high-dose loperamide to control diarrhea. J Natl Cancer Inst. 1994;86:446–449. doi: 10.1093/jnci/86.6.446. [DOI] [PubMed] [Google Scholar]
- 79.Negoro S, Fukuoka M, Masuda N, et al. Phase I study of weekly intravenous infusions of CPT-11, a new derivative of camptothecin, in the treatment of advanced non-small cell lung cancer. J Natl Cancer Inst. 1991;83:1164–1168. doi: 10.1093/jnci/83.16.1164. [DOI] [PubMed] [Google Scholar]
- 80.Pitot HC, Wender D, O’Connell MJ, et al. A phase II trial of CPT-11 (irinotecan) in patients with metastatic colorectal cancer: a North Central Cancer Treatment Group study. Proc Am Soc Clin Oncol. 1994;13:197. [Google Scholar]
- 81.Rothenberg ML, Eckardt JR, Kuhn JG, et al. Phase II trial of irinotecan in patients with progressive or rapidly recurrent colorectal cancer. J Clin Oncol. 1996;14:1128–1135. doi: 10.1200/JCO.1996.14.4.1128. [DOI] [PubMed] [Google Scholar]
- 82.Shimada Y, Yoshino M, Wakui A, et al. Phase II study of CPT-11, a new camptothecin derivative, in metastatic colorectal cancer. J Clin Oncol. 1993;11:909–913. doi: 10.1200/JCO.1993.11.5.909. [DOI] [PubMed] [Google Scholar]
- 83.Cunningham D, Pyrhonen S, James RD, et al. A phase III multicenter randomized study of CPT-11 vs supportive care alone in patients with 5-FU-resistant metastatic colorectal cancer. Proc Am Soc Clin Oncol. 1998;17 [Google Scholar]
- 84.Van Cutsem E, Bajetta E, Niederle N, et al. A phase III multicenter randomized trial comparing CPT-11 to infusional 5-FU regimen in patients with advanced colorectal cancer after 5-FU failure. Proc Am Soc Clin Oncol. 1998;17 [Google Scholar]
- 85.Tait N, Parnes H, Van Echo DA. A phase I, multischedule study of CPT-11, 5FU and leucovorin (LV) in advanced cancer. Ann Oncol. 1996;7(Suppl 1):460. [Google Scholar]
- 86.Saltz L, Kanowitz J, Kemeny N, et al. Phase I trial of irinotecan (CPT-11), 5 fluorouracil (FU) and leucovorin (LV) in patients with advanced solid tumors. Proc Am Soc Clin Oncol. 1995;14:1546. doi: 10.1200/JCO.1996.14.11.2959. [DOI] [PubMed] [Google Scholar]
- 87.Yamao T, Shimada Y, Shirao K, et al. Phase I study of CPT-11 combined with sequential 5FU in metastatic colorectal cancer (CRC) Proc Am Soc Clin Oncol. 1996;15:1527. [Google Scholar]
- 88.Houghton PJ, Cheshire PJ, Hallman JD, et al. Efficacy of topoisomerase I inhibitors, topotecan and irinotecan, administered at low dose levels in protracted schedules to mice bearing xenografts of human tumors. Cancer Chemother Pharmacol. 1995;36:393–403. doi: 10.1007/BF00686188. [DOI] [PubMed] [Google Scholar]
- 89.Becouarn Y, Ychou M, Ducreux M, et al. Phase II trial of oxaliplatin as first-line chemotherapy in metastatic colorectal cancer patients. J Clin Oncol. 1998;16:2739–2744. doi: 10.1200/JCO.1998.16.8.2739. [DOI] [PubMed] [Google Scholar]
- 90.Diaz-Rubio E, Sastre J, Zaniboni A. Oxaliplatin as single agent in previously untreated colorectal carcinoma patients: a phase II multicentric study. Ann Oncol. 1998;9:105–108. doi: 10.1023/a:1008200825886. [DOI] [PubMed] [Google Scholar]
- 91.Machover D, Diaz-Rubio E, de Gramont A, et al. Two consecutive phase II studies of oxaliplatin (L-OHP) for treatment of patients with advanced colorectal carcinoma who were resistant to previous treatment with fluoropyrimidines. Ann Oncol. 1996;7:95–98. doi: 10.1093/oxfordjournals.annonc.a010489. [DOI] [PubMed] [Google Scholar]
- 92.Levi F, Perpint B, Garufi C, et al. Oxaliplatin activity against metastatic colorectal cancer. A phase II study of 5-day continuous venous infusion at circadian rhythm modulated rate. Eur J Cancer. 1993;29A:1280–1284. doi: 10.1016/0959-8049(93)90073-o. [DOI] [PubMed] [Google Scholar]
- 93.Levi FA, Zidani R, Vannetzel JM, et al. Chronomodulated versus fixed infusion rate delivery of ambulatory chemotherapy with oxaliplatin, fluorouracil, and folinic acid (leucovorin) in patients with colorectal cancer metastases: a randomized multi-institutional trial. J Natl Cancer Inst. 1994;86:1608–1617. doi: 10.1093/jnci/86.21.1608. [DOI] [PubMed] [Google Scholar]
- 94.Levi F, Zidani R, Misset JL. Randomised multicentre trial of chronotherapy with oxaliplatin, fluorouracil, and folnic acid in metastatic colorectal cancer. For the Interational Organization for Cancer Chronotherapy. Lancet. 1997;350:681–686. doi: 10.1016/s0140-6736(97)03358-8. [DOI] [PubMed] [Google Scholar]
- 95.Giacchetti S, Zidani R, Perpoint B, et al. Phase III trial of 5-fluorouracil (5-FU) folinic acid (FA), with or without oxaliplatin (OXA) in previously untreated patients (pts) with metastatic colorectal cancer (MCC) Proc Am Soc Clin Oncol. 1997;16 [Google Scholar]
- 96.De Gramont A, Vignoud J, Tournigand C, et al. Oxaliplatin with high-dose leucovorin and 5-fluorouracil 48-hour continuous infusion in pretreated metastatic colorectal cancer. Eur J Cancer. 1997;33:214–219. doi: 10.1016/s0959-8049(96)00370-x. [DOI] [PubMed] [Google Scholar]
- 97.Andre T, Bensmaine MA, Louvet C, et al. Addition of oxaliplatin (Eloxatine) to the same leucovorin (LV) and 5-fluorouracil (5-FU) bimonthly regimens after pgoression in patients (pts) with metastsatic colorectal cancer (MCRC): preliminary report. Eur J Cancer. 1997;33:S165–S166. [Google Scholar]
- 98.Levi F, Misset JL, Brienza S, et al. A chronopharmacologic phase II clinical trial with 5-fluorouracil, folinic acid, and oxaliplatin using an ambulatory multichannel programmable pump: High antitumor effectiveness against metastatic colorectal cancer. Cancer. 1992;69:893–900. doi: 10.1002/1097-0142(19920215)69:4<893::aid-cncr2820690410>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 99.Brienza S, Levi F, Valori VM, et al. Intensified (every 2 weeks) chronotherapy with 5-fluorouracil (5-FU) folinic acid (FA) and oxaliplatin (L-OHP) in previously treated patients (pts) with metastatic colorectal cancer. Proc Am Soc Clin Oncol. 1993;12:100. [Google Scholar]