

Abstract
Aims
To investigate whether or not there is a correlation between failure to respond to typical antipsychotics and CYP2D6 ultrarapid metaboliser status.
Methods
CYP2D6 phenotype (metaboliser status) was assigned following genotyping for gene duplication, as well as for the CYP2D6*3, CYP2D6*4, and CYP2D6*5 null alleles in 235 treatment-refractory patients and 73 nonrefractory patients.
Results
Four (1.7%) of the 235 treatment-refractory subjects were positive on the duplication assay, but, of these, two were found to represent duplications of a null allele (CYP2D6*4), therefore leaving only two (0.85%) positive for duplication of a wild type allele (ultrarapid metabolisers). Three (4.1%) of the nonrefractory subjects had a genotype consistent with ultrarapid metaboliser status. Fisher’s exact test gave a two-tailed P value of 0.091, i.e. a trend towards an excess of ultrarapid metabolisers in the nonrefractory group, which was in the opposite direction to that predicted by our hypothesis.
Conclusions
Although the results show a trend towards an excess of ultrarapid metabolisers in the nonrefractory group, the percentages in the two groups of patients are both within the range for ultrarapid metabolisers in Caucasian populations. Our data are not consistent with ultrarapid metaboliser status being a major cause of failure to respond to typical antipsychotics.
Keywords: CYP2D6, genotype, metabolism, pharmacokinetics, treatment-refractory, typical antipsychotics
Introduction
The cytochrome P450 enzyme CYP2D6 is a polymorphic enzyme which contributes significantly to the pharmacokinetics of most typical antipsychotics [1]. Between 0.5% and 7% of Caucasians have very high enzyme activity [2, 3] and are known as ultrarapid metabolisers, or UMs. This high enzyme activity is due to the presence of 2 or more copies of a functional CYP2D6 allele existing in tandem on the same chromosome [2, 4–6]. At the other end of the metabolic spectrum are individuals who are homozygous for null alleles (poor metabolisers or PMs), representing about 7–9% of most Caucasian populations. The remainder of the population (excluding PMs and UMs) are known as extensive metabolisers (or EMs).
Two patients with ultrarapid metaboliser status have been described for whom tricyclic antidepressants at doses beyond the usual therapeutic range were required in order to achieve a therapeutic response [4]. Up to 30% of patients with schizophrenia who are prescribed typical antipsychotics are treatment-resistant [7]. The term treatment-resistant includes those who are treatment-refractory (show inadequate clinical response) and those who are treatment-intolerant (exhibit adverse responses). We hypothesized that patients with schizophrenia who were refractory to treatment with typical antipsychotics would be more likely to be ultrarapid metabolisers, as compared with patients who responded to typical antipsychotics. If the hypothesis were confirmed, it could form the rationale for a preprescribing genotyping assay to predict patients who would be less likely to respond well to typical antipsychotics at standard doses, and therefore assist the process of clinical dose finding and/or the more rapid progression of such patients on to an atpyical antipsychotic not subject to the CYP2D6 polymorphism.
Methods
Our 235 treatment-refractory subjects came from a sample of 246 patients treated with clozapine in the United Kingdom (UK). All of these patients were resistant to treatment with typical antipsychotics, and had a diagnosis of schizophrenia or schizoaffective disorder (clozapine prescribing restrictions in the UK, UK Clozaril Patient Monitoring Service). Prescribing consultants provided data regarding whether their patients were refractory to typical antipsychotics, intolerant of typical antipsychotics, or both refractory and intolerant of typical antipsychotics. Out of the 246 patients on clozapine there were a total of 235 who were refractory, or refractory and intolerant to typical neuroleptics. The standard definition of treatment-refractory schizophrenia has been provided by Kane et al. [7]: (1) at least three periods of treatment in the preceding 5 years with antipsychotics (from at least two different chemical classes) at doses equivalent to or greater than 1000 mg day−1 of chlorpromazine for a period of 6 weeks, each without significant symptomatic relief, and (2) no period of good functioning within the preceding 5 years. Data regarding which typical antipsychotics had been prescribed were not available. However, the most commonly prescribed typical antipsychotic prior to switching to clozapine in the UK for the period during which our sample was collected was haloperidol (Novartis, personal communication).
Our comparison group, nonrefractory to typical antipsychotics, comprised 73 patients from the Maudsley and Bethlem Royal Hospitals NHS Trust. Sixty-six of these had been treated with various antipsychotics, mostly haloperidol (approximately 60%) or fluphenazine, at doses equivalent to at least 100 mg chlorpromazine daily for at least 12 months prior to assessment and had DSM-IIIR schizophrenia. Information regarding duration of treatment was not available for the remaining seven subjects, but six had been treated with the equivalent of at least 100 mg chlorpromazine daily, while the remaining patient received 15 mg flupenthixol decanoate by depot injection fortnightly, the equivalent of 75 mg chlorpromazine daily. Six of these seven subjects had a clinical diagnosis of schizophrenia; one had a diagnosis of affective psychosis. As the patients were treated with a variety of antipsychotics, we converted all the prescriptions to chlorpromazine equivalents according to British National Formulary guidelines, in order to assess whether or not there was a relationship between the magnitude of the dose and the CYP2D6 genotype.
Ethics Committee approval was obtained for the study on all subjects, and, as there were insufficient numbers of non-Caucasians in the sample for the analysis to be informative, all non-Caucasians were excluded.
DNA was extracted from blood collected in EDTA tubes using the Nucleon II kit (Nucleon Biosciences, UK). CYP2D6 gene duplication was detected by the long-PCR method of Løvlie and colleagues [8], using the Expand Long Template PCR System (Boehringer Mannheim, UK), and primers 5′-TCCCCCACTGAC CCAACTCT-3′ and 5′-CACGTGCAGGGCAC CTAGAT-3′. The PCR was performed in a final volume of 25 μl including 2.5 μl Boehringer buffer 1, 4.5 μl of a 2 mm solution of each dNTP, 0.5 μl of each primer (10 μm solutions), and 0.25 μl of Boehringer enzyme mix (Taq/Pwo). Boehringer buffer 1 contains 20 mm Tris-HCl pH 7.5, 100 mm KCl, 1 mm dithiothreitol, 0.1 mm EDTA, 0.5% (v/v) TweenR 20, 0.5% (v/v) NonidetR P40, 50% glycerol (v/v), and 1.75 mm MgCl2. Cycling conditions were modified as follows: initial denaturation for 2 min at 94° C, 35 cycles of 93° C for 10 s, 60° C for 30 s and 68° C for 5 min, followed by an elongation step of 68° C for 7 min.
As cases have been described in which there are extra copies of a nonfunctional or null CYP2D6 allele [8–10], it is necessary to assay for nonfunctional CYP2D6 alleles as well as for the presence of a duplication event in order to confirm ultrarapid metaboliser status. We therefore assayed for the CYP2D6*3, CYP2D6*4, and CYP2D6*5 null alleles. CYP2D6*4 and CYP2D6*5are the most common and next most common null alleles, respectively; analysis for CYP2D6*4, CYP2D6*5, and CYP2D6*3 should detect 90–95% of null alleles in a European Caucasian population [11–13].
The CYP2D6*3 and CYP2D6*4 point mutation alleles were detected by PCR followed by restriction enzyme digestion as in the method of Smith et al. [14], with minor modifications. For the CYP2D6*3 assay, primers 5′-ATGAGCTGCTAACTGAGCCC-3′ and 5′-CCGA GAGCATACTCGGGAC-3′ were used in a total reaction volume of 25 μl containing 10 mm Tris-HCl pH 8.3, 50 mm KCl, 0.001% (w/v) gelatin, 3 mm MgCl2, 0.2 mm each dNTP, 0.25 μm each primer, and 1.25 U AmpliTaq (Perkin-Elmer, UK). Cycling conditions were: initial denaturation at 94 °C for 3 min, 30 cycles at 95° C for 1 min, 60° C for 30 s, and 72° C for 1 min, followed by final elongation at 72° C for 10 min. PCR products were digested using HpaII, and analysed on a 3% agarose gel, together with a 1kb ladder (Gibco BRL). For the CYP2D6*4 assay, we used primers 5′-GCCTTCGCCAACCACTCCG-3′ and 5′-AAATCCTGCTCTTCCGAGGC-3′ and the same conditions as for CYP2D6*3, except a MgCl2 concentration of 1.5 mm. PCR products were digested with BstNI and analysed on a 3% agarose gel as above.
The CYP2D6*5 gene deletion allele was assayed by long-PCR using the GeneAmp XL PCR kit (Perkin Elmer, UK) by the method of Steen and colleagues [15], using primers 5′-ACCGGGCACCTGTACTCCTCA-3′ and 5′-GCATGAGCTAAGGCACCCAGAC-3′. PCR was performed according to the manufacturer’s instructions using Ampliwax® beads to facilitate a hot start and a 100-μl reaction volume with 200 ng genomic DNA, 1xXL reaction buffer, 0.2 mm each dNTP, 0.3 μm each primer, 1.1 mm Mg(OAc)2 and 2 U of the rTth/Vent® DNA polymerase mixture. The XL reaction buffer contains Tricine, K(OAc), glycerol, and DMSO (concentrations not given by the supplier). Cycling conditions were: initial denaturation at 93° C for 1min, 35 cycles at 93° C for 1 min, 65° C for 30 s, 68° C for 5 min, and a final elongation at 72° C for 10 min.
Two cases were positive on both the CYP2D6*4 and the duplication assays. These were further tested to determine whether the null or the wild type allele was duplicated as described by Sachse et al. [10]: a further duplication assay was performed with primers as described by Johansson et al. [16], giving a 10kb amplicon in casespositive for a duplication, which was then subjected to anested PCR followed by digestion with HphI. Theprimers for the Johansson et al. assay were5′-GCCACCATGGTGTCTTTGCTTTC-3′ and 5′-ACCGGATTCCAGCTGGGAAATG-3′, and we modified the conditions by performing the assay with the Expand Long Template PCR System (Boehringer Mannheim, UK), using a total reaction volume of 25 μl with 2.5 μl of Boehringer buffer 1, 4.5 μl of a 2 mm solution of each dNTP, 0.5 μl of each primer (10 μm solutions), 0.37 μl of the Taq/Pwo enzyme mix, and 200 ng of genomic DNA. Cycling conditions were: initial elongation of 94° C for 2 min; 10 cycles of 93° C for 10 s, 60° C for 30 s, 68° C for 12 min; 20 cycles of 93° C for 10 s, 60° C for 30 s, and 68° C for 12 min with a 15 s increment per cycle; and a terminal elongation step of 68° C for 7 min. The product of this reaction was then diluted 1:5 and subjected to nested PCR using primers 5′-TCAACACAGCAGGTTCA-3′ and 5′-CTGTGGT TTCACCCACC-3′. The reaction volume was 65 μl, containing 10 mm Tris-HCl pH 8.3, 50 mm KCl, 1.5 mm MgCl2, 0.16 mm each dNTP, 0.1 μm each primer, and 0.2 U AmpliTaq. The cycling conditions were: initial elongation at 94°C for 2 min, 25 cycles of 94°C for 30 s, 58°C for 10 s, 72°C for 1 min, and a final elongation step of 72°C for 7 min. The products were digested with HphI and separated on a 3% agarose gel.
The results were analysed using SPSS for Windows and EpiInfo Version 6 (Centers for Disease Control and Prevention, USA and World Health Organization, Switzerland).
Results
CYP2D6 genotype and deduced phenotype for the 235 subjects refractory to treatment with typical antipsychotics vs the 73 responsive to typical antipsychotics are given in Table 1. Of the 235 treatment-refractory subjects, four (1.7%) cases were positive on the duplication assay. However, two cases yielded a CYP2D6*4/wt result with the CYP2D6*4 assay and a positive result with the duplication assay; these cases were found to represent duplications of the CYP2D6*4 allele. As these cases possessed only one functional copy of CYP2D6, they were deduced to be phenotypically equivalent to heterozygous null cases and hence extensive metabolisers, not ultrarapid metabolisers. Therefore only two out of 235 cases (0.9%) were positive for duplication of a wild type allele. In contrast, of the sample of 73 nonrefractory patients, 3 (4.1%) were positive for the duplication assay, of which none was positive for the null alleles tested. The results were therefore in the opposite direction to that predicted by our hypothesis, but they did not reach significance: Fisher’s exact test was performed, comparing the presence or absence of UM status in the two clinical groups, which gave a two-tailed P value of 0.091. (Chi square was not appropriate here, with the number of cases being less than 5; with Fisher’s exact test the one-tailed and two-tailed values were identical, but the value is reported as two-tailed as the results were in the opposite direction to that predicted by the hypothesis).
Table 1.
CYP2D6genotype and deduced phenotype in subjects refractory to treatment with typical antipsychotics and nonrefractory to treatment with typical antipsychotics; numbers of cases given with percentages in parentheses.
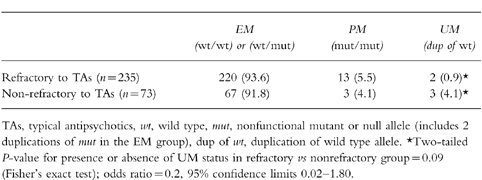
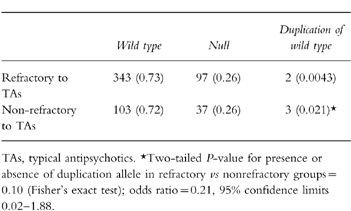
Table 2 shows the distribution of the alleles with allele frequencies in the two clinical groups, with the results being reported under the assumption that the duplication allele is present in the heterozygous state. If this allele were present in a homozygous state in all cases in which it was found, then the frequencies of the wild type and null alleles in the treatment-refractory group would be unchanged, while that of the duplication allele would be doubled at 0.0085; the frequencies of the wild type, null, and duplication alleles in the nonrefractory group would be 0.71, 0.25, and 0.041, respectively. However, as in the paper by Johansson et al. none of their cases with a duplication allele was homozygous for this variant, we have assumed that all of our cases are heterozygous. The frequencies of the CYP2D6*4, CYP2D6*5, and CYP2D6*3 alleles, respectively, in the sample of 235 and 73 were: 0.223, 0.024, and 0.018 (in the sample of 235), and 0.219, 0.02, and 0.02 (in the sample of 73). Comparing the presence or absence of the duplication allele in the two groups, Fisher’s exact test gave a two-tailed P value of 0.10 (Table 2).
Table 2.
Distribution of CYP2D6alleles in the treatment-refractory and nonrefractory groups; allele numbers given, with frequencies in parentheses. The CYP2D6*4×2 allele is included in the null alleles.
Discussion
We did not find an excess of ultrarapid metabolisers in subjects refractory to treatment with typical antipsychotics. On the contrary, only two out of 235 (0.9%) treatment-refractory cases were positive for duplication of a wild-type allele, while three out of 73 (4.1%) nonrefractory cases were genotyped as ultrarapid metabolisers. This gives a trend (P = 0.091, Fisher’s exact test) towards an excess of ultrarapid metabolisers in the nonrefractory group of patients. However, both percentages are within the range for ultrarapid metabolisers in Caucasian populations [2, 3, 6, 10]. Two cases of CYP2D6*4 duplications were found, which is the second time this has been reported in Caucasians, Sachse and colleagues [10] having provided the first report.
The results demonstrate that ultrarapid hydroxylation by CYP2D6 of typical antipsychotics is not a major cause of failure to respond to treatment with these agents. There are at least five possible explanations for this surprising result. Firstly, we could have failed to find a significant result when there is in fact a significant association of ultrarapid hydroxylation either with treatment-refactory status (the direction of the original hypothesis), or with treatment nonrefractory status (the direction of the trend found). The odds ratio (OR) for UM status, counting the treatment-refractory group as the ‘diseased state’ and the nonrefractory group as the ‘nondiseased state’, was 0.2, with exact lower and upper 95% confidence limits of 0.02 and 1.8, respectively (Table 1). This means that ultrarapid metaboliser status is less associated with being treatment-refractory than with being nonrefractory, with the range extending to being more associated with treatment-refractory. Of note the two patient sample groups are unequal in size; if we had had as many in the treatment nonrefractory group and the percentage of ultrarapid metabolisers in this group had remained the same as in our current findings, then we would have found 10 ultrarapid metabolisers in the nonrefractory group, which would have given a chi square of 5.47, a Pvalue of 0.019, and an OR of 0.19, with exact lower and upper limits of 0.02 and 0.92, respectively. In this scenario a significant result in the opposite direction to our original hypothesis would have been found.
Secondly, in both of our groups of subjects, the dose of antipsychotic was titrated by the prescribing consultants according to clinical effect. This could obscure any pharmacogenetic effects, i.e. ultrarapid metabolisers could be receiving doses above or at the upper end of the normal prescribed range, and then respond as if they were extensive metabolisers. However, for the nonrefractory group one of the patients with CYP2D6 duplication was on only 30 mg flupenthixol decanoate 2 weekly (equivalent to 150 mg chlorpromazine daily, i.e. a low dose).
Thirdly, although CYP2D6 is known to contribute to the pharmacokinetics of many typical antipsychotics [1], the specific contribution is different for different antipsychotics, and other cytochromes are involved. CYP2D6 is involved in the first pass metabolism and systemic elimination of perphenazine [17], and the systemic elimination of zuclopenthixol [18]. For these drugs, high CYP2D6 activity would be expected to lead to lower serum levels of the drugs, and hence possible therapeutic resistance. Although inhibition studies demonstrated that CYP2D6 is likely to be involved in the metabolism of chlorpromazine [19, 20], Muralidhan and colleagues [21] showed that CYP2D6 makes a relatively minor contribution to the large interindividual variability seen in plasma chlorpromazine levels.
The systemic elimination of haloperidol has been shown by Llerena and colleagues to be dependent on CYP2D6 activity [22], and although early reports showed that CYP2D6 catalysed the oxidation of reduced haloperidol back to haloperidol [23, 24], other work was not consistent with this [25], and recent reports [26, 27] have demonstrated that CYP3A4 is the primary enzyme involved in this step in the metabolic pathway. The steps in the metabolism of haloperidol in which CYP2D6 is involved are at present unclear, but, consistent with the results of Llerena and colleagues [22], Nyberg et al. showed that a CYP2D6 poor metaboliser had higher concentrations of plasma haloperidol throughout a 4-week treatment period with haloperidol decanoate as compared with 7 CYP2D6 extensive metabolisers [28]. Although none of the subjects in the study of Nyberg et al. was an ultrarapid metaboliser, it would be logical to assume that a UM would have low plasma haloperidol levels. Suzuki and colleagues [29] studied the correlation between CYP2D6 genotype and steady-state plasma concentrations (Css) of haloperidol and reduced haloperidol in a group of 50 Japanese patients with schizophrenia. They found that the mean Css of haloperidol was significantly higher (P<0.05) in the patients with 1 mutant allele compared with those with no mutant alleles, and that the mean Css of reduced haloperidol was significantly higher (P<0.05) in the patients with 1 or 2 mutant alleles compared with those with no mutant alleles. They therefore suggested that the Css of reduced haloperidol was more dependent upon CYP2D6 activity than the Css of haloperidol. However, although they did not find a significant difference between the mean Css of haloperidol in patients with 2 mutant alleles compared to those with no mutant alleles, it is of note that in this study the patients with 2 mutant alleles were either homozygous for the CYP2D6*10 allele (n = 4), which is associated with reduced but not absent CYP2D6 activity, or were compound heterozygotes for the CYP2D6*10 and CYP2D6*5 alleles (n = 2). Hence no patient actually had 2 CYP2D6 null alleles. Lane et al. [30] examined the relationship between CYP2D6 phenotype (as measured by dextromethorphan/dextrorphan metabolic ratio) and haloperidol disposition in 18 newly hospitalized Chinese patients with schizophrenia. Despite the fact that no PMs were found in this study, significant correlations between the metabolic ratio and plasma haloperidol concentration, reduced haloperidol concentration, and reduced haloperidol/haloperidol ratios were found. In a preliminary report of a larger study, Schmider et al. [31] investigated therapeutic drug monitoring data in 178 patients vs CYP2D6 genotype and found that PMs had significantly higher reduced haloperidol but not haloperidol concentrations compared with patients with one or no mutant alleles. The suggestion of Suzuki et al. [29] that CYP2D6 affects reduced haloeperidol levels at steady-state to a greater extent than haloperidol levels might therefore be correct. However, Suzuki et al. also suggested, based on the work of Tyndale and colleagues [24], that CYP2D6 catalyses the oxidation of reduced haloperidol back to haloperidol. As already outlined above, more recent work is not consistent with this [25–27], although the precise step in the metabolism of reduced haloperidol in which CYP2D6 is involved is at present unclear. Young et al. [25] showed that reduced haloperidol was the preferred form in the plasma after the administration of a single dose of either haloperidol or reduced haloperidol to healthy volunteers. A negative correlation between clinical response and reduced haloperidol levels or reduced haloperidol/haloperidol ratios has been observed [32]; it is possible that ultrarapid metabolisers of CYP2D6 could have lower reduced haloperidol levels and hence a better clinical response. This would be consistent with the trend that we have found for an excess of UMs in the nonrefractory group. However, Lane et al. [30] did not find a correlation between response and reduced haloperidol levels, reduced haloperidol/haloperidol ratios, or haloperidol levels. This is consistent with analyses by other authors [33, 34].
In the case of thioridazine, CYP2D6 catalyses the formation of mesoridazine, a metabolite with antipsychotic activity [35], and may be involved in the generation of another active metabolite, sulphoridazine. Extensive metabolisers have been shown to have higher peak levels of mesoridazine and sulphoridazine than poor metabolisers after a single oral dose, with lower levels of thioridazine [35]. The total serum concentrations of substances with antipsychotic activity at steady state will be determined by the relative magnitudes of the equilibrium constants of all the reactions in the metabolic pathway; these equilibrium constants and the relative antipsychotic potencies of the different active metabolites are unknown. It is therefore difficult to predict the effect of ultrarapid metaboliser status on clinical response to thioridazine.
Both of our groups of patients had been treated with various typical antipsychotics; it is therefore possible that we failed to show a correlation in one direction or the other as effects with some antipsychotics vs other antipsychotics cancelled each other out. Furthermore, it is possible that some patients in the treatment-refractory group were treated with agents whose levels are not significantly affected by CYP2D6 genotype (such as chlorpromazine). However, we would emphasize that a significant result would be unlikely to be obscured by either of the above possibilities as the numbers of individuals with duplications is very low in both the treatment-refractory and nonrefractory groups. It is also of note that the subject on zuclopenthixol who is an ultrarapid metaboliser is clinically stable on a low dose.
Fourthly, several of the above studies (especially those on normal volunteers) were single-dose pharmacokinetic analyses; single dose effects may differ markedly from those seen at steady state in a situation of pharmacological adaptation [36]. However, the work of Nyberg et al. [28], Suzuki et al. [29], Lane et al. [30], and Schmider et al. [31] was conducted on patients at steady-state. Furthermore, Jerling et al. [37] conducted a study on patients during continuous treatment and CYP2D6 genotype was shown to predict significantly the oral clearance of perphenazine and zuclopenthixol (patients with 2 CYP2D6 null alleles having a significantly lower clearance than those with one or no mutant alleles).
Finally, other factors may contribute towards nonresponse to medication, including noncompliance, pharmacodynamic factors, other biological factors, and psychosocial factors. Non-compliance occurs in up to 50% of patients on neuroleptics [38]. Pharmacodynamic factors have been implicated in the clinical response to clozapine, an atypical antipsychotic [39]; it may be that other pharmacodynamic factors (e.g. D2-receptor variants) are involved in the response to typical antipsychotics. Lieberman and colleagues [40] and Van Os et al. [41] have reviewed predictors of outcome in psychotic illness and concluded that factors such as longer duration of untreated illness, and structural brain abnormality on CT or MRI predict unfavourable outcome, while living in a low ‘expressed emotion’ environment is one of the predictors of a favourable outcome.
We have not found an association between ultrarapid metaboliser status and being treatment-refractory to typical antipsychotics. On the contrary, we found a trend towards an association between ultrarapid metaboliser status and being nonrefractory to typical antipsychotics, which could have reached significance had our nonrefractory group been equal in size to our refractory group.
Nonetheless, we have not excluded the possibility that ultrarapid metaboliser status could lead to failure to respond to a standard dose of some typical antipsychotics. But as only 0.5% to 7% of Caucasians are ultrarapid metabolisers, unless ultrarapid metaboliser status were associated with the psychotic illnesses for which the antipsychotics were prescribed (in this study, mainly schizophrenia), then as treatment resistance occurs in up to 30% of cases of schizophrenia, this factor would be unlikely to account for the majority of cases of treatment resistance. In summary, our data are not consistent with ultrarapid metaboliser status being a major cause of failure to respond to typical antipsychotics, and are consistent with ultrarapid metaboliser status being weakly associated with response to typical antipsychotics, especially in the case of haloperidol.
Acknowledgments
We thank Dr A.K. Daly for the provision of a CYP2D6*5 positive control. Katherine J. Aitchison is a Wellcome Trust Clinical Training Fellow. Christoph Sachse is supported by BMBF grant 01EC9408.
References
- 1.Dahl M-L, Bertilsson L. Genetically variable metabolism of antidepressants and neuroleptic drugs in man. Pharmacogenetics. 1993;3:61–70. doi: 10.1097/00008571-199304000-00001. [DOI] [PubMed] [Google Scholar]
- 2.Agúndez JAG, Ledesma MC, Ladero JM, Benitez J. Prevalence of CYP2D6 gene duplication and its repercussion on the oxidative phenotype in a white population. Clin Pharmacol Ther. 1995;57:265–269. doi: 10.1016/0009-9236(95)90151-5. [DOI] [PubMed] [Google Scholar]
- 3.Jerling M, Mellé Y, Mentré F, Mallet A. Population pharmacokinetics of nortriptyline during monotherapy and during concomitant treatment with drugs that inhibit CYP2D6—an evaluation with the nonparametric maximum likelihood method. Br J Clin Pharmacol. 1994;38:453–462. doi: 10.1111/j.1365-2125.1994.tb04382.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bertilsson L, Dahl ML, Sjöqvist F, et al. Molecular basis for rational megaprescribing in ultrarapid hydroxylators of debrisoquine: letter. Lancet. 1993;341:63. doi: 10.1016/0140-6736(93)92546-6. [DOI] [PubMed] [Google Scholar]
- 5.Johansson I, Lundqvist E, Bertisson L, Dahl ML, Sjöqvist F, Ingelman-Sundberg M. Inherited amplification of an active gene in the cytocrome P450 CYP2D locus as a cause of ultrarapid metabolism of debrisoquine. Proc Natl Acad Sci USA. 1993;90:11825–11829. doi: 10.1073/pnas.90.24.11825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dahl M-L, Johansson I, Bertilsson L, Ingelman-Sundberg M, Sjöqvist F. Ultrarapid hydroxylation of debrisoquine in a Swedish population. Analysis of the molecular genetic basis. J Pharmacol Exp Ther. 1995;274:516–520. [PubMed] [Google Scholar]
- 7.Kane J, Honigfeld G, Singer J, Meltzer H. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45:789–796. doi: 10.1001/archpsyc.1988.01800330013001. [DOI] [PubMed] [Google Scholar]
- 8.Løvlie R, Daly AK, Molven A, Idle JR, Steen VM. Ultrarapid metabolisers of debrisoquine: characterisation and PCR-based detection of alleles with duplication of the CYP2D6 gene. FEBS Lett. 1996;392:30–34. doi: 10.1016/0014-5793(96)00779-x. [DOI] [PubMed] [Google Scholar]
- 9.Masimirembwa CM, Johansson I, Hasler JA, Ingelman-Sundberg M. Genetic polymorphism of cytochrome P450 CYP2D6 in Zimbabwean population. Pharmacogenetics. 1993;3:275–280. doi: 10.1097/00008571-199312000-00001. [DOI] [PubMed] [Google Scholar]
- 10.Sachse C, Brockmöller J, Bauer S, Roots I. Cytochrome P450 2D6 variants in a Caucasian population: allele frequencies and phenotypic consequences. Am J Hum Genet. 1997;60:284–295. [PMC free article] [PubMed] [Google Scholar]
- 11.Heim M, Meyer UA. Genotyping of poor metabolisers of debrisoquine by allele-specific PCR amplification. Lancet. 1990;336:529–532. doi: 10.1016/0140-6736(90)92086-w. [DOI] [PubMed] [Google Scholar]
- 12.Broly F, Gaedigk A, Heim M, Eichelbaum M, Morike K, Meyer UA. Debrisoquine/sparteine hydroxlation genotype and phenotype: analysis of common mutations and alleles of CYP2D6 in a European population. DNA Cell Biol. 1991;10:545–558. doi: 10.1089/dna.1991.10.545. [DOI] [PubMed] [Google Scholar]
- 13.Dahl M-L, Johansson I, Porsmyr Palmertz M, Ingelman-Sundberg M, Sjöqvist F. Analysis of the CYP2D6 gene in relation to debrisoquin and desipramine hydroxylation in a Swedish population. Clin Pharmacol Ther. 1992;51:12–17. doi: 10.1038/clpt.1992.2. [DOI] [PubMed] [Google Scholar]
- 14.Smith CAD, Gough AC, Leigh PN, et al. Debrisoquine hydroxylase gene polymorphism and susceptibility to Parkinson’s disease: letter. Lancet. 1992;341:63. doi: 10.1016/0140-6736(92)91196-f. [DOI] [PubMed] [Google Scholar]
- 15.Steen VM, Andreassen OA, Daly AK, et al. Detection of the poor metabolizer-associated CYP2D6 (D)gene deletion allele by long-PCR technology. Pharmacogenetics. 1995;5:215–223. doi: 10.1097/00008571-199508000-00005. [DOI] [PubMed] [Google Scholar]
- 16.Johansson I, Lundqvist E, Dahl M-L, Ingelman-Sundberg M. PCR-based genotyping for duplicated and deleted CYP2D6 genes. Pharmacogenetics. 1996;6:351–355. doi: 10.1097/00008571-199608000-00008. [DOI] [PubMed] [Google Scholar]
- 17.Dahl-Puustinen ML, Lidén A, Alm C, Nordin C, Bertilsson L. Disposition of perphenazine is related to polymorphic debrisoquin hydroxylation in human beings. Clin Pharmacol Ther. 1989;46:78–81. doi: 10.1038/clpt.1989.109. [DOI] [PubMed] [Google Scholar]
- 18.Dahl M-L, Ekqvist B, Widén J, Bertilsson L. Disposition of the neuroleptic zuclopenthixol cosegrates with the polymorphic hydroxylation of debrisoquine in humans. Acta Psychiatr Scand. 1991;84:99–102. doi: 10.1111/j.1600-0447.1991.tb01428.x. [DOI] [PubMed] [Google Scholar]
- 19.Inaba T, Jurima M, Mahon WA, Kalow W. In vitro inhibition studies of two isozymes of human liver cytochrome P-450. Mephenytoin p-hydroxylase and sparteine monooxygenase. Drug Metab Dispos. 1985;12:443–448. [PubMed] [Google Scholar]
- 20.Spina E, Martines C, Caputi AO, et al. Debrisoquine oxidation phenotype during neuroleptic monotherapy. Eur J Clin Pharmacol. 1991;41:467–470. doi: 10.1007/BF00626371. [DOI] [PubMed] [Google Scholar]
- 21.Muralidharan G, Cooper JK, Hawes EM, et al. Quinidine inhibits the 7-hydroxylation of chlorpromazine in extensive metabolisers of debrisoquine. Eur J Clin Pharmacol. 1996;50:121–128. doi: 10.1007/s002280050079. [DOI] [PubMed] [Google Scholar]
- 22.Llerena A, Alm C, Dahl M-L, Ekqvist B, Bertisson L. Haloperidol disposition is dependent on debrisoquine hydroxylation phenotype. Ther Drug Monit. 1992;14:92–97. doi: 10.1097/00007691-199204000-00003. [DOI] [PubMed] [Google Scholar]
- 23.Chakraborty BS, Hubbard JW, Hawes EM, et al. Interconversion between haloperidol and reduced haloperidol in healthy volunteers. Eur J Clin Pharmacol. 1989;37:45–48. doi: 10.1007/BF00609423. [DOI] [PubMed] [Google Scholar]
- 24.Tyndale RF, Kalow W, Inaba T. Oxidation of reduced haloperidol to haloperidol: involvement of human P450IID6 (sparteine/debrisoquine monooxygenase) Br J Clin Pharmacol. 1991;31:655–660. doi: 10.1111/j.1365-2125.1991.tb05588.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Young D, Midha KK, Fossler MJ, et al. Effect of quinidine on the interconversion kinetics between haloperidol and reduced haloperidol in humans: implications for the involvement of cytochrome P450IID6. Eur J Clin Pharmacol. 1993;44:433–438. doi: 10.1007/BF00315539. [DOI] [PubMed] [Google Scholar]
- 26.Fang J, Baker GB, Silverstone PH, et al. Involvement of CYP3A4 and CYP2D6 in the metabolism of haloperidol. Cellular Molec Neurobiol. 1997;17:227–233. doi: 10.1023/A:1026317929335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pan LP, De Vriendt C, Belpaire FM. In-vitro characterisation of the cytochrome P450 isoenzymes involved in the back oxidation and N-dealkylation of reduced haloperidol. Pharmacogenetics. 1998;8:383–389. doi: 10.1097/00008571-199810000-00003. [DOI] [PubMed] [Google Scholar]
- 28.Nyberg S, Farde L, Halldin C, et al. D2 dopamine receptor occupancy during low-dose treatment with haloperidol decanoate. Am J Psychiatry. 1995;152:173–178. doi: 10.1176/ajp.152.2.173. [DOI] [PubMed] [Google Scholar]
- 29.Suzuki A, Otani K, Mihara K, et al. Effects of the CYP2D6 genotype on the steady-state plasma concentrations of haloperidol and reduced haloperidol in Japanese schizophrenic patients. Pharmacogenetics. 1997;7:415–418. doi: 10.1097/00008571-199710000-00013. [DOI] [PubMed] [Google Scholar]
- 30.Lane H-Y, Hu OY-P, Jann MW, et al. Dextromethorphan phenotypin and haloperidol disposition in schizophrenic patients. Psychiatry Res. 1997;69:105–111. doi: 10.1016/s0165-1781(96)02999-x. [DOI] [PubMed] [Google Scholar]
- 31.Schmider J, Walter S, Sachse C, et al. Metabolism of antipsychotics and CYP2D6 genotype. Nauyn-Schmiedebergs Arch Pharmacol. 1998;357(4:SS):R459. [Google Scholar]
- 32.Bareggi SR, Mauri M, Cavallaro R, Regazzetti MG, Moro AR. Factors affecting the clinical response to haloperidol therapy in schizophrenia. Clin Neuropharmacol. 1990;13(Suppl 1):S29–S34. doi: 10.1097/00002826-199001001-00003. [DOI] [PubMed] [Google Scholar]
- 33.Chang WH. Reduced haloperidol: a factor in determining the therapeutic benefit of haloperidol treatment? Psychopharmacol. 1992;106:289–296. doi: 10.1007/BF02245407. [DOI] [PubMed] [Google Scholar]
- 34.Altamura AC. A multidimensional (pharmacokinetic and clinical-biological) approach to neuroleptic response in schizophrenia: with particular reference to drug resistance. Schizophr Res. 1993;8:187–198. doi: 10.1016/0920-9964(93)90017-d. [DOI] [PubMed] [Google Scholar]
- 35.von Bahr C, Movin G, Nordin C, et al. Plasma levels of thioridazine and metabolites are influenced by the debrisoquine hydroxylation phenotype. Clin Pharmacol Ther. 1991;49:234–240. doi: 10.1038/clpt.1991.22. [DOI] [PubMed] [Google Scholar]
- 36.Grahame-Smith DG. The Lilly Prize Lecture 1996 ‘Keep on taking the tablets’: pharmacological adaptation during long-term drug therapy. Br J Clin Pharmacol. 1997;44:227–238. doi: 10.1046/j.1365-2125.1997.00578.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jerling M, Dahl M-L, Åberg-Wistedt A, et al. The CYP2D6 genotype predicts the oral clearance of the neuroleptic agents perphenazine and zuclopenthixol. Clin Pharmacol Ther. 1996;59:423–428. doi: 10.1016/S0009-9236(96)90111-3. [DOI] [PubMed] [Google Scholar]
- 38.Bebbington PE. The content and context of compliance. Int Clin Pyschopharmacol. 1995;9:41–50. doi: 10.1097/00004850-199501005-00008. [DOI] [PubMed] [Google Scholar]
- 39.Arranz MJ, Collier DA, Sodhi MS, et al. Association between clozapine response and allelic variation in 5-HT2A receptor gene. Lancet. 1995;345:281–282. doi: 10.1016/s0140-6736(95)92168-0. [DOI] [PubMed] [Google Scholar]
- 40.Lieberman JA, Alvir JM, Koreen A, et al. Psychobiological correlates of treatment response in schizophrenia. Neuroposychopharmacol. 1996;14(3 Suppl):13S–21S. doi: 10.1016/0893-133X(95)00200-W. [DOI] [PubMed] [Google Scholar]
- 41.van Os J, Fahy TA, Jones P, et al. Psychobiological syndromes in the functional psychoses: associations with course and outcome. Psychol Med. 1996;26:161–176. doi: 10.1017/s0033291700033808. [DOI] [PubMed] [Google Scholar]