

Abstract
Aims
Theophylline is a model substrate of cytochrome P4501A2. The ability of the proton pump inhibitors (PPI) omeprazole, lansoprazole and pantoprazole to induce cytochrome P4501A2 has not yet been unequivocally resolved. The aim of this comprehensive study was to compare directly the effect of the three PPI on the absorption and disposition of theophylline.
Methods
Twenty healthy, nonsmoking, male and female volunteers (extensive metabolisers of cytochrome P4502C19 and Helicobacter pylori negative) participated in a randomized, double-blind, four-period, placebo-controlled crossover study. In each of the four periods they received either omeprazole (40 mg), lansoprazole (60 mg), pantoprazole (80 mg) or placebo once daily for 10 days. Sustained release theophylline (350 mg twice daily) was coadministered from day 8–10. Pharmacokinetics of theophylline as well as of all three PPI were determined at steady-state (day 10).
Results
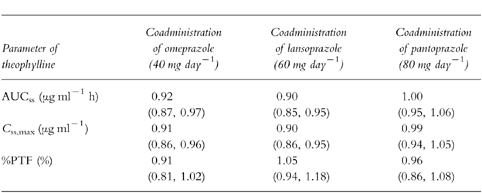
In all periods, point estimates and 90% confidence intervals of the area under the concentration-time curves (AUC), maximum steady-state concentrations and peak-trough fluctuations of theophylline were not altered by PPI pretreatment and met the required limits for bioequivalence. Point estimates (90% confidence intervals) of the AUC ratios of theophylline plus PPI to theophylline alone were 0.92 (0.87–0.97), 0.90 (0.85–0.95) and 1.00 (0.95–1.06) for omeprazole, lansoprazole and pantoprazole, respectively.
Conclusions
Concomitant intake of omeprazole, lansoprazole or pantoprazole at high therapeutic doses does not affect the absorption and disposition of theophylline.
Keywords: cytochrome P4501A2, drug interaction, proton pump inhibitors, theophylline
Introduction
The H+, K+-ATPase (proton pump) inhibitors (PPI) omeprazole, lansoprazole and pantoprazole have been proved effective as monotherapy for gastric or duodenal ulcers and gastro-oesophageal reflux, and in combination regimens for the eradication of Helicobacter pylori [1–3]. These drugs are structurally very similar and they are mainly metabolized by the polymorphically expressed cytochrome P4502C19 (CYP2C19), which means that about 3% of Caucasians (poor metabolisers of CYP2C19) eliminate the compounds more slowly than the majority (extensive metabolisers of CYP2C19) of the Caucasian population [4, 5]. As a class, benzimidazole derivatives show high affinity for cytochrome P450 enzymes and they may act either as inhibitors or inducers of several isoenzymes of cytochrome P450 [6–11]. For example, omeprazole and lansoprazole were shown to be inducers of cytochrome P4501 A2 (CYP1A2) in human liver and alimentary tract in vitro and in vivo [12–14]. The extent of CYP1A2 induction by omeprazole was dependent both on the dose and the CYP2C19 phenotype resulting in a significant effect at lower doses of omeprazole in the subset of poor metabolisers of CYP2C19 [15]. Pantoprazole has been claimed to have less potential than omeprazole and lansoprazole to interact with other drugs metabolized by cytochrome P450 enzymes [16, 17]. In general, putative induction of CYP1A2 by PPI in vivo is highly controversial as clinical studies have shown that low therapeutic doses of PPI have little or no effect on CYP1A2 activity [18–20].
Theophylline is commonly used in patients with chronic obstructive lung diseases [21]. Its metabolism (N-demethylation and 8-hydroxylation) is catalysed primarily by CYP1A2 [22–24] and it can be used as a probe drug for this CYP isoenzyme [25, 26]. Other substrates of human CYP1A2 include caffeine, paracetamol, phenacetin, and tacrine [27]. As theophylline shows concentration-dependent effects with a narrow therapeutic range, induction of CYP1A2 by PPI may compromise clinical efficacy [28].
To date no clinical study has compared directly the effect of all three PPI on steady-state plasma concentrations of theophylline in the same individual. A number of interaction studies have been perfomed between one of the PPI and theophylline [29–35]. However, their interpretation is limited because of several shortcomings such as low dosage, single dose administration or nonrandomization. For example, lack of effect of pantoprazole on the disposition of theophylline is based upon the data of a single study with intravenous drug administration [31]. The most appropriate way to detect interactions on the levels of absorption and disposition is by means of oral drug administration under steady-state conditions. If alterations of the catalytic activity of the cytochrome P450 system are to be detected, dosage and duration of drug administration should be at the upper end of the range therapeutically recommended. Therefore, we designed a comprehensive clinical study that compared directly the effect of all three available PPI (omeprazole, lansoprazole, pantoprazole) on absorption and disposition of theophylline.
Methods
Subject population
Ten healthy females (age 33.0±5.6 years, body mass index 21.9±2.5 kg m−2; mean±s.d.) and 10 healthy males (age 32.0±3.3 years, body mass index 24.3± 0.7 kg m−2; mean±s.d.) participated in the study. The study was approved by the Ethics Committee of the Landesärztekammer Baden-Württemberg, Stuttgart, Germany, according to the ethical guidelines of the 1996 Declaration of Helsinki. Written informed consent was obtained from all volunteers. Before entering the study each subject underwent a complete medical examination including laboratory investigations, vital signs, ECG, and [13C]-urea breath testing for exclusion of Helicobacter pylori infection. The subjects were all Caucasian, nonsmokers and phenotyped as extensive metabolisers of CYP2C19 with the probe drug mephenytoin [36]. No comedication was allowed for 3 weeks before and during the treatment periods.
Study design
The study had a randomized, double-blind, double-dummy, four-period, placebo-controlled crossover design with wash-out periods of at least 11 days. In each of the four periods the volunteers ingested either 40 mg omeprazole (Antra® 20, Astra GmbH, Wedel, Germany, two capsules) or 60 mg of lansoprazole (Agopton®, Takeda Pharma GmbH, Aachen, Germany, two capsules) or 80 mg pantoprazole (Pantozol®, Byk Gulden Lomberg Chemische Fabrik GmbH, Konstanz, Germany, two tablets) or placebo once daily at 07.30 h before breakfast for 10 consecutive days. A dose of 350 mg sustained release theophylline (Bronchoretard® 350, Klinge Pharma GmbH, München, Germany) was coadministered in an open-labelled fashion at 07.30 h and 19.30 h on day 8 and day 9. After an overnight fast the last doses of study medication were taken in the morning of day 10. Blood samples (7.5 ml) for determination of theophylline, omeprazole, lansoprazole and pantoprazole were drawn via an indwelling cannula into tubes containing ethylendiaminetetraacetic acid (EDTA) before and at 0.5, 1, 2, 3, 4, 6, 8, 10, 12 and 24 h after drug administration. A standardized breakfast consisting of two slices of toast, 20 g butter, 25 g jam and 100 ml orange juice, that has been proved not to affect the pharmacokinetic profile of sustained release theophylline [37], was served 2 h after drug intake. For the first 4 h following drug administration, the subjects remained supine. Urine was collected from 0 to 12 h after the last dose of theophylline in each period. Plasma and aliquots of urine were stored at −20° C until analysis. Subjects were not allowed to consume alcohol, methylxanthine-containing foods and beverages (coffee, tea, cola, chocolate) and grapefruit from day 8 through day 12. They were also told to avoid charcoal-grilled meat and certain cruciferous vegetables (broccoli, spinach, turnips, cabbage, cauliflower, brussels sprouts) during the entire study because these factors have been shown to alter CYP1A2 activity [38–40]. Compliance was assessed by daily telephone calls and also directly by unannounced randomized withdrawals of blood at two different occasions in each period.
Determination of theophylline
Theophylline plasma concentrations were assayed by using a fluorescent polarization immunoassay technique (AxSYM Theophylline II, Abbott GmbH, Wiesbaden, Germany). This assay has a sensitivity range of 0.82–40.0 μg ml−1. The within-run coefficients of variation were 2.7% at 7.0 μg ml−1, 2.5% at 12.0 μg ml−1, and 3.8% at 26.0 μg ml−1. The between-day coefficients of variation were 1.2% at 7.0 μg ml−1,2.0% at 12.0 μg ml−1, and 3.8% at 26.0 μg ml−1. Urine analysis for theophylline was carried out by the same technique on one single day following a 5–10 fold dilution with blank plasma before analysis. The within-run coefficients of variation were 4.0% at 7.0 μg ml−1, 3.8% at 12.0 μg ml−1, and 2.0% at 26.0 μg ml−1.
Determination of omeprazole, lansoprazole and pantoprazole
Omeprazole plasma levels were determined by h.p.l.c. as decribed previously [41]. In brief, 0.5 μg of internal standard (lansoprazole) and 10 μl of 0.5 m sodium carbonate were added to 1 ml of plasma and extracted with 6 ml ethyl acetate. The upper organic phase was vaporized under nitrogen and the residue was dissolved in 100 μl of mobile phase (55% methanol +1% triethylamine, pH 7). Using a Hypersil ODS column (5 μm; Shandon Southern Products Ltd, Chesire, U.K.) omeprazole and lansoprazole eluted at 4.7 min and 6.3 min, respectively (flow rate 0.8 ml min−1, wavelength of UV-detection 302 nm). Peak height ratios were used for quantification. Calibration curves ranged from 10 to 500 ng ml−1. Analysis of lansoprazole and pantoprazole was performed by the same assay (calibration range 100 to 1000 ng ml−1 and 10–8000 ng ml−1, respectively) using omeprazole and lansoprazole as internal standards, and 280 nm or 290 nm as recording wavelength, respectively. Based on quality control samples, coefficients of variation for intra-and interassay variability ranged for omeprazole, lansoprazole and pantoprazole between 3 and 6%, 7 and 8.5% and 3.8 and 11.4%, respectively.
Pharmacokinetic analysis
Pharmacokinetic analysis was based on plasma concentrations above the lower limit of quantification. Maximum steady-state plasma concentration during a dosing interval (Css, max), time of Css, max (tmax), and minimum (trough) steady-state plasma concentration at the end of a dosing interval (Css, min) were taken directly from the plasma concentration-time profiles. Area under the curve at steady-state (AUCss) was calculated by the trapezoidal rule over the dosing interval (τ) and apparent terminal elimination half-life (t1/2) was determined after stopping drug administration using standard noncompartmental analysis of the TopFit 2.1 program [42]. Apparent oral clearance (CLo) was derived from the equation CLo = dose/AUCss, average steady-state plasma concentration (Css, av) from Css, av = AUCss/τ, and percentage peak-trough fluctuation (%PTF) as characteristics of rate of absorption from percentagePTF = 100 [Css,max−Css, min]/Css,av. Renal clearance (CLR) was derived from CLR = Aess/AUCss, in which Aess is the amount of drug excreted unchanged into urine during a dosing interval.
Statistical analysis
Data are presented as means with 95% confidence intervals. To assess the effects of PPI on pharmacokinetics of theophylline a repeated measures anova with Bonferroni correction (GraphPad Instad®) was performed. The level of significance was set at α = 0.05. Proving lack or existence of any clinically significant pharmacokinetic interaction was then handled as an equivalence problem [43]. 90% confidence intervals of the log-transformed parameters AUCss, Css,max and percentagePTF of theophylline during treatment with PPI relative to coadministration of placebo were derived from the residual variance in multifactor anova (STATGRAPHICS Plus®) with effects for treatment, period, sequence and subject nested within sequence. No interaction was concluded if the 90% confidence intervals of the differences were in the equivalence range of 0.8–1.25 (AUCss) and 0.7–1.43 (Css,max, %PTF), respectively. Stratification by gender is not necessary for pharmacokinetic investigations of substrates for CYP1 A2 since this is not a determinant of their disposition [44].
Results
All subjects were compliant since PPI could be detected in all blood samples. One subject dropped out of the study because of a sport accident. He was replaced by another subject, who completed the study. Figure 1 shows the mean (±s.d.) steady-state plasma concentration-time profiles for theophylline when administered with placebo, omeprazole, lansoprazole or pantoprazole, respectively. When theophylline and pantoprazole were coadministered the curve was superimposable on the curve of theophylline given together with placebo. Table 1 summarizes the pharmacokinetic parameters of theophylline with results of repeated measures anova. Mean AUCss of theophylline was reduced by 7% and 10% after coadministration with omeprazole and lansoprazole, respectively, and mean Css, min of theophylline was lower by 6% and 11%, respectively. CLR of theophylline was not affected by comedication with any of the PPI. Table 2 summarizes equivalence assessment. The 90% confidence intervals as well as the point estimates of AUCss, Css,max and percentagePTF were within the required limits for bioequivalence. Thus, a clinically significant pharmacokinetic interaction on the levels of absorption and disposition could be excluded between theophylline and omeprazole, lansoprazole or pantoprazole, respectively.
Figure 1.

Mean (±s.d.) plasma steady-state theophylline concentration-time curves (350 mg twice daily) in 20 subjects following coadministration of placebo (□), omeprazole 40 mg day−1 (▵), lansoprazole 60 mg day−1 (▿) or pantoprazole 80 mg day−1 [○].
Table 1.
Effect of PPI treatment on the pharmacokinetics of theophylline (350 mg twice daily) at steady-state in 20 subjects. Values are given as means with 95% confidence intervals.
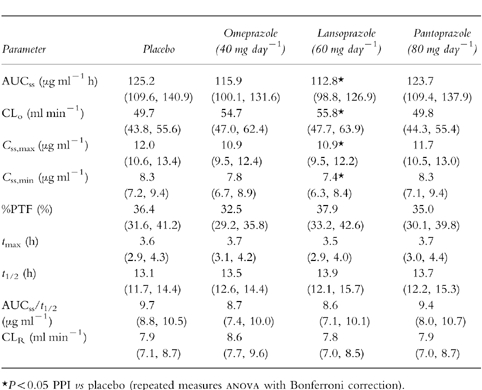
Table 2.
Summary of equivalence assessment in 20 subjects: pharmacokinetic characteristics of theophylline (350 mg twice daily) at steady-state. Data are point estimates with 90 percnt; confidence intervals in parentheses.
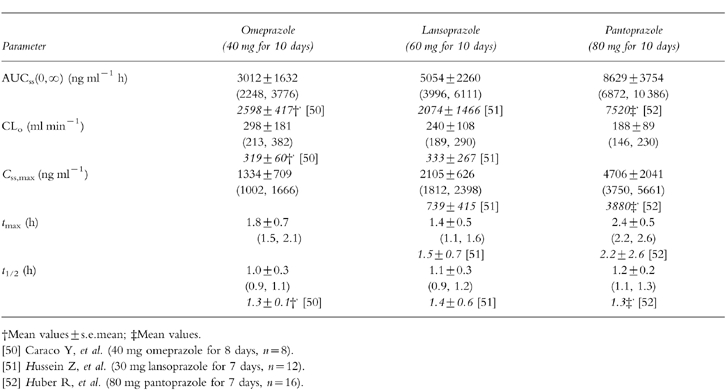
Mean (±s.d.) plasma concentration-time profiles for omeprazole, lansoprazole and pantoprazole following multiple doses (last day of treatment concomitantly with theophylline) are presented in Figure 2. Based on lag time and plasma concentrations one hour after ingestion lansoprazole appears to be absorbed more rapidly than omeprazole or pantoprazole. There were no gender-related differences in the pharmacokinetic parameters of PPI listed in Table 3.
Figure 2.

Mean (±s.d.) plasma concentration-time curves of omeprazole 40 mg day−1, lansoprazole 60 mg day−1 and pantoprazole 80 mg day−1 following multiple doses in 20 subjects during treatment with theophylline (350 mg twice daily).
Table 3.
Pharmacokinetic parameters of PPI following multiple doses during treatment with theophylline (350 mg twice daily) in 20 subjects. Data are mean values±s.d. (95% confidence intervals). For comparison pharmacokinetic parameters of PPI without comedication (reference data) are given in italics.
Discussion
In extensive metabolisers of CYP2C19 coadministration of the PPI omeprazole (40 mg day−1), lansoprazole (60 mg day−1), or pantoprazole (80 mg day−1) for 10 days did not alter the steady-state disposition of theophylline administered as a sustained release formulation. Clinical efficacy and toxicity of theophylline are closely related to its plasma concentration and a range between 10 and 20 μg ml−1 is generally accepted as the therapeutic window in asthmatics [45]. However, some have suggested a lower target range between 8 and 15 μg ml−1 to avoid toxicity [46]. As a clinically significant drug interaction could be exluded from our data, coadministration of PPI in therapeutically recommended doses does not require additional therapeutic drug monitoring of theophylline.
A drug interaction on the level of absorption, as reported in the case of omeprazole plus itraconazole [47], was considered a priori to be unlikely, because the pharmacokinetics of the sustained release theophylline dosage form was not sensitive to major changes in gastric pH induced by ranitidine [48]. In agreement, the ratio AUCss/t1/2 of theophylline which characterizes the extent of absorption [49] did not differ during treatment with PPI.
As already mentioned drug interaction between PPI and theophylline cannot be excluded definitely if the drugs are administered intravenously. Therefore, some studies with intravenous administration of pantoprazole and omeprazole and/or intravenous administration of theophylline claiming lack of interaction are of limited value [29–31]. Since induction of CYP1A2 activity is regarded as the most likely mechanism of possible interaction between PPI and theophylline, and since the extent of induction depends on the dose of the inductive compound previous studies with lower doses of omeprazole (20 mg day−1) demonstrating no significant influence on the pharmacokinetics of theophylline are not surprising in retrospect [32, 33].
The metabolic capacity of subjects with regard to CYP2C19, that determines plasma concentrations of PPI is another important factor of the extent of CYP1A2 induction. However, it was not taken into account in any of the clinical studies with PPI and theophylline. Especially, results from Asian populations with a high proportion of poor metabolisers of CYP2C19 may differ from others if the study populations had not been phenotyped. A Japanese study without phenotyped subjects and a lower dose of lansoprazole (30 mg day−1) given for 11 days found no significant alteration in the metabolism of theophylline [34]. An investigation with higher doses of lansoprazole (60 mg day−1) given for 10 days reported a pharmacokinetic interaction between lansoprazole and theophylline, but the observed effect (13% decrease in AUC of theophylline) was not considered as clinically significant [35]. The 11% reduction in mean AUCss of theophylline after coadministration with lansoprazole observed in our study is in very good agreement with this investigation.
Caffeine breath test and urinary caffeine metabolite ratios are commonly used to determine CYP1A2 activity in clinical studies. A recent clinical study concluded that none of the three PPI (7-day treatment with low therapeutic doses) is an inducer of caffeine metabolism, neither in poor nor in extensive metabolisers of CYP2C19 [18]. However, it should be noted, that the results of only two poor metabolisers were regarded as representative for this group of individuals. Two other investigations using low therapeutic doses of omeprazole (20 mg day−1), lansoprazole (30 mg day−1) and pantoprazole (40 mg day−1) also demonstrated lack of induction of CYP1A2 activity measured by means of urinary caffeine metabolite ratios [19, 20].
As only very limited data could be found in the literature on disposition of PPI following multiple higher therapeutic oral doses we assessed also their pharmacokinetic properties (see Table 3). Although interpretation of the PPI pharmacokinetic data is difficult since measurements were made in the presence of theophylline, there appeared to be no effect of theophylline on absorption and disposition of PPI. The pharmacokinetic parameters of omeprazole, lansoprazole and pantoprazole given together with theophylline were similar to those values reported in some previous studies with PPI [50–52].
In conclusion, even in therapeutically recommended higher doses the CYP1A2 inductive potential of the PPI omeprazole, lansoprazole and pantoprazole is too small to influence significantly the pharmacokinetics of theophylline in extensive metabolisers of CYP2C19.
Acknowledgments
This work was supported by the Robert Bosch Foundation (Stuttgart, Germany), Takeda Pharma GmbH (Aachen, Germany) and Klinge Pharma GmbH (München, Germany). Mr Z. Zheng was supported by a scholarship from the Chinese government. We are grateful to Mrs A. Godel and Mrs A. Riebe for their excellent technical assistance.
References
- 1.Howden CW. Clinical pharmacology of omeprazole. Clin Pharmacokinet. 1991;20:38–49. doi: 10.2165/00003088-199120010-00003. [DOI] [PubMed] [Google Scholar]
- 2.Fitton A, Wisemann L. Pantoprazole. A review of its pharmacological properties and therapeutic use in acid-related disorders. Drugs. 1996;51:460–482. doi: 10.2165/00003495-199651030-00012. [DOI] [PubMed] [Google Scholar]
- 3.Langtry HD, Wilde MI. Lansoprazole. An update of its pharmacological properties and clinical efficacy in the management of acid-related disorders. Drugs. 1997;54:473–500. doi: 10.2165/00003495-199754030-00010. [DOI] [PubMed] [Google Scholar]
- 4.Andersson T. Pharmacokinetics, metabolism and interactions of acid pump inhibitors. Focus on omeprazole, lansoprazole and pantoprazole. Clin Pharmacokinet. 1996;31:9–28. doi: 10.2165/00003088-199631010-00002. [DOI] [PubMed] [Google Scholar]
- 5.Bertilsson L. Geographical/interracial differences in polymorphic drug oxidation. Current state of knowledge of cytochromes P450 (CYP) 2D6 and 2C19. Clin Pharmacokinet. 1995;29:192–209. doi: 10.2165/00003088-199529030-00005. [DOI] [PubMed] [Google Scholar]
- 6.Andersson T. Omeprazole drug interaction studies. Clin Pharmacokinet. 1991;21:195–212. doi: 10.2165/00003088-199121030-00004. [DOI] [PubMed] [Google Scholar]
- 7.Humphries TJ. Clinical implications of drug interactions with the cytochrome P-450 enzyme system associated with omeprazole. Dig Dis Sci. 1991;36:1665–1669. doi: 10.1007/BF01296606. [DOI] [PubMed] [Google Scholar]
- 8.Ko J, Sukhova N, Thacker D, Chen P, Flockhart DA. Evaluation of omeprazole and lansoprazole as inhibitors of cytochrome P450 isoforms. Drug Metab Dispos. 1997;25:853–862. [PubMed] [Google Scholar]
- 9.Zomorodi K, Houston JB. Diazepam–omeprazole inhibition interaction: an in vitro investigation using human liver microsomes. Br J Clin Pharmacol. 1996;42:157–162. doi: 10.1046/j.1365-2125.1996.03563.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tucker GT. The interaction of proton pump inhibitors with cytochromes P450. Aliment Pharmacol Ther. 1994;8:33–38. doi: 10.1111/j.1365-2036.1994.tb00233.x. [DOI] [PubMed] [Google Scholar]
- 11.Andersson T, Cederberg C, Edvardsson G, Heggelund A, Lundborg P. Effect of omeprazole treatment on diazepam plasma levels in slow versus normal rapid metabolizers of omeprazole. Clin Pharmacol Ther. 1990;47:79–85. doi: 10.1038/clpt.1990.12. [DOI] [PubMed] [Google Scholar]
- 12.Diaz D, Fabre I, Daujet M, et al. Omeprazole is an aryl hydrocarbon-like inducer of human hepatic cytochrome P450. Gastroenterology. 1990;99:737–747. doi: 10.1016/0016-5085(90)90963-2. [DOI] [PubMed] [Google Scholar]
- 13.McDonnell WM, Scheimann JM, Traber PG. Induction of cytochrome P450IA genes (CYP1A) by omeprazole in the human alimentary tract. Gastroenterology. 1992;103:1509–1516. doi: 10.1016/0016-5085(92)91171-y. [DOI] [PubMed] [Google Scholar]
- 14.Curi-Pedrosa R, Daujet M, Pichard L, et al. Omeprazole and lansoprazole are mixed inducers of CYP1A and CYP3A in human hepatocytes in primary culture. J Pharmacol Exp Ther. 1994;269:384–392. [PubMed] [Google Scholar]
- 15.Rost KL, Roots I. Accelerated caffeine metabolism after omeprazole treatment is indicated by urinary metabolite ratios: Coincidence with plasma clearance and breath test. Clin Pharmacol Ther. 1994;55:402–411. doi: 10.1038/clpt.1994.49. [DOI] [PubMed] [Google Scholar]
- 16.Meyer UA. Metabolic interactions of the proton-pump inhibitors lansoprazole, omeprazole and pantoprazole with other drugs. Eur J Gastroenterol Hepatol. 1996;8:S21–S25. doi: 10.1097/00042737-199610001-00005. [DOI] [PubMed] [Google Scholar]
- 17.Steinijans VW, Huber R, Hartmann M, et al. Lack of pantoprazole drug interactions in man: an updated review. Int J Clin Pharmacol Ther. 1996;34:243–262. [PubMed] [Google Scholar]
- 18.Andersson T, Holmberg J, Röhss K, Walan A. Pharmacokinetics and effect on caffeine metabolism of the protonpump inhibitors, omeprazole, lansoprazole, and pantoprazole. Br J Clin Pharmacol. 1998;45:369–375. doi: 10.1046/j.1365-2125.1998.t01-1-00702.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rizzo N, Padoin C, Palombo S, Scherrmann JM, Girre C. Omeprazole and lansoprazole are not inducers of cytochrome P4501A2 under conventional therapeutic conditions. Eur J Clin Pharmacol. 1996;49:491–495. doi: 10.1007/BF00195936. [DOI] [PubMed] [Google Scholar]
- 20.Hartmann M, Bliesath H, Zech K, et al. Lack of induction of CYPIA2 activity in man by pantoprazole. Gut. 1995;37:32. [PubMed] [Google Scholar]
- 21.Vassallo R, Lipsky JJ. Theophylline: Recent advances in the understanding of its mode of action and uses in clinical practice. Mayo Clin Proc. 1998;73:346–354. doi: 10.1016/S0025-6196(11)63701-4. [DOI] [PubMed] [Google Scholar]
- 22.Sarkar MA, Jackson BJ. Theophylline N-demethylations as probes for P4501A1 and P4501A2. Drug Metab Dispos. 1994;22:827–834. [PubMed] [Google Scholar]
- 23.Zhang Z, Kaminsky LS. Characterisation of human cytochromes P450 involved in theophylline 8-hydroxylation. Biochem Pharmacol. 1995;50:205–211. doi: 10.1016/0006-2952(95)00120-o. [DOI] [PubMed] [Google Scholar]
- 24.Tjia JF, Colbert J, Back DJ. Theophylline metabolism in human liver microsomes: inhibition studies. J Pharmacol Exp Ther. 1996;276:912–917. [PubMed] [Google Scholar]
- 25.Vesell ES. The model drug approach in clinical pharmacology. Clin Pharmacol Ther. 1991;50:239–248. doi: 10.1038/clpt.1991.132. [DOI] [PubMed] [Google Scholar]
- 26.Kivistö KT, Kroemer HK. Use of probe drugs as predictors of drug metabolism in humans. J Clin Pharmacol. 1997;37:40S–48S. doi: 10.1177/009127009703700121. [DOI] [PubMed] [Google Scholar]
- 27.Meyer UA. Overview of enzymes of drug metabolism. J Pharmacokinet Biopharm. 1996;24:449–459. doi: 10.1007/BF02353473. [DOI] [PubMed] [Google Scholar]
- 28.Jonkman JHG, Upton RA. Pharmacokinetic drug interactions with theophylline. Clin Pharmacokinet. 1984;9:309–334. doi: 10.2165/00003088-198409040-00002. [DOI] [PubMed] [Google Scholar]
- 29.Gugler R, Jensen JC. Drugs other than H2-receptor antagonists as clinically important inhibitors of drug metabolism in vivo. Pharmac Ther. 1987;33:133–137. doi: 10.1016/0163-7258(87)90041-6. [DOI] [PubMed] [Google Scholar]
- 30.Oosterhuis B, Jonkman JHG, Andersson T, Zuiderwijk PBM. No influence of single intravenous doses of omeprazole in theophylline elimination kinetics. J Clin Pharmacol. 1992;32:470–475. doi: 10.1002/j.1552-4604.1992.tb03864.x. [DOI] [PubMed] [Google Scholar]
- 31.Schulz HU, Hartmann M, Steinijans VW, et al. Lack of influence of pantoprazole on the disposition kinetics of theophylline in man. Int J Clin Pharmacol Ther Toxicol. 1996;34:S51–S57. [PubMed] [Google Scholar]
- 32.Pilotto A, Franceschi M, Lagni M, et al. The effect of omeprazole on serum concentrations of theophylline, pepsinogens A and C, and gastrin in elderly duodenal ulcer patients. Am J Ther. 1995;2:43–46. doi: 10.1097/00045391-199501000-00008. [DOI] [PubMed] [Google Scholar]
- 33.Taburet AM, Geneve J, Bocquentin M, Simoneau G, Caulin C, Singlas E. Theophylline steady state pharmacokinetics is not altered by omeprazole. Eur J Clin Pharmacol. 1992;42:343–345. doi: 10.1007/BF00266361. [DOI] [PubMed] [Google Scholar]
- 34.Kokufu T, Ihara N, Sugioka N, et al. Effects of lansoprazole on pharmacokinetics and metabolism of theophylline. Eur J Clin Pharmacol. 1995;48:391–395. doi: 10.1007/BF00194956. [DOI] [PubMed] [Google Scholar]
- 35.Grannemann GR, Karol MD, Locke CS, Cavanaugh JH. Pharmacokinetic interaction between lansoprazole and theophylline. Ther Drug Monit. 1995;17:460–464. doi: 10.1097/00007691-199510000-00005. [DOI] [PubMed] [Google Scholar]
- 36.Sanz EJ, Villen T, Alm C, Bertilsson L. S-Mephenytoin hydroxylation phenotypes in a Swedish population determined after coadministration with debrisoquin. Clin Pharmacol Ther. 1989;45:495–499. doi: 10.1038/clpt.1989.63. [DOI] [PubMed] [Google Scholar]
- 37.Fuchs WS, von Nieciecki A, Molz KH, et al. Effect of gallbladder contraction induced cholagogia on the pharmacokinetic profile of a sustained-release theophylline formulation. Drug Res. 1996;46:1120–1126. [PubMed] [Google Scholar]
- 38.Conney AH, Pantuck EJ, Hsiao KC, et al. Enhanced phenacetin metabolism in human subjects fed charcoal-broiled beef. Clin Pharmacol Ther. 1976;20:633–642. doi: 10.1002/cpt1976206633. [DOI] [PubMed] [Google Scholar]
- 39.Pantuck EJ, Pantuck CB, Garland WA, et al. Stimulatory effect of brussels sprouts and cabbage on human drug metabolism. Clin Pharmacol Ther. 1979;25:88–95. doi: 10.1002/cpt197925188. [DOI] [PubMed] [Google Scholar]
- 40.Vistisen K, Poulsen HE, Loft S. Foreign compound metabolism capacity in man measured from metabolites of dietary caffeine. Carcinogenesis. 1992;13:1561–1568. doi: 10.1093/carcin/13.9.1561. [DOI] [PubMed] [Google Scholar]
- 41.Treiber G, Ammon S, Klotz U. Age-dependent eradication of Helicobacter pylori with dual therapy. Aliment Pharmacol Ther. 1997;11:711–718. doi: 10.1046/j.1365-2036.1997.00210.x. [DOI] [PubMed] [Google Scholar]
- 42.Heinzel G, Woloszczak R, Thomann P. TopFit, Version 2.0: Pharmacokinetic and Pharmacodynamic Data Analysis System for the PC. New York: Gustav Fischer; 1993. [Google Scholar]
- 43.Steinijans VW, Hartmann M, Huber R, Radtke HW. Lack of pharmacokinetic interaction as an equivalence problem. Int J Clin Pharmacol Ther Toxicol. 1991;29:323–328. [PubMed] [Google Scholar]
- 44.Kashuba ADM, Bertino JS, Kearns G, et al. Quantification of three-month intraindividual variability and influence of sex and menstrual cycle phase on CYP1A2, N-acetyltransferase-2, and xanthine oxidase activity determined with caffeine phenotyping. Clin Pharmacol Ther. 1998;63:540–551. doi: 10.1016/S0009-9236(98)90105-9. [DOI] [PubMed] [Google Scholar]
- 45.Weinberger M, Hendeles L. Theophylline in asthma. N Engl J Med. 1996;334:1380–1388. doi: 10.1056/NEJM199605233342107. [DOI] [PubMed] [Google Scholar]
- 46.Skorodin MS. Pharmacotherapy for asthma and chronic obstructive pulmonary disease. Arch Intern Med. 1993;153:814–828. [PubMed] [Google Scholar]
- 47.Jaruratanasirikul S, Sriwiriyajan S. Effect of omeprazole on the pharmacokinetics of itraconazole. Eur J Clin Pharmacol. 1998;54:159–161. doi: 10.1007/s002280050438. [DOI] [PubMed] [Google Scholar]
- 48.Wilson CG, Washington N, Greaves JL, et al. Effect of pretreatment with ranitidine on the pharmacokinetics and gastrointestinal transit of a sustained release theophylline preparation. Drug Res. 1991;41:1154–1159. [PubMed] [Google Scholar]
- 49.Schall R, Hundt HKL, Luus HG. Pharmacokinetic characteristics for extent of absorption and clearance in drug/drug interaction studies. Int J Clin Pharmacol Ther. 1994;32:633–637. [PubMed] [Google Scholar]
- 50.Caraco Y, Lagerstrom PO, Wood AJJ. Ethnic and genetic determinants of omeprazole disposition and effect. Clin Pharmacol Ther. 1996;60:157–167. doi: 10.1016/S0009-9236(96)90131-9. [DOI] [PubMed] [Google Scholar]
- 51.Hussein Z, Grannemann GR, Mukherjee D, et al. Age-related differences in the pharmacokinetics and pharmacodynamics of lansoprazole. Br J Clin Pharmacol. 1993;36:391–398. doi: 10.1111/j.1365-2125.1993.tb00386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huber R, Hartmann M, Bliesath H, Lühmann R, Steinijans VW, Zech K. Pharmacokinetics of pantoprazole in man. Int J Clin Pharmacol Ther. 1996;34:S7–S16. [PubMed] [Google Scholar]