

Abstract
Aims
To investigate the pharmacokinetics and safety of tolterodine and tolterodine metabolites after single-and multiple-dose administration in the absence and presence of ketoconazole, an inhibitor of cytochrome P450 (CYP) 3A4, in healthy volunteers with deficient CYP2D6 activity, i.e. poor metabolisers of debrisoquine.
Methods
Eight healthy volunteers received single oral doses (2 mg) of tolterodine l-tartrate. Following a wash-out period of about 3 months, six of the subjects participated in a multiple-dose (1 mg twice daily) phase of the study. Ketoconazole 200 mg was given once daily for 4–4.5 days during both the single and multiple dose tolterodine administration phases. Blood samples were drawn and the pharmacokinetics of tolterodine and its metabolites were determined.
Results
A decrease (P <0.01) in apparent oral clearance of tolterodine, from 10– 12 l h−1 to 4.3–4.7 l h−1, was obtained during concomitant administration of ketoconazole, yielding at least a two-fold increase in the area under the serum concentration-time curve after single as well as after multiple doses following single dose administration of tolterodine. The mean (±s.d.) terminal half-life increased by 50% from 9.7±2.7 h to 15±5.4 h in the presence of ketoconazole.
Conclusions
CYP3A4 is the major enzyme involved in the elimination of tolterodine in individuals with deficient CYP2D6 activity (poor metabolisers), since oral clearance of tolterodine decreased by 60% during ketoconazole coadministration. This inhibition resulted in 2.1-fold increase in AUC.
Keywords: antimuscarinic, CYP2D6, CYP3A4, drug interaction, ketoconazole, tolterodine, urinary incontinence
Introduction
Tolterodine is a new antimuscarinic drug for the treatment of patients with overactive bladder presenting with urinary frequency, urgency and urge incontinence [1–3]. In vivo, tolterodine exhibits functional selectivity for the urinary bladder over salivary glands, a profile that cannot be explained in terms of selectivity for a single muscarinic receptor subtype [4]. In humans, tolterodine is rapidly absorbed, and the systemically available drug is mainly eliminated by two different oxidative metabolic pathways [5], hydroxylation catalysed by cytochrome P450 (CYP) 2D6 and N-dealkylation, catalysed by CYP3A (Figure 1) [6, 7]. Preclinical studies have shown that the pharmacologically equipotent 5-hydroxymethyl metabolite (5-HM; PNU-200577) has similar functional bladder selectivity in vivo as tolterodine [8]. Further oxidation of 5-HM yields the carboxylic acid of tolterodine and its N-dealkylated form, along with N-dealkylated 5-HM [5]. The importance of the CYP2D6 polymorphism in drug metabolism is widely recognized for a number of drugs particularly certain antidepressant and antipsychotic agents [9]. Regarding tolterodine, the mean systemic clearance in poor metabolisers of debrisoquine is five times lower than in extensive metabolisers and results in levels of 5-HM that are not quantifiable among poor metabolisers [6]. However, the large differences in pharmacokinetics between extensive and poor metabolisers is of minor importance for the antimuscarinic effect because the absence of the 5-HM is balanced by higher levels of parent drug, along with a 10-fold difference between tolterodine and 5-HM in terms of serum protein binding [6, 10].
Figure 1.
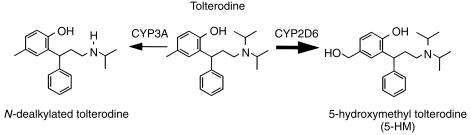
Tolterodine (R)-N,N-diisopropyl-3-(2-hydroxy-5-methylphenyl)-3-phenylpropanamine, and the primary metabolic pathways.
Ketoconazole was the first orally active azole antifungal introduced for the treatment of a number of important mycoses. Ketoconazole was subsequently identified as an inhibitor of hepatic oxidative drug metabolism [11] and more recently as a potent and selective inhibitor of CYP3A activity in vitro [12]. Clinical consequences of these observations are recommendations to avoid coadministration of ketoconazole with drugs that are extensively metabolized by CYP3A4, e.g., terfenadine and triazolam, to avoid serious interactions [13, 14].
The high CYP2D6 specificity of tolterodine metabolism suggests that extensive metabolisers are unlikely to be affected by a metabolic interaction with a CYP3A4 inhibitor, since the mean oral clearance of tolterodine in poor metabolisers is less than 3% of that in extensive metabolisers. In contrast, individuals devoid of CYP2D6 activity (poor metabolisers) probably N-dealkylate tolterodine by CYP3A to some extent which has been suggested from in vitro studies [7]. However the importance of this metabolic route has not been investigated. Ketoconazole was selected as a potent CYP3A4 inhibitor to investigate the effect on the pharmacokinetics of tolterodine in poor metabolisers. The objective of the present study was accordingly to investigate the pharmacokinetics and safety of tolterodine and tolterodine metabolites after single-and multiple-dose administration in the absence and presence of ketoconazole in healthy volunteers with deficient CYP2D6 activity.
This study was presented in part at the 16th Workshop on Drug Metabolism, Copenhagen, Denmark, June 21– 26, 1998.
Methods
Subjects
Four male and four female healthy volunteers were recruited for the study. All volunteers were investigated and assessed as healthy according to biochemical and haematological parameters, 12-lead ECG, and medical investigation including blood pressure. The subjects were previously classified as poor metabolisers by debrisoquine phenotyping (all had metabolic ratios >12.6). The subjects had also been genotyped for the most frequent mutated alleles (CYP2D6*3, *4 and *5) and found to be homozygous poor metabolisers (all except subject nos. 6 and 8 who were CYP2D6*3 and *4 mutated, respectively, in combination with an unidentified mutation). Additionally, the subjects had also been phenotyped with mephenytoin (CYP2C19) and all were found to be extensive metabolisers except subject no. 1 who was a poor metaboliser. The mean (±s.d.) demographic characteristics of female volunteers were as follows: age, 29±6.8 years; body weight, 58±13 kg; and height, 1.64±0.26 m. The mean demographic characteristics of male volunteers were as follows: age, 30±7.7 years; body weight, 77±6.7 kg; and height, 1.84± 0.76 m. The study was approved by the ethics committee of the Huddinge University Hospital, and each volunteer gave written informed consent before the study.
Study design
This open, nonrandomised crossover study was divided into two phases: single-and multiple-dose administration of tolterodine in the absence and presence of ketoconazole, respectively. Doses of tolterodine are expressed as the l-tartrate salt. Eight volunteers received single oral doses of tolterodine during the first phase of the study, while six out of the eight volunteers participated in the multiple-dose phase of the study. There was a wash-out period of about 3 months between the two phases, during which the single-dose pharmacokinetics of tolterodine were determined. Due to the anticipated inhibitory effect of ketoconazole on the metabolism of tolterodine, tolterodine 1 mg twice daily (half the recommended therapeutic dosage) was to be used to obtain steady-state. Adverse effects related to ketoconazole treatment (e.g. gastrointestinal disturbances) were to be expected, and therefore the length of the ketoconazole administration phase was made as short as possible.
Volunteers were initially given a 2 mg single oral dose of tolterodine (Detrusitol®, Pharmacia & Upjohn). After a wash-out period of at least 4 days, each volunteer then received ketoconazole (Fungoral®, Janssen-Cilag) 200 mg once daily for 4 days, and tolterodine as a 2 mg single dose on day 2.
In order to avoid unnecessary adverse events only five doses of ketoconazole were given during the multiple-dose phase of the study. The six volunteers were each given tolterodine 1 mg twice daily (12 h apart) for 4.5 days. On day 3, blood samples were drawn to determine the steady-state pharmacokinetics of tolterodine, and in the evening a 2×1 mg loading dose was given along with a ketoconazole dose to rapidly obtain the new steady-state level of tolterodine. On days 4 and 5, ketoconazole 200 mg was given concomitantly with tolterodine, and on days 6 and 7 in the absence of tolterodine. The dosage regimen, using a loading dose, had been simulated using the data and considering the difference in half-life (in absence and presence of ketoconazole) obtained following the single-dose phase of the study.
Drugs with known CYP3A4 or other liver isoenzyme inducing or inhibitory properties were strictly forbidden. Intake of alcohol-containing beverages around the study days was not allowed, and intake of grapefruit juice was also forbidden.
Assessment
Venous blood samples were drawn at 15, 30 and 45 min, and at 1, 1.5, 2, 4, 6, 8, 12, 24, 26 and 32 h, after the single doses of tolterodine. During coadministration of ketoconazole, the 26 h sample was omitted and measurements at 48 and 56 h were added. During the multiple-dose investigations, blood samples were drawn on days 3 and 5 immediately before tolterodine administration and at 15, 30 and 45 min, and at 1, 1.5, 2, 4, 6, 8 and 12 h, after dose. Additional blood samples were drawn on days 6 and 7 at 24, 32, 48 and 56 h after the last tolterodine dose on day 5.
Adverse events were assessed by spontaneous reports, observations and questioning at regular intervals. Laboratory parameters (P-AGP, P-creatinine, P-ASAT, P-ALAT, P-cholesterol, B-haemoglobin, B-thrombocytes, B-leukocytes, U-glucose, U-erythrocytes, U-haematology, U-protein and U-ketones) were assessed prior to and at the end of the study and vital signs data were screened for trends.
Analytical methods
Quantification of tolterodine and 5-HM in serum was performed using a specific and sensitive capillary gas chromatography-mass spectrometry assay [15]. The limit of quantification (LOQ) was set to 0.4 nm. The interassay accuracy for both analytes varied between 97 and 101% for serum over the range 0.9–60 nm. The precision was better than 10%.
Serum samples were assayed for N-dealkylated and carboxyl containing tolterodine metabolites using a liquid chromatography-electrospray ionization-mass spectrometry/mass spectrometry (LC-ESI-MS/MS) technique [16]. The accuracy for all analytes varied between 96 and 106% for serum over the range 3–100 nm with a precision better than 15%.
Plasma concentrations of ketoconazole were determined using a high performance liquid chromatography system in combination with u.v. detection [17]. The LOQ was approximately 0.50 μm with an accuracy for all analytes from 100% to 104% and a precision from 5.5% to 7.7%.
Data analysis
All data are expressed as mean±s.d.. Non-compartmental analysis based on serum concentrations of tolterodine and its metabolites was performed using WinNonlin (Scientific Consulting, Inc., Apex, USA). The area under the serum concentration-time curve (AUC) was obtained by linear trapezoidal approximation [18] with extrapolation to infinity by dividing the last calculated data point by the terminal slope (λz) derived from concentration points sampled from 1.8 to 56 h postdose for tolterodine. At steady-state AUC(0,12h) was used: Serum concentration values below the limit of quantification at early time points (lag-time) were treated as zero. Terminal concentration points below the limit of quantification were omitted from the analysis. The apparent oral clearance (CL/F, where CL represents total clearance and F represents bioavailability [i.e. dose/AUC]) and terminal half-life (t1/2,z [i.e. ln 2/terminal slope]) were estimated according to standard equations [19]. The statistical analysis for pharmacokinetic variables was performed using Wilcoxon one sample test on log-transformed data. Differences were considered to be significant at P <0.05.
The apparent unbound fraction (fu) of tolterodine, deemed to be the active moiety in poor metabolisers (as opposed to the sum of unbound tolterodine+5-HM in extensive metabolisers) [6], was calculated for each volunteer using the individual concentration of α1-acid glycoprotein (Pt) and the respective association constant (Ka) of tolterodine to α1-acid glycoprotein using the following relationship [20]:
The association constant of tolterodine to α1-acid glycoprotein, the major binding protein in serum, has previously been determined in man, 2.1×106 m−1 [10]. A good correlation has been reported between observed mean fu and the mean predicted value for tolterodine based on AGP concentrations and Ka value [10]. The α1-acid glycoprotein levels were determined when tolterodine was given separately as well as during ketoconazole interaction.
Results
Eight healthy volunteers entered the study, all of whom completed the single-dose investigations. As planned in the protocol six of these subjects also completed the multiple-dose part of the study. The most frequently reported adverse events were headache (five subjects), dry mouth (three subjects) and nausea (three subjects), events which mainly occurred during coadministration of the drugs. One volunteer showed pathological ECG changes (unspecific T-wave changes) during multiple-dose administration of tolterodine such that the last two doses of ketoconazole were omitted. In a follow-up investigation without drug administration performed by a cardiologist, the subject was found to have hypertension with pathological ECGs in combination with a family history of cardiovascular disease.
The individual plasma concentration-time curves for ketoconazole during single-and multiple-dose administration of tolterodine are presented in Figure 2. There was a high intra (up to six-fold difference) and interindividual (up to six-fold difference) variability in the ketoconazole AUC(0,4h).
Figure 2.
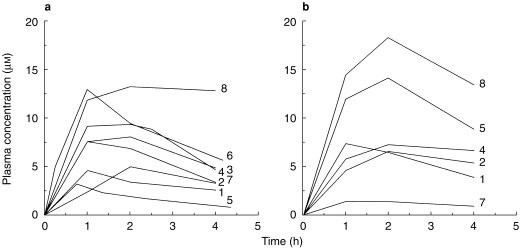
Plasma concentration-time profiles of ketoconazole after single- (a) and multiple-dose (b) administration of tolterodine l-tartrate (2 mg and 1 mg twice daily, respectively) to subjects with deficient CYP2D6 activity. The identification of individual subjects is indicated in the graph.
Tolterodine
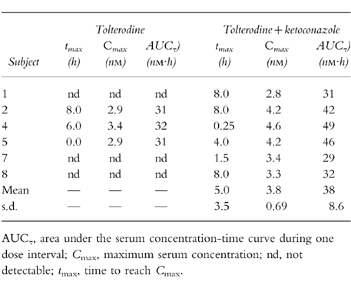
The pharmacokinetic parameters of tolterodine and the active moiety (unbound tolterodine) following both single- and multiple-dose administration in the absence and presence of ketoconazole are given in Tables 1 and 2, respectively.
Table 1.
Pharmacokinetic parameters of tolterodine and active moiety (unbound tolterodine) after 2 mg single administration of tolterodine l-tartrate in the the absence and presence of ketoconazole in subjects with deficient CYP2D6 activity.
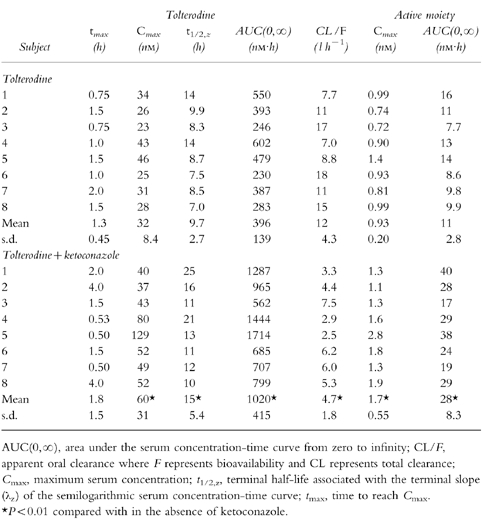
Table 2.
Pharmacokinetic parameters of tolterodine and active moiety (unbound tolterodine) at steady-state during 1 mg twice-daily administration of tolterodine l-tartrate for 2.5 days in the absence and presence of ketoconazole in subjects with deficient CYP2D6 activity.
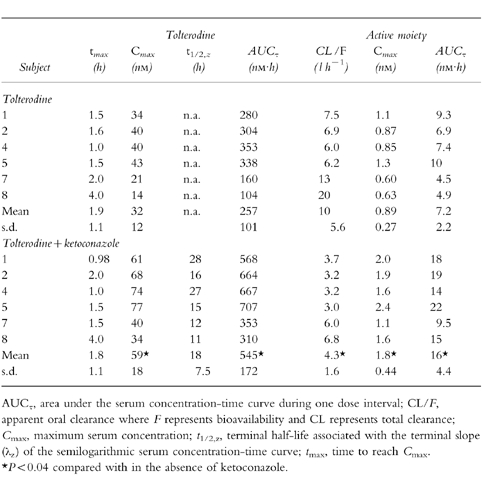
The individual serum concentration-time profiles of tolterodine at steady-state in the absence and presence of ketoconazole are shown in Figure 3. These levels correlated well with the simulations from single-dose data. The Cmax of tolterodine increased 1.8–2-fold (P <0.01) in the presence of ketoconazole without any significant effect on time to Cmax (tmax). A 60–61% decrease (P <0.01) in CL/F of tolterodine was obtained during concomitant administration of ketoconazole, yielding a 2.1–2.6-fold increase in AUC. The individual steady-state CL/F values were similar to the corresponding single-dose values in the absence and presence of ketoconazole. The two subjects with lowest CL/F were subject no. 1, a poor metaboliser of mephenytoin, and subject no. 4, a likely heterozygote with respect to CYP2C19 activity (mephenytoin S/R ratio 0.54). Following single doses of tolterodine, the mean t1/2,z increased by 50% from 9.7±2.7 h to 15±5.4 h during the interaction.
Figure 3.
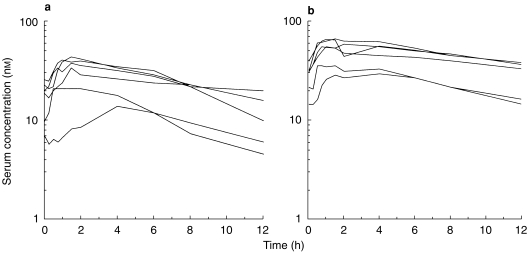
Steady-state serum concentration-time profiles of tolterodine in the absence (a) and presence (b) of ketoconazole, 200 mg once daily, after 1 mg twice-daily administration of tolterodine l-tartrate to subjects with deficient CYP2D6 activity.
Although the steady-state t1/2,z during tolterodine administration could not be determined with high precision because blood samples were only collected for 12 h postdose, but during ketoconazole coadministration the t1/2,z (tolterodine) were similar.
It was found that the values obtained after multiple-dose administration were similar to those obtained after single doses of tolterodine during ketoconazole coadministration. Correction for differences α1-acid glycoprotein levels to obtain levels of the active moiety did not change the previously mentioned magnitude of inhibition by ketoconazole. The subject (no. 8) with the highest CL/F also showed the lowest levels of α1-acid glycoprotein.
N-dealkylated tolterodine and other metabolites
Serum levels of the active metabolite (5-HM) and other metabolites of tolterodine were not quantifiable in any of the volunteers after single-dose administration. The individual steady-state serum concentration-time profiles of N-dealkylated tolterodine in the absence and presence with ketoconazole are shown in Figure 4, and the individual pharmacokinetic parameters are given in Table 3. Only three volunteers had detectable levels of N-dealkylated tolterodine at steady-state. During the interaction with ketoconazole, however, all six volunteers had measurable levels of the metabolite. AUC values in the three volunteers with available data in the absence and presence of ketoconazole increased by 35% to 53% during the interaction. The shape of the serum concentration-time profiles were extremely flat, and Cmax occurred during the 0–24 h sampling interval. The levels at 12 h postdose did not deviate from those before the last dose.
Figure 4.
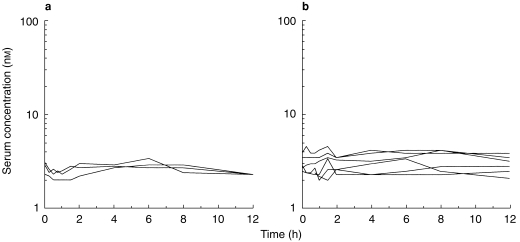
Steady-state serum concentration-time profiles of N-dealkylated tolterodine in the absence (a) and presence (b) of ketoconazole, 200 mg once daily, after 1 mg twice-daily administration of tolterodine l-tartrate to subjects with deficient CYP2D6 activity.
Table 3.
Pharmacokinetic parameters of N-dealkylated tolterodine after 1 mg twice daily administration of tolterodine l-tartrate for 2.5 days in the absence and during concomitant administration of ketoconazole in subjects with deficient CYP2D6 activity.
Levels of N-dealkylated 5-HM, tolterodine acid and N-dealkylated tolterodine acid were below the limit of quantification at steady-state, irrespective of ketoconazole coadministration. None of the metabolites was quantifiable after single-dose administration.
Discussion
The present study shows that CYP3A4 catalyses the predominant route of elimination of tolterodine in poor metabolisers, i.e. N-dealkylation, since approximately 60% of CL/F was inhibited during ketoconazole coadministration. Despite the high variability (both intra- and interindividual) in levels of ketoconazole, the systemic inhibition of tolterodine disposition, as seen in change in CL/F, was of the same magnitude following single-and multiple-dose administration of tolterodine. This suggests that the CYP3A4 activity among the poor metabolisers was markedly inhibited, and signifying no time dependent feature of the interaction.
The two-fold increase in Cmax of tolterodine in the presence of ketoconazole was an unexpected finding, since tolterodine has a high absolute bioavailability of around 80% among poor metabolisers [6] and little is eliminated before reaching tmax. With tmax not being materially affected by ketoconazole, one explanation is that ketoconazole substantially decreases the volume of distribution of tolterodine, probably by decreasing the tissue binding. Indeed, decrease in the volume of distribution of other drugs by ketoconazole has been described for antipyrine [21], chlordiazepoxide [11] and methylprednisolone [22]. Another possible explanation to this increase in Cmax could be an inhibition of the plasma membrane transporter P-glycoprotein, of which ketoconazole is an inhibitor, in the intestine [23]. However, a difference tmax of tolterodine was not seen which suggests that the absorption was not altered. Furthermore, the high bioavailability of tolterodine among poor metabolisers implies that the major part the ketoconazole inhibition is systemic and consequently only systemic CYP3A4 inhibitors (e.g. other azole antifungals and macolide antibiotics) [24] are likely to significantly affect the pharmacokinetics of tolterodine. Therefore, the effect of CYP3A4 inhibitors that only affect the gastrointestinal tract, such as grapefruit juice components [25], is likely to be marginal.
Little information regarding the pharmacokinetics of other tolterodine metabolites among poor metabolisers has been reported [6]. The present study shows that poor metabolisers N-dealkylate tolterodine to a major extent. The extremely flat serum concentration-time curves for this metabolite suggest an almost immediate elimination, i.e. a possible direct conjugation after dealkylation in combination with biliary and renal excretion or further metabolism to other metabolites. At steady-state, N-dealkylated tolterodine was only quantifiable in those volunteers with the lowest CL/F values (<7.0 l h−1), but during the interaction with ketoconazole the levels of this metabolite increased and were quantifiable in all volunteers (CL/F was then <7.0 l h−1 in all volunteers). These observations imply that ketoconazole not only decreases the formation clearance of this metabolite but also that it may possibly decrease the clearance of N-dealkylated tolterodine, suggesting that this metabolite is further metabolized, at least in part, by CYP3A4.
Additional metabolism of tolterodine, possibly by other CYP enzymes, was also indicated in the present study by the slightly longer t1/2,z in two subjects irrespective of CYP3A4 inhibition. In vitro studies with microsomes from cells specifically expressing CYP2C9, 2C19 and 3A4 suggested that N-dealkylation of tolterodine primarily correlated to CYP3A4 activity, taking into account the high in vivo expression of CYP3A4 relative to CYP2C9 and 2C19 [7]. When comparing the intrinsic clearance for the two CYP2C isoenzymes, CYP2C19-mediated N-dealkylation was seven times larger than that for CYP2C9. Interestingly, one of the two volunteers with the longest t1/2,z values of tolterodine, both in the absence and presence of ketoconazole, was a poor metaboliser of mephenytion (volunteer no. 1), a probe drug of CYP2C19 activity. The other volunteer (no. 4) had relatively low CYP2C19 activity (S/R ratio of mephenytoin was 0.54), which might suggest an in vivo relation to CYP2C19 activity. However, the relative contribution of CYP2C enzymes to the metabolism of tolterodine in CYP2D6 deficient metabolisers is minor since CYP3A4 accounted for 60% of CL/F in such subjects.
Although N-dealkylated tolterodine is the major metabolite in poor metabolisers, the contribution to the pharmacological effect of tolterodine is negligible (approximately 3% relative to that of the active moiety, unbound tolterodine). This can be concluded from the 20 times less potency of the metabolite (the racemate) with regard to inhibition of carbachol-induced contraction of guinea-pig isolated urinary bladder (personal communication, L. Nilvebrant, July 1998) and considering the 4-fold higher unbound fraction of the metabolite, i.e. fu = 14% [10].
During potent CYP2D6 inhibition by fluoxetine, CL/F of tolterodine decreased by 91% in extensive metabolisers [26]. In the present study, the decrease in CL/F of tolterodine was 60% during CYP3A4 inhibition in subjects with no functional CYP2D6 genes. Tolterodine is a drug with a high extraction ratio in extensive metabolisers and a low extraction ratio drug in poor metabolisers [6]. Thus the clinical importance of the two interactions differs. In extensive metabolisers the interaction with fluoxetine resulted in similar exposure (active moiety) as prior to the interaction, because the increased tolterodine concentrations were compensated for by decreased 5-HM levels [26]. A comparison of those results with the present study, where AUC increased 2.1-fold to 2.6-fold during ketoconazole coadministration, demonstrates that potent CYP3 4 inhibition during tolterodine treatment is enough to warrant a clinical consideration. However, the safety concerns for a 2-fold increase in tolterodine exposure may be of lesser significance [27] compared with ketoconazole interactions with drugs that are highly extracted by CYP3A4, e.g. terfenadine or triazolam [13, 14].
In conclusion, CYP3A4 catalyses the predominant metabolic pathway of tolterodine in CYP2D6 deficient metabolisers, i.e. N-dealkylation, since 60% of CL/F was inhibited by ketoconazole. This inhibition resulted in 2.1-fold increase in AUC.
Acknowledgments
This study was supported by Pharmacia & Upjohn AB, Sweden. We would like to thank P. Blomström. M.D. for his generous help with the follow-up ECG investigation and E. Götharson, RN, K. Andersson, RN, and M-L. Odell, BSc, for their expert assistance. G. Tybring, PhD, is cordially thanked for her analytical expertise in conducting the ketoconazole assay.
References
- 1.Rentzhog L, Stanton SL, Cardozo L, Fall M, Nelson E, Abrams P. Efficacy and safety of tolterodine in patients with detrusor instability: a dose ranging study. Br J Urol. 1998;81:42–48. doi: 10.1046/j.1464-410x.1998.00501.x. [DOI] [PubMed] [Google Scholar]
- 2.Jonas U, Höfner K, Madersbacher H, Holmdahl TH. and the participants of the international study group. Efficacy and safety of two doses of tolterodine versus placebo in patients with detrusor overactivity and symptoms of frequency, urge incontinence, and urgency: urodynamic evaluation. World J Urol. 1997;15:144–151. doi: 10.1007/BF02201987. [DOI] [PubMed] [Google Scholar]
- 3.Van Kerrebroeck PEVA, Amarenco G, Thüroff JW, et al. Dose-ranging study of tolterodine in patients with detrusor hyperreflexia. Neurourol Urodyn. 1998;17:499–512. doi: 10.1002/(sici)1520-6777(1998)17:5<499::aid-nau6>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 4.Nilvebrant L, Andersson K-E, Gillberg P-G, Stahl M, Sparf B. Tolterodine—a new bladder-selective antimuscarinic agent. Eur J Pharmacol. 1997;327:195–207. doi: 10.1016/s0014-2999(97)89661-6. [DOI] [PubMed] [Google Scholar]
- 5.Brynne N, Stahl MMS, Hallén B, et al. Pharmacokinetics and pharmacodynamics of tolterodine in man: a new drug for the treatment of urinary bladder overactivity. Int J Clin Pharmacol Ther. 1997;35:287–295. [PubMed] [Google Scholar]
- 6.Brynne N, Dalén P, Alván G, Bertilsson L, Gabrielsson J. Influence of CYP2D6 polymorphism on the pharmacokinetics and pharmacodynamics of tolterodine. Clin Pharmacol Ther. 1998;63:529–539. doi: 10.1016/S0009-9236(98)90104-7. [DOI] [PubMed] [Google Scholar]
- 7.Postlind H, Danielsson Å, Lindgren A, Andersson SHG. Tolterodine, a new muscarinic receptor antagonist, is metabolized by cytochromes P450 2D6 and 3A in human liver microsomes. Drug Metab Dispos. 1998;26:289–293. [PubMed] [Google Scholar]
- 8.Nilvebrant L, Gillberg P-G, Sparf B. Antimuscarinic potency and bladder selectivity of PNU-200577, a major metabolite of tolterodine. Pharmacol Toxicol. 1997;81:169–172. doi: 10.1111/j.1600-0773.1997.tb02064.x. [DOI] [PubMed] [Google Scholar]
- 9.Bertilsson L, Dahl M-L. Polymorphic drug oxidation—Relevance to the treatment of psychiatric disorders. CNS Drugs. 1996;5:200–223. [Google Scholar]
- 10.Påhlman I, Gozzi P. Protein binding of tolterodine and its major metaboltes in human and several animal species. Biopharm Drug Dispos. 1999;20:91–99. doi: 10.1002/(sici)1099-081x(199903)20:2<91::aid-bdd162>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 11.Brown MW, Maldonado AL, Meredith CG, Speeg KV. Effect of ketoconazole on hepatic oxidative drug metabolism. Clin Pharmacol Ther. 1985;3:290–297. doi: 10.1038/clpt.1985.42. [DOI] [PubMed] [Google Scholar]
- 12.Maurice M, Pichard L, Daujat M, et al. Effects of imidazole derivatives on cytochromes P450 from human hepatocytes in primary culture. FASEB J. 1992;6:752–758. doi: 10.1096/fasebj.6.2.1371482. [DOI] [PubMed] [Google Scholar]
- 13.Vahre A, Olkkola KT, Neuvonen PJ. Oral triazolam is potentially hazardous to patients receiving systemic antimycotics ketoconazole or itraconazole. Clin Pharmacol Ther. 1994;56:601–607. doi: 10.1038/clpt.1994.184. [DOI] [PubMed] [Google Scholar]
- 14.Pohjola-Sintonen S, Viitasalo M, Toivonen L, Neuvonen PJ. Itraconazole prevents terfenadine metabolism and increases risk of torsades de pointes ventricular tachycardia. Eur J Clin Pharmacol. 1993;45:191–193. doi: 10.1007/BF00315505. [DOI] [PubMed] [Google Scholar]
- 15.Palmér L, Andersson L, Andersson T, Stenberg U. Determination of tolterodine and the 5-hydroxymethyl metabolite in plasma, serum and urine using gas chromatography-mass spectrometry. J Pharm Biomed Anal. 1997;16:155–165. doi: 10.1016/s0731-7085(97)00023-x. [DOI] [PubMed] [Google Scholar]
- 16.Andersson T, Svanström C, Palmér L. 8th International Symposium on Pharmaceutical and Biomedical Analysis. Orlando: 1997. Quantification of tolterodine metabolites in serum using electrospray ionisation tandem mass spectrometry (LC-EC-MS/MS) (Abstract) May. [Google Scholar]
- 17.Riley CM, James MO. Determination of ketoconazole in the plasma, liver, lung and adrenal of the rat by high-performance liquid chromatography. J Chromatogr. 1986;377:287–294. doi: 10.1016/s0378-4347(00)80784-7. [DOI] [PubMed] [Google Scholar]
- 18.Gibaldi M, Perrier D. Pharmacokinetics. 2. New York: Marcel Dekker; 1982. [Google Scholar]
- 19.Rowland M, Tozer TN. Clinical Pharmacokinetics: Concepts and Applications. 3. Philadelphia: Williams & Wilkins; 1995. [Google Scholar]
- 20.Tozer T. Implications of altered plasma protein binding in disease states. In: Benet LZ, Massoud N, Gambertoglio JG, editors. Pharmacokinetic basis for drug treatment. New York: Raven Press; 1984. [Google Scholar]
- 21.Blyden GT, Abernethy DR, Greenblatt DJ. Ketoconazole does not impair the antipyrine clearance in humans. Int J Clin Pharmacol Ther Toxicol. 1986;24:225–226. [PubMed] [Google Scholar]
- 22.Glynn AM, Slaughter RL, Brass C, Dámbrosio R, Jusko WJ. Effects of ketoconazole in methylprednisolone pharmacokinetics and cortisol secretion. Clin Pharmacol Ther. 1986;39:654–659. doi: 10.1038/clpt.1986.114. [DOI] [PubMed] [Google Scholar]
- 23.Salphati L, Benet LZ. Effects of ketoconazole on digoxin absorption and disposition in rat. Pharmacology. 1998;56:308–313. doi: 10.1159/000028214. [DOI] [PubMed] [Google Scholar]
- 24.Wilkinson GR. Cytochrome P4503A (CYP3A) metabolism: prediction of in vivo activity in humans. J Pharmacokinet Biopharm. 1996;24:475–490. doi: 10.1007/BF02353475. [DOI] [PubMed] [Google Scholar]
- 25.Ameer B, Weintraub R. Drug interactions with grapefruit juice. Clin Pharmacokinet. 1997;33:103–121. doi: 10.2165/00003088-199733020-00003. [DOI] [PubMed] [Google Scholar]
- 26.Brynne N, Svanström C, Åberg-Wistedt A, Hallén B, Bertilsson L. Fluoxetine inhibits the metabolism of tolterodine-pharmacokinetic implications and proposed clinical relevance. Br J Clin Pharmacol. 1999;48:553–563. doi: 10.1046/j.1365-2125.1999.00051.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Larsson G, Hallén B, Nilvebrant L. Tolterodine in the treatment of overactive bladder: analysis of the pooled phase II efficacy and safety data. Urol. 1999;53:990–998. doi: 10.1016/s0090-4295(98)00629-3. [DOI] [PubMed] [Google Scholar]