

Abstract
Aims
To investigate the change in disposition of tolterodine during coadministration of the potent cytochrome P450 2D6 (CYP2D6) inhibitor fluoxetine.
Methods
Thirteen patients received tolterodine l-tartrate 2 mg twice daily for 2.5 days, followed by fluoxetine 20 mg once daily for 3 weeks and then concomitant administration for an additional 2.5 days. They were characterized as extensive metabolizers (EM1 with one functional CYP2D6 gene, EM2 with two functional genes) or poor metabolizers (PM).
Results
Nine patients, three EM2 and four EM1 and two PM, completed the trial. Following tolterodine administration, the area under the serum concentration–time curve (AUC) of tolterodine was 4.4-times and 30-times higher among EM1 and PM, respectively, compared with EM2. The AUC of the 5-hydroxymethyl metabolite (5-HM) was not quantifiable in PM. Fluoxetine significantly decreased (P < 0.002) the oral clearance of tolterodine by 93% in EM2 and by 80% in EM1. The AUC of 5-HM increased in EM2 and decreased in EM1. However, the exposure to the active moiety (unbound tolterodine +5-HM) was not significantly increased in the two phenotypes. The subdivision of the EM group showed a 2.1-fold increase in active moiety in EM2 but the exposure was still similar to EM1 compared with before the interaction.
Conclusions
The study suggests a difference in the pharmacokinetics of tolterodine and its 5-hydroxymethyl metabolite depending on the number of functional CYP2D6 genes. Fluoxetine significantly inhibited the hydroxylation of tolterodine. Despite the effect on the pharmacokinetics of tolterodine in extensive metabolizers, the clinical effect is expected to be within normal variation.
Keywords: antimuscarinic, CYP2D6, CYP3A4, drug interaction, drug metabolism, PNU-200577, tolterodine, urinary incontinence
Introduction
Tolterodine (R)-N,N-diisopropyl-3-(2-hydroxy-5-methylphenyl)-3-phenylpropanamine, is a new antimuscarinic drug for the treatment of patients with overactive bladder presenting with urinary frequency, urgency and urge incontinence [1–3]. In vitro, tolterodine has high affinity and specificity for muscarinic receptors and shows a selectivity for the urinary bladder over salivary glands in vivo [4]. In humans, tolterodine is rapidly absorbed, exhibits high first-pass extraction and the systemically available drug is mainly eliminated by metabolism [5]. Two different oxidative metabolic pathways, hydroxylation and N-dealkylation, have been identified in man (Figure 1) [5]. Hydroxylation to the pharmacologically active 5-hydroxymethyl metabolite (5-HM) (PNU-200577) is catalysed by cytochrome P450 (CYP) 2D6 [6, 7], while the N-dealkylation pathway is catalysed by CYP3A [7]. Preclinical studies have shown that 5-HM is equipotent compared with tolterodine in vitro, and has similar functional bladder selectivity in vivo [8]. Further oxidation of 5-HM yields the carboxylic acid of tolterodine and its N-dealkylated form, along with N-dealkylated 5-HM. Following oral administration of tolterodine, the majority of the administered dose is excreted renally as the two carboxylic acids [5]. The isoenzymes catalysing these reactions are given in Figure 1 [7]. With the exception of 5-HM, the pharmacokinetics of other metabolites of tolterodine have yet to be characterized.
Figure 1.
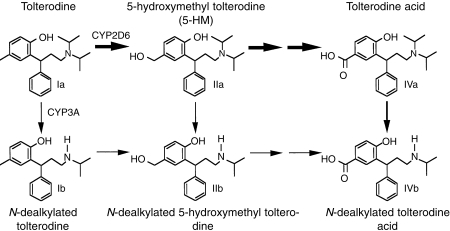
Metabolic pathways of tolterodine (R)-N,N-diisopropyl-3-(2-hydroxy-5-methylphenyl)-3-phenylpropanamine in man.
It is well established that CYP2D6 is subject to genetic polymorphism [9], with important implications for drugs that are metabolized by this enzyme. In extensive metabolizers (EM) of debrisoquine, a probe drug for CYP2D6 activity, the mean systemic clearance of tolterodine was found to be 44 l h−1 yielding a half-life of 2–3 h [6]. In contrast, PM have 5-times lower clearance and a mean half-life of 9 h, which results in a 7-fold higher maximum serum concentration of tolterodine at steady-state. The levels of 5-HM are similar to those of tolterodine in EM and not quantifiable in PM [6]. However, while large differences in pharmacokinetics exist between EM and PM, this is of minor importance for the antimuscarinic effect because of additive action of parent drug and active metabolite, along with a 10-fold difference between tolterodine and 5-HM in terms of protein binding [6, 10].
The selective serotonin re-uptake inhibitors (SSRIs) comprise a relatively novel class of compounds with antidepressant properties. Fluoxetine is world-wide the most used of these agents and is prescribed for a variety of psychopathological conditions including mood and eating disorders, obsessive-compulsive disorders, depression and dysthymia [11–14]. Among the SSRIs, fluoxetine, its active metabolite (norfluoxetine) and paroxetine are the most potent, albeit not selective, inhibitors of CYP2D6 activity in vitro [15, 16]. Pharmacokinetic interactions with the SSRIs have been described to lead frequently, but not generally, to adverse effects [17]. Since the prevalence of urinary incontinence increases with age and is highest among women [18], populations frequently treated with antidepressants, the potential exists for drug–drug interaction between tolterodine and fluoxetine.
The objectives of the present study were to determine the pharmacokinetics of tolterodine in psychiatric patients with subjective symptoms of urinary incontinence after coadministration of fluoxetine, and to quantify N-dealkylated and carboxylic metabolites of tolterodine in serum and urine.
This study was presented in part at the Second European Association of Clinical Pharmacology and Therapeutics Meeting, Berlin, Germany, September 21–25, 1997.
Methods
Subjects
Thirteen female patients with depression and/or anxiety syndrome and subjective symptoms of urinary incontinence were included in the study. All patients were medically investigated and healthy according to laboratory tests, ECG, and blood pressure. The mean (±s.d.) demographic characteristics were as follows: age, 63±9.4 years; body weight, 70±9.6 kg; and height, 1.65±0.05 m. Each patient was genotyped with respect to CYP2D6 (*1, *3 and *4 alleles) during the study. The study was approved by the ethics committee of the Karolinska Hospital, and each patient gave written informed consent before the study.
Study design
The study had an open and non-randomised crossover design. Patients were treated with tolterodine l-tartrate (Detrusitol® [Detrol®], Pharmacia & Upjohn) 2 mg twice daily (12 h apart) for 2.5 days. Thereafter, patients commenced treatment with fluoxetine (Fontex® [Prozac®], Eli Lilly) 20 mg once daily orally for 3 weeks, following which fluoxetine and tolterodine were given concomitantly for an additional 2.5 days. Each patient fasted overnight before the tolterodine and tolterodine+fluoxetine steady-state assessment days (Days 3 and 27, respectively). Intake of alcohol-containing beverages around the study period was not allowed. Administration of all drugs known to be potent inhibitors of CYP2D6 (e.g. antidepressants and neuroleptic agents) was strictly forbidden during the study period.
Assessment
On the two tolterodine steady-state days (Days 3 and 27) venous blood samples were drawn before administration of tolterodine and at 10, 20, 30 and 45 min and at 1, 1.5, 2, 4, 6, 8, 10 and 12 h post dose. An additional blood sample was drawn for the determination of α1-acid glycoprotein (AGP) levels, since this is the major binding protein of tolterodine and 5-HM in serum [10]. Venous blood samples were also drawn before administration and at 1 and 2 h after the fluoxetine dose on Day 24. On Days 3 and 27 each patient emptied the bladder before the administration of tolterodine. Urine was subsequently collected quantitatively during the 12 h dose interval.
Adverse events were assessed by spontaneous reports, observations and questioning at regular intervals. Laboratory parameters and vital signs were assessed prior to and at the end of the study.
Analytical methods
Quantification of tolterodine and 5-HM in serum and urine was performed using a specific and sensitive capillary gas chromatography-mass spectrometry assay [19]. The limit of quantification was set to 0.4 and 1.5 nm for serum and urine, respectively. The accuracy and precision of the method were continuously examined during the study by analysing spiked quality control samples. The interassay accuracy for both analytes varied between 97 and 101% for serum over the range 0.9–60 nm and for urine between 91 and 107% over the range 1.5–135 nm. Precision was better than 10% and 11% for serum and urine, respectively.
Serum and urine samples were assayed for dealkyl and carboxyl tolterodine using a liquid chromatography-electrospray ionization-mass spectrometry/mass spectrometry (LC-ESI-MS/MS) technique. The metabolites were extracted from the matrix by solid phase extraction. Deuterium-labelled 5-HM was used as internal standard. Urinary concentrations of the metabolites were also assessed following incubation of samples with β-glucuronidase (Sigma Chemical Company, St Louis, USA). The metabolites were synthesized by the Department of Medicinal Chemistry, Pharmacia & Upjohn AB, Uppsala, Sweden. The analytes in serum were extracted from a volume of 0.05–1.0 ml, using C18 columns (Isolute IST, Hengoed, UK). After evaporation of the eluent, the residue was reconstituted in 0.5 ml 0.1% formic acid in distilled water. Aliquots of 5 μl were injected into the high performance liquid chromatography system. Separation was achieved on a 5 μm 2.1×50 mm Zorbax SB-CN column (Rockland Technologies Inc., Newport, USA) and a CN opti-guard guard column (Optimize Technologies Inc., Oregon City, USA) at a temperature of 30° C, using a mobile phase consisting of a mixture of 10 mm ammonium acetate, methanol and acetonitrile (6:3:1 v/v) and 0.1% (v/v) formic acid. The flow rate was 0.2 ml min−1. Detection was performed with a Micromass Quattro II mass spectrometer using multiple reaction monitoring. Aliquots of 0.05–0.2 ml of urine samples were extracted on C2 columns (Isolute IST, Hengoed, UK) and the analytes eluted from the column with the mobile phase. Separation and detection were performed as for serum analysis. Calibration curves prepared in serum and urine were linear over the range 1.6–125 nm and 16–950 nm, respectively. The limit of quantification was set to 1.6 nm for serum (1 ml sample volume) and 16 nm for urine (0.1 ml sample volume). The accuracy and precision of the method were continuously examined during the study by analysing spiked quality control samples. The accuracy for all analytes varied between 96 and 106% for serum over the range 3–100 nm and for urine between 94 and 108% over the range 110–670 nm. Precision was better than 15% and 6% for serum and urine, respectively.
Plasma concentrations of fluoxetine and norfluoxetine were determined using a high performance liquid chromatography system and u.v.-detection [20]. The limit of quantification was approximately 20 nm. The interassay accuracy for the analytes varied from 95% to 110% over the 50–1600 nm range. The precision was better than 8.0%.
Genotyping was performed by means of polymerase chain reaction followed by cleavage with restriction enzymes and polyacrylamide electrophoresis [21]. The wild-type CYP2D6*1 allele and the mutated CYP2D6*3 and CYP2D6*4 alleles were determined to characterize the patients as homozygous or heterozygous EM (EM2 and EM1, respectively) or homozygous PM.
Data analysis
Non-compartmental analysis based on serum concentrations of tolterodine and its metabolites was performed using PCNonlin (version 4.2) [22]. The area under the serum concentration–time curve (AUC) was obtained by linear trapezoidal approximation [23] with extrapolation to 12 h by dividing the last calculated data point by the terminal slope (λz) derived from concentrations obtained at 1.8–12 h for tolterodine and 5-HM, and at 3–12 h for the remaining metabolites. Concentrations below the limit of quantification at early time points (lag-time) were treated as zero. Terminal concentrations and concentrations in the middle of the serum concentration–time profile below the limit of quantification were omitted from the analysis. Oral clearance (CL/F, where F indicates bioavailability [i.e. dose/AUC]) and terminal half-life (t1/2,z [i.e. ln 2/terminal slope]) were estimated according to standard formulae [23, 24].
The effects of fluoxetine on tolterodine pharmacokinetics were analysed by sign test using all three genotypes. The separation between the three genotypes, prior to the interaction, was analysed by Exact Kruskal Wallis permutation tests. The separation between EM2 and EM1 was tested by Exact Wilcoxon. Differences were considered to be significant at P < 0.05.
The unbound fractions (fu) of tolterodine and 5-HM were calculated for each patient using the individual concentration of AGP (Pt) and the respective affinity constants (Ka) of tolterodine and 5-HM to AGP using the following relationship [25].
| 1 |
The affinities of tolterodine and 5-HM to AGP, the major binding protein in serum, are 2.1×106 m−1 and 13×106 m−1, respectively [10]. A good correlation has been reported between observed mean fu and the mean predicted value for both tolterodine and 5-HM based on AGP concentrations and Ka values [10]. The active moiety after tolterodine administration, defined as the sum of unbound tolterodine+5-HM, were calculated on Days 3 and 27 using the AGP levels for these particular days.
Results
Thirteen patients entered the study; three were withdrawn due to adverse events (headache) during the fluoxetine treatment period and one patient completed the study at a reduced fluoxetine dosage because of headache. These four patients were omitted from the analysis. Of the nine evaluable patients, three were homozygous EM with two functional genes (EM2), four were heterozygous EM with one functional gene (EM1) and the remaining two patients were PM with no functional gene of CYP2D6.
The individual plasma concentrations of fluoxetine and norfluoxetine for the three time points (0, 1 and 2 h postdose), taken in absence and in presence of tolterodine, are presented in Figure 2. The two PM had higher plasma concentrations of fluoxetine, and lower plasma levels of norfluoxetine, compared with EM. One EM1 patient (number 3) had similar plasma levels of norfluoxetine to those of PM.
Figure 2.
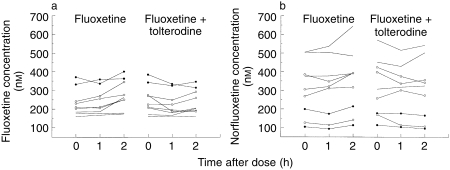
Plasma concentration–time profiles of fluoxetine (a) and norfluoxetine (b) taken before, 1 and 2 h after administration of fluoxetine and during coadministration of tolterodine in extensive (EM2[———]; EM1 [○]) and PM (•).
Tolterodine
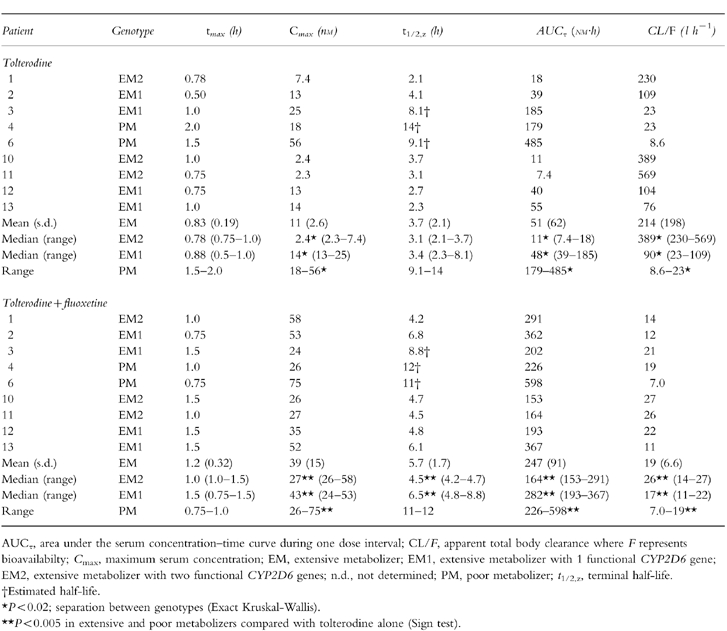
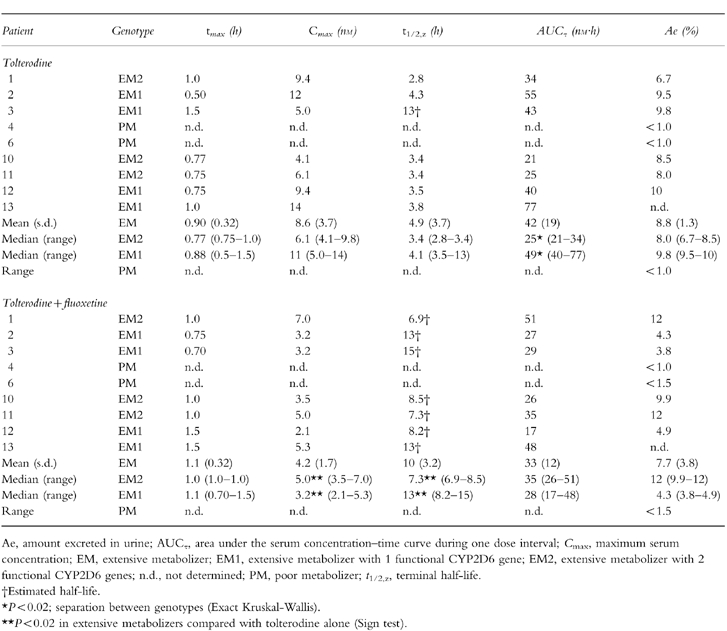
The individual serum concentration–time profiles of tolterodine, 5-HM and the other metabolites during tolterodine treatment are shown in Figure 3. There was a distinct difference in serum tolterodine concentrations between EM and PM although one EM1 patient (number 3) showed levels of tolterodine similar to PM. The pharmacokinetic parameters of tolterodine in the absence and presence of fluoxetine are given in Table 1. A significant (P < 0.02) separation between the three phenotypes was apparent for both AUC and CL/F, but not for t1/2,z. The Exact Wilcoxon test between EM2 and EM1 was almost significant (P < 0.06) although the number of subjects was only 7. The median AUC of tolterodine was 4.4 times larger in EM1 and 30 times larger in PM, respectively, compared with the value in EM2.
Figure 3.
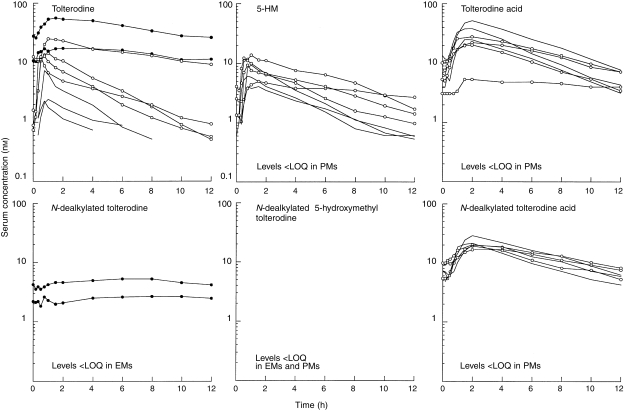
Serum concentration–time profiles of tolterodine and its metabolites after treatment with tolterodine (2 mg twice daily) for 2.5 days in EM2 [———]; EM1 [○]) and PM (•). 5-HM, 5-hydroxymethyl tolterodine; LOQ, limit of quantification; PM, poor metabolizers; EM, extensive metabolizers.
Table 1.
Pharmacokinetic parameters of tolterodine following treatment with tolterodine [as l-tartrate salt] 2 mg twice daily for 2 5 days in the absence and presence of fluoxetine (20 mg once daily for 27 days).
The individual serum concentration–time profiles of tolterodine, 5-HM and the other metabolites during coadministration of tolterodine and fluoxetine are shown in Figure 4. Fluoxetine significantly inhibited (P < 0.005) the metabolism of tolterodine in EM and the difference between the three genotypes in terms of serum tolterodine levels almost disappeared. CL/F values decreased by 93% in EM2, 81% in EM1 and by 18% in PM during fluoxetine coadministration, resulting in a 1.5-fold increase in t1/2,z in EM2 and a 1.9-fold increase in EM1 while no apparent effect on t1/2,z was seen in PM. The median AUC of tolterodine was 1.7 times larger in EM1 and 2.5 times larger in PM, respectively, compared with the value in EM2. The mean excretion of unchanged drug in urine (β-glucuronidase-treated samples) was <5% in all evaluable patients, irrespective of concomitant treatment with fluoxetine.
Figure 4.
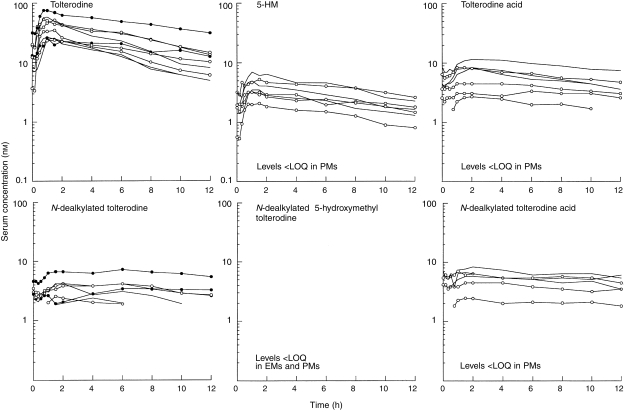
Serum concentration–time data of tolterodine and its metabolites after treatment with tolterodine (2 mg twice daily) for 2.5 days in EM2 [———]; EM1 [○]) and PM (•) during coadministration of fluoxetine (20 mg once daily for 27 days). 5-HM, 5-hydroxymethyl tolterodine; LOQ, limit of quantification; PM, poor metabolizers; EM, extensive metabolizers.
5-hydroxymethyl tolterodine
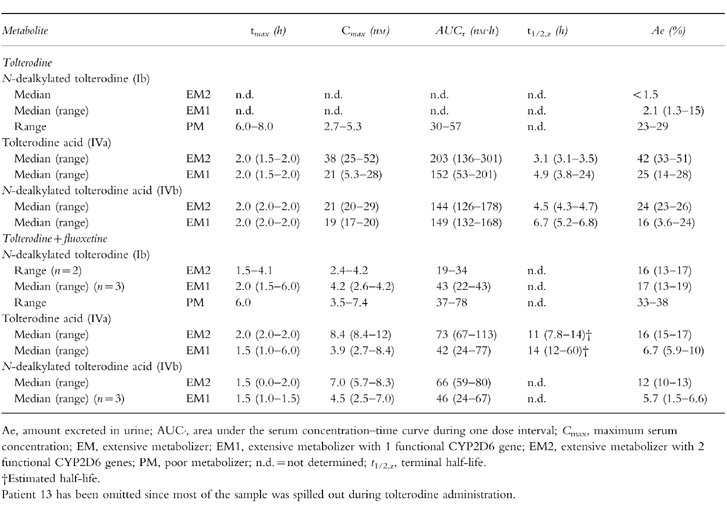
During tolterodine treatment the median AUC of 5-HM was 2.0 times larger in EM1 compared with the value in EM2 (P < 0.02), while the levels of 5-HM were below the limit of quantification in PM. The pharmacokinetic parameters of 5-HM in the absence and presence of fluoxetine are given in Table 2. The t1/2,z was longer in the EM1 group compared with the EM2 group.
Table 2.
Pharmacokinetic parameters of the 5-hydroxymethyl metabolite of tolterodine following treatment with tolterodine [as l-tartrate salt] 2 mg twice daily for 2.5 days in the absence and presence of fluoxetine (20 mg once daily for 27 days).
Fluoxetine did not significantly change the AUC of 5-HM for all EM combined, since an increase was seen in EM2 while a decrease was seen in EM1. Serum 5-HM levels in the two PM were still below the limit of quantification. A significant (P < 0.005) decrease in peak 5-HM serum concentration (Cmax) was observed in both EM2 and EM1 patients (decreases of 20% and 60%, respectively) along with 1.3-fold and 1.9-fold increases, respectively, in t1/2,z. The median AUC of 5-HM was 20% smaller in EM1 compared with the value in EM2. The median amount excreted as 5-HM in urine increased from 8.0% to 12% in EM2 during fluoxetine coadministration, while a decrease from 9.8% to 4.3% was observed in EM1. In PM, the excretion of 5-HM was <1.5% irrespective of concomitant treatment with fluoxetine.
Other metabolites
The pharmacokinetic parameters of N-dealkylated tolterodine, tolterodine acid and N-dealkylated tolterodine acid, in the absence and presence of fluoxetine, are given in Table 3. The flat serum concentrations time curves of N-dealkylated tolterodine were only seen in PM (Figure 3). During coadministration with fluoxetine, similar levels of N-dealkylated tolterodine to those of PM were seen in most EM (Figure 4). N-dealkylated tolterodine was the major metabolite in urine (β-glucuronidase-treated samples) in PM and EM during fluoxetine treatment. The serum concentration of N-dealkylated 5-hydroxymethyl tolterodine was below the limit of quantification in all patients. Tolterodine acid and N-dealkylated tolterodine acid were only found in serum of EM. The median AUC of tolterodine acid was 18-fold larger than tolterodine in EM2 and 3.1-fold higher in EM1. The corresponding relationship between N-dealkylated tolterodine acid was similar to that of tolterodine acid. The two metabolites decreased by 50–70% during fluoxetine treatment. The amount of tolterodine metabolites and parent tolterodine excreted in urine was lower in EM1 and PM, respectively, compared with EM2. These amounts decreased in EM2 and EM1 during the interaction with fluoxetine.
Table 3.
Pharmacokinetic parameters of tolterodine metabolites following treatment with tolterodine [as l-tartrate salt] 2 mg twice daily for 2.5 days in the absence and presence of fluoxetine (20 mg once daily for 27 days).
Active moiety
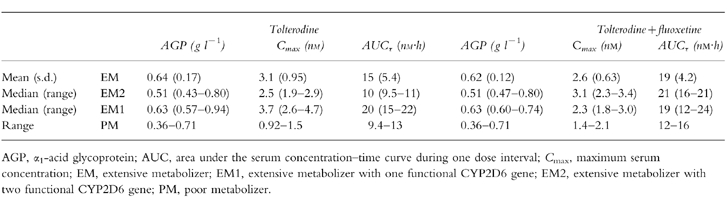
The AGP levels, along with Cmax and AUC values for the active moiety (unbound tolterodine +5-HM) in the three genotypes are given in Table 4. There was no significant change in AGP level during treatment with fluoxetine. One PM (number 4) had a very low concentration of AGP (close to pathological) that resulted in a higher clearance and lower levels of tolterodine. The AUC of the active moiety was similar in EM2 and PM, while the values were 2.0-fold higher in EM1 compared with EM2. Concomitant treatment with fluoxetine increased the AUC value 2.1-fold in EM2 without any apparent difference in EM1. Fluoxetine increased the unbound AUC of tolterodine in PM by 25%.
Table 4.
Pharmacokinetic parameters of the active moiety (unbound tolterodine+the 5-hydroxymethyl metabolite) following treatment with tolterodine [as l-tartrate salt] 2 mg twice daily for 2.5 days in the absence and presence of fluoxetine (20 mg once daily for 27 days).
Discussion
This study confirms that tolterodine shows a high CYP2D6 specificity and that approximately 85% of the systemic clearance of tolterodine in EM is due to the biotransformation of tolterodine to 5-HM [6]. Despite the limited number of patients the present study, by analysing CYP2D6*3 and *4, expands the isozyme specificity, and suggests a separation between EM with one or two functional CYP2D6 genes in terms of the serum levels of both tolterodine and 5-HM. Thus, contribution of the CYP2D6*5 allele which was not analysed can not be excluded. However, if the high frequency of mutated alleles in the 9 subjects (8 out 18) are considered it seems unlikely that additional mutated alleles should be present. The 4-fold difference in CL/F, without overlap between EM2 and EM1, was almost expected considering the 38-fold difference between a panel of EM, proposed to be EM1 (metabolic ratio debrisoquine <1.0), and a panel of PM [6]. The potent inhibition of CYP2D6 activity by fluoxetine and norfluoxetine decreased CL/F by 81% in EM1 and 93% in EM2 without total inhibition of 5-HM formation, which also supports the suggested difference between genotypes. Previously it has been concluded that at least 80% of the biotransformation of tolterodine occurs in the liver during first-pass [6]. In the present study, the reduction in CL/F during fluoxetine coadministration was associated with only a 1.5-fold to 1.9-fold increase in t1/2,z, in EM2 and EM1, respectively, implying that most of the inhibition occurred during first-pass.
Several indications of a CYP2D6 dependent clearance of 5-HM (to some extent) were also revealed when the number of functional genes was taken into account in the analysis. Lower Cmax levels of 5-HM in combination with a shorter t1/2,z among EM2 compared with EM1 were observed. The t1/2,z of 5-HM was 1.1-fold to 1.2-fold longer among EM1 than in EM2 and this difference increased during fluoxetine treatment to 1.6-fold to 1.7-fold. The AUC differences between EM1 and EM2 was 2-fold higher prior to the interaction, indicating that lower CYP2D6 activity increases the 5-HM levels. Further despite the large decrease in CL/F of tolterodine in EM2 in the presence of fluoxetine, the AUC of 5-HM increased by 37% while a 45% decrease was seen in EM1. Preclinical in vitro studies of the metabolism of 5-HM in microsomes, containing an overexpressed human CYP2D6, have shown a correlation between CYP2D6 activity and the formation of the tolterodine aldehyde (Postlind H., personal communication) an intermediate metabolite, prior to tolterodine acid formation, which is not quantifiable in serum.
The present data show that N-dealkylation of tolterodine is an important metabolic pathway in PM and contributes to the elimination of tolterodine in EM during inhibition of CYP2D6. The extremely flat serum concentration–time curves for N-dealkylated tolterodine, with similar levels predose and at 12 h, suggest formation rate-limited elimination kinetics, i.e. the serum curve show the formation of the metabolite rather than the elimination. Fluoxetine has been shown to inhibit the metabolism of alprazolam and carbamazepine [26, 27], a substrate and a substrate/inducer of CYP3A4, respectively. The approximate 22% decrease in CL/F of tolterodine during fluoxetine treatment in the two PM is in agreement with the in vitro observation that CYP3A4 is involved in the N-dealkylation of tolterodine [7]. This was confirmed in a recently performed study of the effect of ketoconazole, a potent CYP3A4 inhibitor [28, 29], on the metabolism of tolterodine in subjects with deficient CYP2D6 activity. That study showed a significant inhibition of the metabolism of tolterodine, resulting in an approximate 2.5-fold increase in AUC [30].
The serum levels of the two tolterodine acids were higher than both tolterodine and 5-HM (Figure 4) and were measurable soon after the parent compound was administered, consistent with the high rate of biotransformation. Tolterodine acid and its N-dealkylated form are the two major metabolites in urine, accounting for approximately 60% of the excretion of a given tolterodine dose [5]. In combination with fluoxetine the AUC values of the two acid metabolites decreased by 62–63%, resulting in a corresponding decrease in the amount in urine. In the view of the 6- and 15-fold increase of tolterodine AUC, an additional inhibitory effect of fluoxetine on CYP3A4 activity in EM cannot be excluded. However, the relative importance is difficult to interpret since the total amount of tolterodine-derived metabolites in urine closely followed the decreased CYP2D6 activity. This indicates a change in the route of elimination in favour of biliary excretion depending on the number of functional genes.
To estimate the clinical importance of inhibition of tolterodine disposition during coadministration of CYP2D6 inhibitors, the active moiety was calculated using AGP levels to derive the unbound fraction. We have reported that most of the antimuscarinic activity in EM (presumably EM2 genotype) correlates to the unbound concentrations of tolterodine +5-HM, while a similar effect is obtained in PM due to higher unbound concentrations of the parent compound [6]. Studies in vitro have shown that AGP is the determinant for the unbound fraction and that there is a good correlation between unbound fraction determinations in vitro and ex vivo [10]. Further the low molar concentrations of tolterodine, its metabolites, fluoxetine and norfluoxetine relative the total amount of AGP does not suggest saturation of the protein.
Using genotyping to differentiate EM2 from EM1 makes it possible to explain the variability among EM. This study suggests an approximate two-fold variability (in terms of AUC) among EM treated with a clinically effective dosage of tolterodine, and that PM receive an exposure to active moiety within the extensive metabolizer interval. The inhibition of tolterodine metabolism by fluoxetine increased the active moiety in EM2 as a result of higher levels of 5-HM, whereas effective concentrations (active moiety) in EM1 were unchanged because decreased levels of 5-HM were compensated for by higher levels of tolterodine. Thus, exposure to active moiety among EM during CYP2D6 inhibition with fluoxetine is similar to that observed prior to coadministration. Stronger inhibition will most likely convert EM into phenotypical PM, who are receiving a similar exposure to active moiety as EM. On the basis of these findings, tolterodine dosage adjustment in patients treated with drugs with CYP2D6 inhibiting properties (e.g. antidepressants, neuroleptics or antiarrhythmics) is not necessary.
In conclusion, the present data suggest a separation in the pharmacokinetics of tolterodine and 5-HM depending on the number of functional CYP2D6 genes. The interaction with fluoxetine was associated with a potent inhibition of tolterodine metabolism in EM2 and EM1 as well as in PM. Overall, the changes in exposure to active moiety during fluoxetine coadministration were within normal variation. It is therefore unlikely that CYP2D6 inhibition will lead to clinically important changes in the therapeutic effect of tolterodine.
Acknowledgments
We would like to thank M. Lexmark, MD, for her help in identifying the patients, and A-C. Ågren, RN, and M-L. Odell, BSc, for their expert assistance. H. Björk, BSc, is thanked for his analytical expertise in conducting the fluoxetine and norfluoxetine assays, and M. Vågerö, B.Sc. for his generous help with the statistical evaluation. This study was supported by Pharmacia & Upjohn AB, Sweden.
References
- 1.Rentzhog L, Stanton SL, Cardozo L, Fall M, Nelson E, Abrams P. Efficacy and safety of tolterodine in patients with detrusor instability: a dose ranging study. Br J Urol. 1998;81:42–48. doi: 10.1046/j.1464-410x.1998.00501.x. [DOI] [PubMed] [Google Scholar]
- 2.Abrams P, Freeman R, Anderstrom C, Mattiasson A. Tolterodine, a new antimuscarinic agent: as effective but better tolerated than oxybutynin in patients with an overactive bladder. Br J Urol. 1998;81:801–810. doi: 10.1046/j.1464-410x.1998.00717.x. [DOI] [PubMed] [Google Scholar]
- 3.Van Kerrebroeck PEVA, Amarenco G, Thüroff JW, et al. Dose-ranging study of tolterodine in patients with detrusor hyperreflexia. Neurourol Urodyn. 1998;17:499–512. doi: 10.1002/(sici)1520-6777(1998)17:5<499::aid-nau6>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 4.Nilvebrant L, Andersson K-E, Gillberg P-G, Stahl M, Sparf B. Tolterodine—a new bladder-selective antimuscarinic agent. Eur J Pharmacol. 1997;327:195–207. doi: 10.1016/s0014-2999(97)89661-6. [DOI] [PubMed] [Google Scholar]
- 5.Brynne N, Stahl MMS, Hallén B, Edlund PO, Palmér L, Gabrielsson J. Pharmacokinetics and pharmacodynamics of tolterodine in man: a new drug for the treatment of urinary bladder overactivity. Int J Clin Pharmacol Ther. 1997;35:287–295. [PubMed] [Google Scholar]
- 6.Brynne N, Dalén P, Alván G, Bertilsson L, Gabrielsson J. Influence of CYP2D6 polymorphism on the pharmacokinetics and pharmacodynamics of tolterodine. Clin Pharmacol Ther. 1998;63:529–539. doi: 10.1016/S0009-9236(98)90104-7. [DOI] [PubMed] [Google Scholar]
- 7.Postlind H, Lindgren A, Andersson SHG. Tolterodine, a novel muscarinic receptor antagonist, is metabolized by cytochromes P450 2D6 and 3A in human liver microsomes. Drug Metab Dispos. 1998;26:289–293. [PubMed] [Google Scholar]
- 8.Nilvebrant L, Gillberg P-G, Sparf B. Antimuscarinic potency and bladder selectivity of PNU-200577, a major metabolite of tolterodine. Pharmacol Toxicol. 1997;81:169–172. doi: 10.1111/j.1600-0773.1997.tb02064.x. [DOI] [PubMed] [Google Scholar]
- 9.Alván G, Bechtel P, Iselius L, Gundert-Remy U. Hydroxylation polymorphisms of debrisoquine and mephenytoin in European populations. Eur J Clin Pharmacol. 1990;39:533–537. doi: 10.1007/BF00316090. [DOI] [PubMed] [Google Scholar]
- 10.Påhlman I, Gozzi P. Protein binding of tolterodine and its major metaboltes in human and several animal species. Biopharm Drug Dispos. 1999;20:91–99. doi: 10.1002/(sici)1099-081x(199903)20:2<91::aid-bdd162>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 11.Altamura AC, De Novellis F, Guercetti G, Invernizzi G, Percudani M, Montgomery SA. Fluoxetine compared with amitriptyline in elderly depression: a controlled clinical trial. Int J Clin Pharmacol Res. 1989;9:391–396. [PubMed] [Google Scholar]
- 12.Altamura AC, Mauri MC. Aspects of treatment of elderly depression: the fluoxetine experience. In: Freeman HL, editor. The Use of Fluoxetine in Clinical Practice. Vol. 183. London: Royal Society of Medicine Services; 1991. pp. 53–59. [Google Scholar]
- 13.Benfield P, Heel RC, Lewis SP. Fluoxetine: a review of its pharmacodynamic and pharmacokinetic properties, and therapeutic efficacy in depressive illness. Drugs. 1986;32:481–508. doi: 10.2165/00003495-198632060-00002. [DOI] [PubMed] [Google Scholar]
- 14.Rosenthal J, Hemlock C, Hellerstein DJ, et al. A preliminary study of serotonergic antidepressants in treatment of dysthymia. Prog Neuropsychopharmacol Biol Psychiatry. 1992;16:933–941. doi: 10.1016/0278-5846(92)90111-q. [DOI] [PubMed] [Google Scholar]
- 15.Crewe HK, Lennard MS, Tucker GT, Woods FR, Haddock RE. The effect of selective serotonin re-uptake inhibitors on cytochrome P4502D6 (CYP2D6) activity in human liver microsomes. Br J Clin Pharmacol. 1992;34:262–265. doi: 10.1111/j.1365-2125.1992.tb04134.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Otton SV, Wu D, Joffe RT, Cheung SW, Sellers EM. Inhibition by fluoxetine of cytochrome P450 2D6 activity. Clin Pharmacol Ther. 1992;53:401–409. doi: 10.1038/clpt.1993.43. [DOI] [PubMed] [Google Scholar]
- 17.Richelson E. Pharmacokinetic drug interactions of new antidepressants: a review of the effects on the metabolism of other drugs. Mayo Clin Proc. 1997;72:835–847. doi: 10.4065/72.9.835. [DOI] [PubMed] [Google Scholar]
- 18.Thomas TM, Plymatt KR, Blannin J, Meade TW. Prevalence of urinary incontinence. Br Med J. 1980;281:1243–1245. doi: 10.1136/bmj.281.6250.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Palmér L, Andersson L, Andersson T, Stenberg U. Determination of tolterodine and the 5-hydroxymethyl metabolite in plasma, serum and urine using gas chromatography-mass spectrometry. J Pharm Biomed Anal. 1997;16:155–165. doi: 10.1016/s0731-7085(97)00023-x. [DOI] [PubMed] [Google Scholar]
- 20.Björk H, Bengtsson F. A simple method for routine TDM of the SSRI fluoxetine and norfluoxetine in serum by HPLC. Eur J Clin Pharmacol. 1997;52(Suppl):A156. [Google Scholar]
- 21.Dahl ML, Johansson I, Palmertz MP, Ingelman-Sundberg M, Sjöqvist F. Analysis of the CYP2D6 gene in relation to debrisoquin and desipramine hydroxylation in a Swedish population. Clin Pharmacol Ther. 1992;51:12–17. doi: 10.1038/clpt.1992.2. [DOI] [PubMed] [Google Scholar]
- 22.Statistical Consultants Inc. PCNonlin and Nonlin84: Software for the statistical analysis of nonlinear models. Am Stat. 1986;40:52. [Google Scholar]
- 23.Gibaldi M, Perrier D. Pharmacokinetics. 2. New York: Marcel Dekker; 1982. [Google Scholar]
- 24.Rowland M, Tozer TN. Clinical pharmacokinetics: Concepts and Applications. 3. Philadelphia: Williams & Wilkins; 1995. [Google Scholar]
- 25.Tozer T. Implications of altered plasma protein binding in disease states. In: Benet LZ, Massoud N, Gambertoglio JG, editors. Pharmacokinetic basis for drug treatment. New York: Raven Press; 1984. pp. 173–193. [Google Scholar]
- 26.Greenblatt DJ, Preskorn SH, Cotreau MM, Horst WD, Harmatz JS. Fluoxetine impairs clearance of alprazolam but not of clonazepam. Clin Pharmacol Ther. 1992;52:479–486. doi: 10.1038/clpt.1992.175. [DOI] [PubMed] [Google Scholar]
- 27.Grimsley SR, Jann MW, Carter JG, D’Mello AP, D’Souza MJ. Increased carbamazepine plasma concentrations after fluoxetine coadministration. Clin Pharmacol Ther. 1991;50:10–15. doi: 10.1038/clpt.1991.98. [DOI] [PubMed] [Google Scholar]
- 28.Maurice M, Pichard L, Daujat M, et al. Effects of imidazole derivatives on cytochromes P450 from human hepatocytes in primary culture. FASEB J. 1992;6:752–758. doi: 10.1096/fasebj.6.2.1371482. [DOI] [PubMed] [Google Scholar]
- 29.Vahre A, Olkkola KT, Neuvonen PJ. Oral triazolam is potentially hazardous to patients receiving systemic antimycotics ketoconazole or itraconazole. Clin Pharmacol Ther. 1994;56:601–607. doi: 10.1038/clpt.1994.184. [DOI] [PubMed] [Google Scholar]
- 30.Brynne N, Forslund C, Hallén B, Gustafsson LL, Bertilsson L. Ketoconazole inhibits the metabolism of tolterodine in subjects with deficient CYP2D6 activity. Exp Toxicol Pathol. 1998;50:96. doi: 10.1046/j.1365-2125.1999.00053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]