

Abstract
Aims
To examine the effect of timing of a risedronate dose relative to food intake on the rate and extent of risedronate absorption following single-dose, oral administration to healthy male and female volunteers.
Methods
A single-dose, randomized, parallel study design was conducted with volunteers assigned to four treatment groups (31 or 32 subjects per group, 127 subjects total). Each subject was orally administered 30 mg risedronate. Group 1 was fasted for 10 h prior to and 4 h after dosing (fasted group); Groups 2 and 3 were fasted for 10 h and were dosed 1 and 0.5 h, respectively, before a high-fat breakfast; and Group 4 was dosed 2 h after a standard dinner. Blood and urine samples were collected for 168 h after dosing. Pharmacokinetic parameters were estimated by simultaneous analysis of risedronate serum concentration and urinary excretion rate-time data.
Results
Extent of risedronate absorption (AUC and Ae) was comparable (P = 0.4) in subjects dosed 2 h after dinner and 0.5 h before breakfast; however, a significantly greater extent of absorption occurred when risedronate was given 1 or 4 h prior to a meal (1.4- to 2.3-fold greater). Administration 0.5, 1, or 4 h prior to a meal resulted in a significantly greater rate of absorption (Cmax 2.8-, 3.5-, and 4.1-fold greater, respectively) when compared with 2 h after dinner.
Conclusions
The comparable extent of risedronate absorption when administered either 0.5–1 h before breakfast or 2 h after an evening meal support previous clinical studies where risedronate was found to have similar effectiveness using these dosing regimens. This flexibility in the timing of risedronate administration may provide patients an alternative means to achieve the desired efficacy while maintaining their normal daily routine.
Keywords: absorption, bioavailability, bisphosphonate, food, osteoporosis, Paget’s disease, pharmacokinetics, risedronate
Introduction
Risedronate sodium (1-hydroxy-2-[3-pyridinyl] ethylidene bisphosphonic acid monosodium salt) is a pyridinyl bisphosphonate developed for the treatment of osteoporosis and other metabolic bone disorders, and has recently been approved for the treatment of Paget’s disease by the United States Food and Drug Administration (FDA). It is a highly potent antiresorptive agent that binds to hydroxyapatite in bone and inhibits osteoclast-mediated bone resorption [1]. Risedronate has been shown to decrease bone turnover and increase bone mass at the hip and spine in early postmenopausal women [2], reduce pain and normalize biochemical indicators of disease activity in patients with Paget’s disease of bone [3–5], and prevent bone loss and fractures associated with corticosteroid therapy for rheumatoid arthritis [6, 7].
Previous clinical pharmacokinetic studies have described risedronate absorption as relatively rapid (tmax~1 h) and occurring throughout the upper gastrointestinal tract when administered to different sites [8]. The serum concentration–time and urinary excretion rate–time profiles are multiphasic, and the rate and extent of risedronate absorption is linear over a dose range of 2.5–30 mg [9].
Orally administered bisphosphonates have demonstrated low bioavailability (<1%) [10]. Absorption of bisphosphonates is significantly inhibited by food [10, 11], and the dosing instructions for some bisphosphonates specify that they be taken with water at least 30 min before food intake, following an overnight fast [12]. However, the recommended dosing regimen for risedronate in phase II clinical studies was at least 2 h from any meal, with evening dosing 2 h after dinner being particularly convenient [2]. The regimen for the phase III clinical studies is dosing from 0.5 to 1 h before breakfast [13]. Therefore, the purpose of this study was to compare the rate and extent of risedronate absorption in healthy volunteers following single-dose, oral administration of a 30 mg dose using regimens in which the dose was given 2 h after dinner followed by an overnight fast, or 0.5, 1 or 4 h before a meal, after an overnight fast.
Methods
Study design
This was a single-dose, randomized, parallel-design study that followed Good Clinical Practice guidelines, Declaration of Helsinki, and was approved by a local ethics review committee (Besselaar Institutional Review Board, Madison, WI), with written informed consent obtained from each subject prior to enrolment. The study population consisted of healthy 18–40 year-old male and female volunteers. Subjects were assigned at random to one of four treatment groups: Group 1 fasted for 10 h prior to risedronate administration and received the dose 4 h prior to lunch; Group 2 fasted for 10 h prior to drug administration and received a high-fat breakfast 1 h after risedronate administration; Group 3 fasted for 10 h prior to risedronate administration and received a high-fat breakfast 0.5 h after drug administration; and Group 4 received risedronate 2 h after a standard dinner. The high-fat breakfast consisted of two slices of white toast, two pats of butter, two eggs fried in butter, two slices of bacon, 57 g of hash brown potatoes, and 226 g of whole milk. This breakfast contained approximately 30 g of protein, 46 g of fat, 50 g of carbohydrates, and 3066 kJ [14]. The lunch comprised 283 g of vegetable and beef soup with crackers, 85 g of smoked turkey on whole wheat bread with lettuce, 15 ml of mayonnaise, 142 g of tossed salad with 12 g of light salad dressing, 2 canned peach halves, and 283 g of skimmed milk. This lunch contained approximately 38 g of protein, 19 g of fat, 104 g of carbohydrates, and 2999 kJ [14]. The dinner consisted of 113 g of baked boneless chicken breast, 28 g of light gravy, one baked potato, one pat of margarine, 0.5 cup of carrot rounds, 0.5 cup of apple sauce, one large peanut butter cookie, and 283 g of lemonade. The dinner contained approximately 40 g of protein, 16 g of fat, 103 g of carbohydrates, and 2919 kJ [14]. Nutrient calculations were performed using the Minnesota Nutrition Data System (NDS) software developed by the Nutrition Coordinating Center (NCC), University of Minnesota, Minneapolis, Minnesota, USA (food database version 6A; nutrient database version 21). All subjects received a single oral dose of 30 mg risedronate (3×10 mg cellulose film-coated tablets, (Procter and Gamble Pharmaceuticals, Cincinnati, OH, USA) with 240 ml of water.
Blood (serum) and urine samples were collected for analysis of laboratory markers (i.e. clinical chemistry, haematology, and urinalysis). Electrocardiograms were performed at screening, prior to drug administration, and 7 days after dosing. Vital signs were assessed at screening, prior to drug administration, and during the final physical examination (day 8). Subjects were monitored continuously for adverse events. All reported and observed adverse events, including clinically significant abnormalities in laboratory values, were followed to resolution or until discharge from follow-up was warranted.
Venous blood was obtained from each subject immediately prior to drug administration and at 0.5, 1, 1.5, 2, 3, 4, 5, 6, 8, 10, 12, 14, 16, 20, 24, 28, 32, 40 and 48 h, and then every 12 h until 168 h after the dose. Serum was harvested from the collected blood. A control urine specimen was obtained from subjects prior to the start of drug administration. Urine was pooled over time intervals from 0–4, 4–8, 8–12, 12–16, 16–24, 24–32, 32–40, and 40–48 h, and then at 12 h intervals until 168 h after the dose. Urine specimens were refrigerated at 4° C until the entire specimen was obtained. Serum and urine samples were stored at −20° C until assayed for risedronate concentration.
Serum and urine risedronate concentrations were determined using a solid-phase extraction procedure coupled with an enzyme-linked immunosorbent assay (ELISA, procedures on file at Procter & Gamble Pharmaceuticals). In this method, 1 ml of serum or urine is acidified with dilute hydrochloric acid, processed through a cation exchange column, and the column eluate subjected to ELISA. The ELISA is based on competitive inhibition between a solid-phase antigenic risedronate equivalent and risedronate for the binding sites on a constant amount of primary antibody. Using a secondary antibody, the primary antibody is quantified using absorbance detection of colour development. The quantification range of the four-parameter standard curve was 0.15–6.0 ng ml−1 for both serum and urine. At the lower limit of quantification (0.15 ng ml−1), the coefficients of variation for the quality control samples were 19% for serum and 9% for urine. Serum or urine quality control samples at three risedronate concentrations (0.20, 1.0, 5.0 ng ml−1) were included in each analysis of study samples. Quality control interassay coefficient of variation ranged 18–24% and 10–18% for serum and urine assays, respectively.
Pharmacokinetic analysis
Risedronate serum concentration–time and urinary excretion rate–time data were analysed simultaneously using PCNONLIN (version 4.2) software [15]. Data were analysed using the following equations:
| 1 |
| 2 |
| 3 |
where C is the serum concentration of risedronate at time t, dAe/dt is the urinary excretion rate occurring at the midpoint of the collection interval, tmid is the midpoint time of the urine collection interval, tlag is the lag time before onset of drug absorption, n is the number of exponents necessary to characterize serum concentration-time and urinary excretion rate-time profiles, Ci is the ith coefficient, i is the ith exponent, CL R is the renal clearance of risedronate, and Cn is the coefficient associated with λn. Predicted serum concentration and urinary excretion rate weights were used in the analysis (1, 1/p, or 1/p2, where p is the predicted value for that function). Decisions on appropriate weighting and number of exponents required to characterize the serum concentration–time and urinary excretion rate–time profiles were based on randomness of scatter of observed data about the best-fit line and sum of weighted squared residuals [16].
Estimated maximum serum concentration (Cmax) and time of occurrence of Cmax (tmax) were derived using equation 1. Area under the serum concentration–time curve (AUC), terminal exponential half-life (t1/2,z),oral clearance (CLO), and terminal volume of distribution uncorrected for bioavailability (Vz/F) were calculated from estimated coefficients and exponents using standard equations [17, 18]. Cumulative urinary excretion (Ae) of risedronate was obtained from AUC and renal clearance (CLR).
Statistical analysis
Treatment group sample size was based on the between subject variability observed in the pharmacokinetic parameters from a previous study [Procter & Gamble Pharmaceuticals, data on file]. The inclusion of at least 30 subjects per treatment group (120 subjects total) was selected to provide at least 80% power to detect a difference of 0.48 for the mean AUC or Cmax on the log scale (a factor of 1.62 on the raw scale) at a 0.05 significance level using analysis of variance (anova). This allowed for detection of pairwise differences of at least 0.402 on the log scale (a factor of 1.50 on the raw scale) with 80% power using Fisher’s least significance difference (LSD) test. In this study, the observed between subject standard deviation (on the log scale) was 0.47 for AUC and 0.46 for Cmax and at least 31 subjects in each treatment group were included in the analysis. This resulted in at least 90% power to detect a difference of 0.48 on the log scale using anova and at least 90% power to detect pairwise differences of at least 0.402 on the log scale using Fisher’s LSD test.
Data were assessed for adherence to normality using the Shapiro-Wilk normality test and were log-transformed as required to satisfy these assumptions. Data were analysed using anova. If the overall test for treatment effect was significant (P<0.05), Fisher’s LSD multiple comparison test was used for all pairwise comparisons.
Non-parametric analyses were performed if the normality assumptions were not met for both raw and log-transformed data. The Kruskall–Wallis test was used to assess treatment effects; if treatment effects were significant, the nonparametric analogue of Fisher’s protected LSD was used to perform multiple comparisons [19]. The Wilcoxon–Mann–Whitney test was used to assess gender effects.
Results
Study population
There were no statistically significant differences among the treatment groups in terms of demographic parameters (age, height, gender, body weight) or creatinine clearance.
Study completion
Of the 127 subjects who entered the study, 126 completed the study. The one subject (Group 3) who did not complete the study experienced an adverse event of intermittent leg pain that was judged by the investigator as having a doubtful relationship to the drug. Nevertheless, data from this subject were included in the analysis of pharmacokinetic parameters. Risedronate concentrations were quantifiable in serum and urine in all 127 subjects, indicating that risedronate was absorbed after all dosing conditions. However, one subject in Group 3 who completed the study was not included in the pharmacokinetic analyses due to an inadequate amount of urine data with quantifiable risedronate concentrations. This subject’s urine output was 5–9 l day−1, compared with 1.5–2.5 l day−1 for the other subjects.
Pharmacokinetic parameters
Median serum risedronate concentration–time profiles are shown in Figure 1 and the median risedronate urinary excretion rate–time profiles are depicted in Figure 2. These profiles illustrate the dependence of Cmax and tmax on the time of dosing relative to meals (Figure 1) and the multiphasic elimination of risedronate (Figure 2). Serum concentration–time and urinary excretion rate–time profiles for individual subjects were adequately characterized by a 3- or 4-exponential function when fitted simultaneously with a weighting of 1, 1/p, or 1/p2. The observed and predicted serum concentration–time and urinary excretion rate–time profiles were in good agreement.
Figure 1.
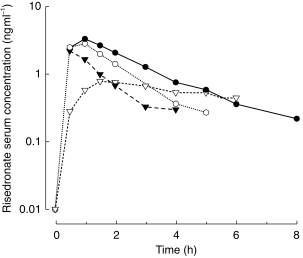
Median risedronate serum concentration-time profile following single-dose oral administration of 30 mg risedronate to healthy volunteers, 4 h prior to a meal (Group 1; •), 1 h prior to a meal (Group 2; ○); 0.5 h prior to a meal (Group 3; ▾); and 2 h after dinner (Group 4; ▿).
Figure 2.
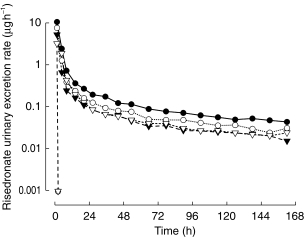
Median risedronate urinary excretion rate-time profile following single-dose oral administration of 30 mg risedronate to healthy volunteers, 4 h prior to a meal (Group 1; •), 1 h prior to a meal (Group 2; ○); 0.5 h prior to a meal (Group 3; ▾); and 2 h after dinner (Group 4; ▿).
Significant differences in AUC and Ae were observed between treatment groups (P<0.0001; Table 1), although there was considerable variability within the treatment groups. The values of AUC and Ae for subjects receiving the dose 4 h before lunch and 1 h before breakfast (Groups 1 and 2, respectively) were significantly larger than for subjects who received the dose 0.5 h before breakfast and 2 h after dinner (Groups 3 and 4, respectively). Additionally, AUC and Ae for subjects who received the dose 4 h before lunch (Group 1) were significantly greater than for subjects who received the dose 1 h before breakfast (Group 2). Median CLR was not significantly different among the four groups (P = 0.123).
Table 1.
Risedronate pharmacokinetic parameters after single-dose oral administration of 30 mg risedronate (n = 31–32 per group).
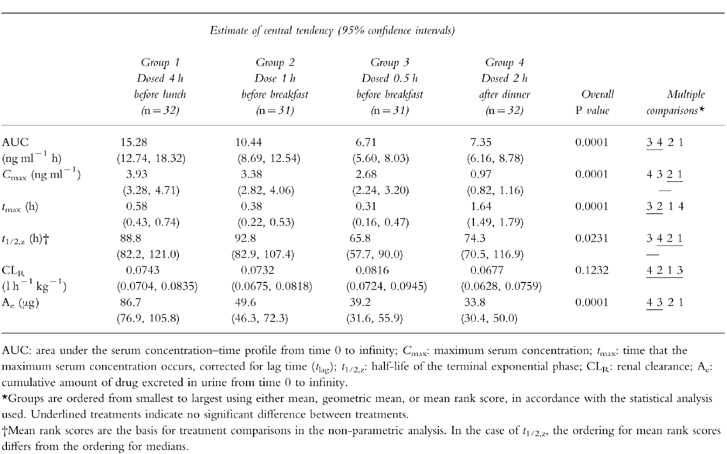
Differences in Cmax among the four treatment groups were also statistically significant (P<0.0001; Table 1). In common with AUC and Ae, considerable variability was also observed with Cmax. The Cmax values for subjects who received the dose before a meal (Groups 1–3) were significantly larger than for subjects who received the dose 2 h after dinner (Group 4). In addition, Cmax for subjects who received the dose 4 h before lunch (Group 1) was significantly larger than for subjects who received the dose 0.5 h before breakfast (Group 3).
The tmax for subjects who received the dose 2 h after dinner (Group 4) was significantly longer than for the other treatment groups (Groups 1–3) (Table 1). Additionally, the tmax for subjects who received the dose 4 h before lunch (Group 1) was significantly longer than for subjects who received the dose 0.5 and 1 h before breakfast (Groups 2 and 3, respectively).
The t1/2,z was significantly different among the treatment groups (P = 0.0231; Table 1). The t1/2,z for subjects who received the dose 0.5 h before breakfast (Group 3) was significantly shorter than for those subjects who received the dose 1 or 4 h prior to a meal (Groups 2 and 1, respectively). The observed difference in t1/2,z may have been due to the inability to quantify risedronate for equivalent time periods after dosing in serum or urine samples in order to provide an accurate assessment of this half-life.
No significant differences in gender were observed for any of the pharmacokinetic parameters (AUC, Cmax, tmax, CLR, Vz/F, Ae), except within one treatment group (Group 3) for half-life. Since the difference in half-life was not consistently observed across the other treatment groups (Groups 1, 2 and 4), nor was there a trend towards a difference in half-lives within the other treatment groups, it was concluded that no gender difference existed in half-life.
Adverse events
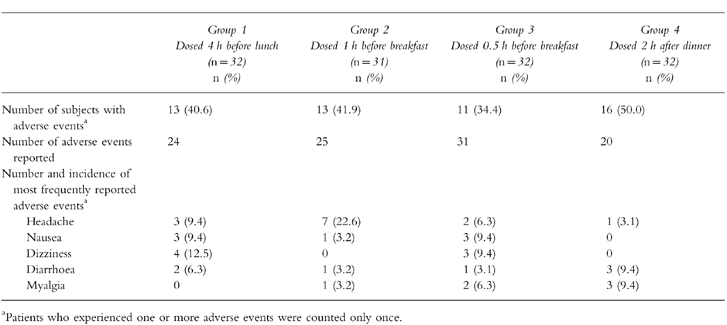
Fifty-three of the 127 enrolled subjects (42%) experienced one or more adverse events. One subject did not complete the study due to intermittent leg pain. This event was judged by the investigator to have a doubtful relationship to the drug.
The four treatment groups were comparable with respect to the number of participants reporting clinical adverse events (Table 2). Overall, the most frequently reported adverse events were headache, nausea, dizziness, diarrhoea and myalgia. These were primarily mild in nature. No clinically relevant changes were observed in clinical chemistry parameters.
Table 2.
Summary of adverse events.
Discussion
Bisphosphonates have generally demonstrated low bioavailability [10], which is significantly inhibited by food. The purpose of this study was to compare the rate and extent of risedronate absorption in healthy volunteers following oral administration of a single 30 mg dose using regimens of dosing 2 h after dinner followed by an overnight fast, or an overnight fast followed by dosing 0.5, 1 or 4 h before a meal.
The value of Cmax was significantly lower (approximately threefold) when risedronate was given 2 h after dinner (Group 4) than when given prior to a meal (Groups 1, 2 and 3, respectively). These results are similar to those reported previously for tiludronate [20]. The 75% reduction in Cmax of risedronate when given 2 h after dinner (Group 4) as compared with 4 h before lunch (Group 1) was similar to the 80% reduction in Cmax observed for tiludronate given 2 h after a meal as compared with fasting [20]. However, the significant (32%) reduction in Cmax for risedronate when administered 0.5 h before breakfast with respect to 4 h prior to a meal was not as large as that reported for tiludronate given just before breakfast (80% decrease in Cmax) [20].
The tmax was three to five-fold greater when subjects received the dose 2 h after dinner (Group 4) compared with the other three treatment groups. Since similar rates of absorption of risedronate solutions administered to stomach, duodenum and terminal ileum have been reported [8], the greater tmax is probably due to slower absorption in the presence of food (Figure 1). In contrast to these results, administration of tiludronate 2 h after a normal meal is reported to result in a shorter tmax [20].
Based on AUC and Ae, the extent of risedronate absorption for subjects receiving a dose 0.5 h before breakfast and 2 h after dinner (Groups 3 and 4, respectively) was reduced by 55% when compared with subjects given the dose 4 h before lunch (Group 1) and by 30–35% when compared with subjects given the dose 1 h before breakfast (Group 2). These results suggest that the change in the dosing regimen will result in a similar systemic exposure to risedronate if administered approximately 0.5 h before breakfast or 2 h after an evening meal. However, if risedronate is administered 1 h before breakfast, an approximately 1.5-fold increase in systemic drug exposure could occur relative to dosing 0.5 h before breakfast or 2 h after dinner. The decrease, relative to fasting, in extent of absorption of risedronate administered 0.5 h before breakfast is similar to that reported for alendronate administered 0.5 and 1 h before a meal [21]. Administration of oral risedronate 2 h after an evening meal results in a 50% decrease in the extent of absorption relative to 4 h prior to a meal. In contrast, the extent of alendronate absorption has been reported as essentially zero following oral administration 2 h after a meal [22], and tiludronate absorption has been shown to be reduced by 80% relative to fasting [20].
The implications of these results have been demonstrated in clinical studies where similar safety and efficacy profiles in the treatment of Paget’s disease were observed using dosing regimens of 2 h after meals (phase II) [3–5] and 0.5–1 h before breakfast (phase III) [13]. These studies demonstrated the same percentage decrease (~80%) in excess alkaline phosphatase (a pharmacodynamic marker elevated in Paget’s disease), relative to baseline, when risedronate was administered 2 h after a meal or in the morning, 0.5–1 h before breakfast. Since the pharmacodynamic results correlate with the extent of absorption (AUC and Ae), these results suggest that the amount of drug absorbed, not the peak serum concentrations (Cmax), is related to the efficacy of risedronate in the treatment of Paget’s disease [23].
The median t1/2,z for the four treatment groups in this study ranged 66–93 h. Half-lives for other bisphosphonates include 12.8 h for clodronate [24, 25], 27.2 h for pamidronate [26], 50 h for tiludronate [27, 28], 17 days for etidronate [29] and 10 years for alendronate [30]. Estimation of a t1/2,z for many bisphosphonates is difficult due to an inadequate duration of drug quantification in serum, plasma, or urine samples, and the use of more conventional methods of analysis. Therefore, many pharmacokinetic parameters reported for bisphosphonates should be viewed with caution until the analytical methodology improves to allow monitoring of serum or urine drug concentrations for a time period equal to two or three half-lives, or alternatively by using simultaneous analysis if serum concentrations are not quantifiable for two or three half-lives [31].
As seen in previous clinical trials, risedronate was well tolerated [3–6, 8, 32, 33]. The number of adverse events was similar among the four risedronate dosing regimens. Only one subject in the present study discontinued risedronate therapy. This subject reported intermittent leg pain that was judged to have a doubtful relationship to the drug.
In conclusion, the comparable extent of risedronate absorption when administered either 0.5–1 h before breakfast or 2 h after an evening meal support previous clinical studies where risedronate was found to have similar effectiveness using these dosing regimens. This flexibility in the timing of risedronate administration may provide patients an alternative means to achieve the desired efficacy while maintaining their normal daily routine.
Acknowledgments
The authors wish to thank Frank van den Ouweland MD, PhD for his assistance in the monitoring and evaluation of safety data. This study was supported by Procter & Gamble Pharmaceuticals, Mason, Ohio, USA.
References
- 1.Sietsema WK, Ebetino FH, Salvagno AM, Bevan JA. Antiresorptive dose–response relationships across three generations of bisphosphonates. Drugs Exp Clin Res. 1989;15:389–396. [PubMed] [Google Scholar]
- 2.Mortensen L, Charles P, Bekker PJ, DiGennaro J, Johnston CC. Risedronate increases bone mass in an early postmenopausal population: two years of treatment plus one year of follow-up. J Clin Endocrinol Metab. 1998;83:396–402. doi: 10.1210/jcem.83.2.4586. [DOI] [PubMed] [Google Scholar]
- 3.Hosking DJ, Eusebio RA, Chines AA. Paget’s disease of bone: reduction of disease activity with oral risedronate. Bone. 1998;22:51–55. doi: 10.1016/s8756-3282(97)00222-6. [DOI] [PubMed] [Google Scholar]
- 4.Siris ES, Chines AA, Altman RD, et al. Risedronate in the treatment of Paget’s disease of bone: an open label, multicenter study. J Bone Miner Res. 1998;13:1032–1038. doi: 10.1359/jbmr.1998.13.6.1032. [DOI] [PubMed] [Google Scholar]
- 5.Singer FR, Clemens TL, Eusebio RA, Bekker PJ. Risedronate, a highly effective oral agent in the treatment of patients with severe Paget’s disease. J Clin Endocrinol Metab. 1998;83:1906–1910. doi: 10.1210/jcem.83.6.4871. [DOI] [PubMed] [Google Scholar]
- 6.Eastell R, Devogelaer JP, Peel NFA, et al. Risedronate therapy prevents bone loss in glucocorticoid-treated rheumatoid arthritis patients: a three year study. Inflammopharmacology. 1997;5:186–188. [Google Scholar]
- 7.Reid D, Cohen S, Pack S, Chines A, Ethgen D. Risedronate reduces the incidence of vertebral fractures in patients on chronic corticosteroid therapy. Arthritis Rheum. 1998;41:S136. [Google Scholar]
- 8.Mitchell DY, Eusebio RA, Dunlap LE, et al. Risedronate gastrointestinal absorption is independent of site and rate of administration. Pharm Res. 1998;15:228–232. doi: 10.1023/a:1011910517200. [DOI] [PubMed] [Google Scholar]
- 9.Mitchell DY, Eusebio RA, Pallone KA, et al. Single dose linearity of risedronate following oral administration of 2.5, 5, or 30mg to healthy volunteers. Pharm Res. 1997;14:S–609. [Google Scholar]
- 10.Lin J. Bisphosphonates: a review of their pharmacokinetic properties. Bone. 1996;18:75–85. doi: 10.1016/8756-3282(95)00445-9. [DOI] [PubMed] [Google Scholar]
- 11.Mitchell DY, Eusebio RA, Dunlap LE, et al. Bioavailability of immediate-release and delayed-release risedronate formulations upon oral administration to healthy male subjects in fasted and fed state. Pharm Res. 1996;14:S609. [Google Scholar]
- 12.Gertz BJ, Holland SD, Kline WF, Matuszewski BK, Porras AG. Clinical pharmacology of alendronate sodium. Osteoporosis Int. 1993;3:S13–S16. doi: 10.1007/BF01623002. [DOI] [PubMed] [Google Scholar]
- 13.Miller PD, Adachi JD, Brown JP, et al. Risedronate vs. etidronate Paget’s study: durable remission with only two months of 30 mg risedronate. J Bone Miner Res. 1997;12:S269. [Google Scholar]
- 14.Schakel S, Sievert Y, Buzzard I. Sources of data for developing and maintaining a nutrient database. J Am Diet Assoc. 1988;88:1268–1271. [PubMed] [Google Scholar]
- 15.Statistical Consultants Inc. PCNONLIN and NONLIN84: software for the statistical analysis of nonlinear models. Am Stat. 1986;40:52. [Google Scholar]
- 16.Boxenbaum HG, Riegelman S, Elashoff RM. Statistical estimations in pharmacokinetics. J Pharmacokinet Biopharm. 1974;2:123–148. doi: 10.1007/BF01061504. [DOI] [PubMed] [Google Scholar]
- 17.Wagner JG. Linear pharmacokinetic equations allowing direct calculations of many needed pharmacokinetic parameters from the coefficients and exponents of polyexponential equations which have been fitted to the data. J Pharmacokinet Biopharm. 1976;4:443–467. doi: 10.1007/BF01062831. [DOI] [PubMed] [Google Scholar]
- 18.Gibaldi M, Perrier D. Pharmacokinetics. 2. New York: Marcel Dekker Inc; 1982. [Google Scholar]
- 19.Conover WJ. Practical Nonparametric Statistics. 2. New York: John Wiley & Sons; 1980. [Google Scholar]
- 20.Necciari J, Mougeot V, Pinquer JL, Arnoux P, Delauture D. 5th World Conference on Clinical Pharmacology and Therapeutics. Yokohama, Japan: 1992. Influence of food intake (hypocalcic or normal meal) on the bioavailability of tiludronate; pp. 305–309. [Google Scholar]
- 21.Gertz BJ, Holland SD, Kline WF, et al. Studies of the oral bioavailability of alendronate. Clin Pharmacol Ther. 1995;58:288–298. doi: 10.1016/0009-9236(95)90245-7. [DOI] [PubMed] [Google Scholar]
- 22.Fogelman I, Smith L, Mazess R, Wilson MA, Bevan JA. Absorption of oral diphosphonate in normal subjects. Clin Endocrinol. 1986;24:57–62. doi: 10.1111/j.1365-2265.1986.tb03254.x. [DOI] [PubMed] [Google Scholar]
- 23.Shah VP, Yacobi A, Barr WH, et al. Workshop report: evaluation of orally administered highly variable drugs and drug formulations. Pharm Res. 1996;13:1590–1594. doi: 10.1023/a:1016468018478. [DOI] [PubMed] [Google Scholar]
- 24.Yakatan GJ, Poynor WJ, Talbert RL, et al. Clodronate kinetics and bioavailability. Clin Pharmacol Ther. 1982;31:402–410. doi: 10.1038/clpt.1982.51. [DOI] [PubMed] [Google Scholar]
- 25.Pentikäinen P, Elomaa I, Nurmi A-K, Kärkkäinen S. Pharmacokinetics of clodronate in patients with metastatic breast cancer. Int J Clin Pharmacol Ther Toxicol. 1989;27:222–228. [PubMed] [Google Scholar]
- 26.Redalieu E, Coleman J, Chan K, et al. Urinary excretion of aminohydroxyproline bisphosphonate in cancer patients after single intravenous infusions. J Pharm Sci. 1993;82:665–667. doi: 10.1002/jps.2600820624. [DOI] [PubMed] [Google Scholar]
- 27.Necciari J, Kiefer G, Maillard D. Pharmacokinetics of (4-chlorophenyl) thiomethylene bisphosphonic acid after single and repeated administration in healthy volunteers. Calcif Tissue Int. 1989;44:S107. [Google Scholar]
- 28.Jacob F, Debry G, Lascombes F, Necciari J. Tiludronate: pharmacokinetic dose proportionality after intravenous administration. Absolute bioavailability in healthy volunteers. J Bone Miner Res. 1996;11:S347. [Google Scholar]
- 29.Gural R, Dittert L. Pharmacokinetics of etidronate in man. Am Pharm Assoc. 1976;6:93. [Google Scholar]
- 30.Rodan G, Balena R. Bisphosphonates in the treatment of metabolic bone diseases. Ann Med. 1993;25:373–378. doi: 10.3109/07853899309147299. [DOI] [PubMed] [Google Scholar]
- 31.Mitchell DY, Eusebio RA, Pallone KA, et al. Bisphosphonate pharmacokinetics: use of simultaneous modelling of urine and serum data to determine parameters. Clin Pharmacol Ther. 1997;61:155. [Google Scholar]
- 32.Bekker P, Licata A, Harris S, et al. Risedronate dose response in prevention of early postmenopausal bone loss. J Bone Miner Res. 1996;11:S347. [Google Scholar]
- 33.Delmas PD, Balena R, Confravreux E, et al. Bisphosphonate risedronate prevents bone loss in women with artificial menopause due to chemotherapy of breast cancer: a double-blind, placebo-controlled study. J Clin Oncol. 1997;15:955–962. doi: 10.1200/JCO.1997.15.3.955. [DOI] [PubMed] [Google Scholar]