

Abstract
Aims
To investigate the rate of excretion and routes of metabolism of tolcapone, a novel inhibitor of catechol-O-methyltransferase (COMT).
Methods
Six healthy male volunteers were given 200 mg [14C]-tolcapone (approximately 50 μCi) orally. To assess excretion balance and to identify metabolites, urine and faeces were collected before administration and until radioactivity fell below 75 d min−1 ml−1 (urine) and 100 d min−1 mg−1 (faeces). Blood samples were collected frequently before and after administration to determine plasma radioactivity, to identify tolcapone metabolites and to measure plasma tolcapone and its methylated derivative 3-O-methyltolcapone (3-OMT).
Results
The mean proportion of the dose excreted in urine was 57.3% and in faeces 40.5%. Excretion was almost complete (more than 95%) in all participants after 9 days. The major early metabolite present in plasma was the 3-O-β,d-glucuronide conjugate, which was detectable within 2 h after dosing. The major late metabolite in plasma was 3-OMT. The 3-O-β,d-glucuronide was also the most abundant metabolite in urine and faeces, accounting for 27% and 33%, respectively, of the total radioactivity excreted by these routes and for 26% of the original dose. Reduction of the nitro moiety yields an amine derivative, detected in both urine and faeces, with subsequent modifications, such as acetylation of the amine group and conjugation with glucuronic acid or sulphate, or both. Oxidative reactions due to cytochrome P450 enzymes are of small significance, as is 3-O-methylation by COMT.
Conclusions
Tolcapone is almost completely metabolized and excreted in urine and faeces (only 0.5% of tolcapone was excreted unchanged); glucuronidation is the most important route of metabolism. The relatively long duration of excretion is caused by the long half-life of 3-OMT.
Keywords: 3-O-methyltolcapone, catechol-O-methyltransferase, cytochrome P450, glucuronidation, methylation, oxidative metabolism, tolcapone
Introduction
Tolcapone (3,4-dihydroxy-4′-methyl-5-nitrobenzophenone; Ro 40–7592; Figure 1) is a novel, reversible, orally active inhibitor of the enzyme catechol-O-methyltransferase (COMT) that has been developed for use as an adjunct to levodopa therapy. Levodopa (3,4-dihydroxy-l-phenylalanine) is currently the most important treatment for patients with Parkinson’s disease. Levodopa metabolism by COMT leads to rapid elimination and this is believed to be responsible in part for the end-of-dose motor fluctuations seen during long-term treatment of Parkinson’s disease with levodopa [1, 2]. Inhibition of COMT extends levodopa exposure and thereby reduces the risk of motor fluctuations and other complications during long-term treatment [3–7, 9].
Figure 1.
Structure of tolcapone (Ro 40–7592). *Position of the 14C isotope.
The pharmacological action of tolcapone arises from its function as a substrate for COMT [8]. In addition to the catechol structure, tolcapone contains two electron-withdrawing substituents and easily delivers a proton (pKa = 4.5); the resulting anion has a high affinity for COMT (IC50 in rat liver 36 nm [8]) and displaces other catechols (such as catecholamines and levodopa) from the catalytic centre of the enzyme. Consequently, O-methylation is prevented.
The present study was part of the drug development program for tolcapone and served as the basis for the rational design of subsequent drug–disease interaction studies and the evaluation of tolcapone’s potential for drug–drug interactions. The study determined the rate and route of elimination of tolcapone in humans and identified the principal metabolites in plasma, urine and faeces.
Methods
Study design
Six healthy males, aged 19–30 years, participated in this open-label single-dose excretion balance study after giving written informed consent. Ethics committee approval was obtained and the study was conducted in conformity with the principles of the Declaration of Helsinki and its amendments.
After an overnight fast, each participant received a gelatine capsule containing 200 mg 14C-labelled tolcapone (mean radioactivity 46.0±0.53 μCi per capsule, radiochemical purity 99.45%) with 200 ml water. The participants then fasted in upright position for 4 h. Subsequent standardized meals were given according to a fixed schedule. The volunteers remained in the centre for the first 11 days of the study.
The tolcapone dose of 200 mg, chosen for this study, is the highest dose used in therapeutic practice. The radioactivity (approximately 50 μCi) was considered to be sufficient to allow detection and quantification of metabolites. The position of the radioactive label is identified in Figure 1.
Sample collection
Venous blood samples were collected into glass tubes containing EDTA before administration of [14C]-tolcapone and at 10, 20, 30, 45, 60 and 90 min after administration. Further samples were taken at 2, 4, 8, 12, 18, 24, 30, 36, 48, 72, 96, 120 and 144 h after administration and after collection of the last urine and faeces samples. Plasma was separated by centrifugation within 30 min of collection (2500 g; 10 min) and stored at −20° C.
Urine samples were collected during the following intervals (relative to [14C]-tolcapone intake): −12–0 h (control), 0–4 h (morning day 1), 4–8 h, 8–12 h, 12–24 h, each 12 h period thereafter until day 10 and then every 24 h until urine radioactivity fell below the threshold of 75 dpm ml−1. The samples were stored at 4° C during each collection period, after which they were frozen at −20° C.
A faecal sample was collected from each participant before administration of tolcapone and all subsequent faeces were collected until faecal radioactivity had fallen below 100 d min−1 g−1. The samples were frozen and stored at −20° C.
Measurement of radioactivity
Plasma
Plasma radioactivity was measured by addition of aliquots (100 μl for samples collected up to 12 h after [14C]-tolcapone administration and 500 μl for later samples) to 4 ml Ultima Gold and counting directly. The limit of quantification (LOQ) was 60 d min−1 ml−1 before correction for blank values (approximately 15 dpm 500 μl−1).
Urine
Urinary radioactivity was determined in a similar way, using 500 μl urine aliquots. The LOQ was set at 60 d min−1 ml−1, before correction for blank values (approximately 15 d min−1 500 μl−1).
Faeces
Samples were homogenized with 1.5–3 volumes water using a Sud mixing device (Kinematica, Polytron, Kinematica AG, Lucerne, Switzerland); 400–550 mg homogenate, equivalent to 200 mg faeces, was combusted in a sample oxidizer (Model 306, Packard Instruments) with automatic addition of the absorption (8 ml Carbosorb II, Packard Instruments) and scintillation solutions (10 ml Permafluor V, Packard Instruments). The LOQ was set at 20 d min−1 g−1, before correction for blank values (approximately 5 d min−1 500 mg−1).
Measurement of tolcapone and 3-O-methyltolcapone
Plasma
Plasma concentrations of tolcapone and its methylated derivative 3-O-methyltolcapone (3-OMT, Ro 40–7591); were measured in all samples by reverse-phase h.p.l.c. with ultraviolet detection as described elsewhere [10, 11]. The LOQ was 0.05 μg ml−1 for both compounds.
Urine
Urinary tolcapone and 3-OMT were measured by the same technique as for plasma, except that, when the 10 μl samples were treated, the deproteinization step was omitted. The LOQ was 0.25 μg ml−1 for both compounds.
Pharmacokinetic evaluation
Pharmacokinetic parameters of tolcapone and 3-OMT (Cmax, tmax, AUC(0, ∞) and t1/2,z) were calculated from plasma concentration–time data using noncompartmental methods [12].
Identification of metabolites
Metabolites in plasma, urine and faeces were detected by h.p.l.c. with liquid scintillation counting. Metabolites were identified by comparing their elution times with those of chemically synthesized reference compounds and by mass spectrometry after liquid chromatography.
Plasma
Plasma pools were obtained by combining equal volumes from each participant from samples taken 2, 4 and 12 h after intake of [14C]-tolcapone. The 1 ml samples were acidified by adding 1 m hydrochloric acid (400 μl) and extracted three times by rotating for 10 min with 6 ml ethyl acetate, followed by centrifugation (1000 g; 10 min). The extracts were combined and evaporated at 30° C and the dried residue was reconstituted in methanol (15 μl) and centrifuged for 10 min at 1000 g, if necessary. The average recovery of total radioactivity was 92% (range 87–100%).
Reversed–phase h.p.l.c. was performed at a flow rate of 1 ml min−1. The stationary phase involved a guard column (Supelguard LC-ABZ, 5 μm, 20×4.6 mm, Supelco, Bellefonte, Pennsylvania, USA) and an analytical column (Supelcosil LC-ABZ, 5 μm, 250×4.6 mm, Supelco). The two mobile phases consisted of water containing 0.05 m ammonium acetate and 0.01 m citric acid, acidified to pH 2.3 with 0.4% trifluoroacetic acid (mobile phase A) and acetonitrile (mobile phase B). The A:B elution gradient varied from 75:25% to 45:55% over 55 min, followed by a 15 min elution with 75:25%. Liquid-scintillation counting was performed in 15 s fractions of the eluate; the total recovery of radioactivity was on average 94% (range 91–98%).
Mass spectrometry was performed with an API-III (PE Sciex, Rotkreuz, Switzerland). Chromatographic separation was performed on a Supelcosil ABZ ODS-25 μm×4.6 mm column with 25 mm ammonium acetate, acidified to pH 2.5 with formic acid, and acetonitrile as mobile phases. The ammonium acetate:acetonitrile gradient varied linearly from 70:30 to 30:70 over 15 min; the flow rate was 1 ml min−1.
Urine
A pool of urine samples collected over 72 h after tolcapone administration was prepared by combining appropriate volumes (on a percentage of volume basis) of the corresponding fractions from all participants. The pooled urine was filtered and a 10 ml sample analysed by h.p.l.c. The h.p.l.c. conditions were as described for plasma, except that the A:B elution gradient varied from 85:15% to 100:0% over 70 min.
Faeces
A representative pool of faeces was prepared by combination of appropriate aliquots (on a percentage of weight basis) of faecal extracts of all six volunteers. For each collection period up to 125.8 h after administration, two samples of faeces (approximately 11 ml) from each participant were weighed and mixed with 5n hydrochloric acid (2 ml) and methanol (40 ml). The samples were sonicated for 10 min and centrifuged (2700 g; 10 min). The pellets were extracted with 35 ml methanol and volumes proportional to the corresponding weight of faeces were pooled for analysis. The pH of the pooled sample was adjusted to approximately 6 by addition of ammonium acetate and the sample was concentrated to approximately 20 ml by evaporation at 30° C. After filtration through a cellulose filter (Minisart RC, Sartorius, Berlin, Germany), 6 ml samples were analysed by h.p.l.c. as described above for urine. The recovery of radioactivity was 96–103%.
Results
Plasma
Pharmacokinetics
The pharmacokinetic parameters of tolcapone and 3-OMT, determined from plasma concentrations, are summarized in Table 1 and illustrated for one participant in Figure 2a.
Table 1.
Pharmacokinetics of tolcapone and 3-O-methyltolcapone (3-OMT) after administration of 200 mg [14C]-tolcapone in healthy volunteers.

Figure 2.
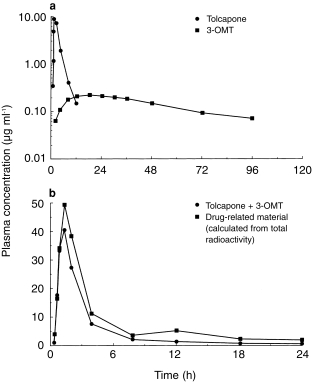
Plasma concentrations of a) tolcapone and 3-O-methyltolcapone (3-OMT) and b) drug-related material after administration of 200 mg [14C]-tolcapone. Data are from one participant; results from all other participants were similar.
Radioactivity
Plasma radioactivity increased rapidly within 1–3 h after administration of tolcapone and then decreased to low levels over the next 6 h. During the first few hours, plasma radioactivity was almost completely accountable as tolcapone and 3-OMT (Figure 2b). A second peak of radioactivity, which could not be attributed to these compounds (Figure 2b), occurred at 12 h in all participants. The t1/2,z of plasma radioactivity (44.5±9.2 h) is not necessarily representative of a single compound but concurred with the t1/2,z of 3-OMT (Table 1). This is consistent with a significant contribution of 3-OMT to the drug-related material in plasma, particularly at later time points.
Identification of metabolites
All major drug-related peaks accounting for more than 3% of the radioactivity in plasma were structurally elucidated and most of the minor metabolites were tentatively identified.
The metabolite pattern from the plasma pool collected 2 h after intake (radiochromatogram after gradient h.p.l.c.: Figure 3a) shows that unchanged tolcapone accounted for 59.3% of plasma radioactivity at this point. The major metabolite, the 3-O-β,d-glucuronic acid conjugate of tolcapone (Ro 61–1448), accounted for 18.6% of plasma radioactivity. The three minor metabolites were identified as 3-OMT (2.1% of radioactivity), an acid metabolite (Ro 47–1669; 1.9% of radioactivity) and an alcohol metabolite (Ro 47–1868; 0.6% of radioactivity).
Figure 3.
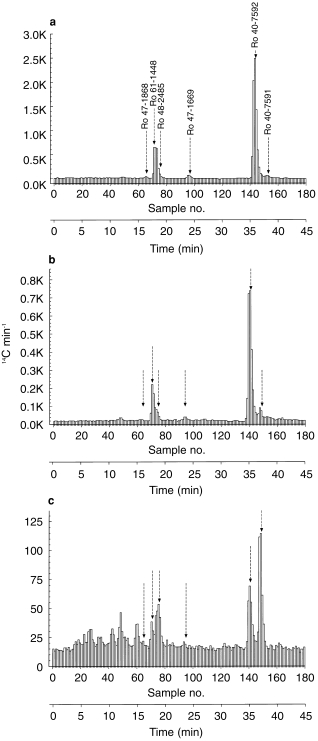
Time profile of the metabolite pattern in human plasma: radiochromatogram of pooled samples collected a) 2 h, b) 4 h and c) 12 h after administration of 200 mg [14C]-tolcapone (50 μCi). Ro 47–1868, primary alcohol metabolite; Ro 61–1448, 3-O-β,d-glucuronide; Ro 48–2485, N-acetylamine (secondary) metabolite; Ro 47–1669, carboxylic acid (secondary metabolite); Ro 40–7592, unchanged tolcapone; Ro 40–7591, 3-O-methyltolcapone (primary metabolite).
At 4 h after administration (Figure 3b), tolcapone and its 3-O-β,d-glucuronide (Ro 61–1448) were still the most prominent compounds in plasma. A slight increase was seen in the relative concentration of 3-OMT and the N-acetylamino derivative of tolcapone (Ro 48–2485) was in some evidence. By 12 h (Figure 3c), the detection limit of the method was nearly reached. Nevertheless, 3-OMT had become the predominant metabolite in plasma. The glucuronide concentration had decreased and the N-acetylamino compound had increased. Unchanged tolcapone remained detectable in plasma for at least 12 h after administration.
Excretion into urine and faeces
Excretion balance
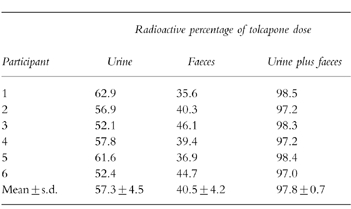
The mean cumulative excretion of radioactivity in urine and faeces was 39.5% 24 h after administration of tolcapone and was almost complete (more than 95%) in all participants after 9 days. Overall, 57.3% of the tolcapone dose was excreted in urine and 40.5% in faeces (Table 2).
Table 2.
Excretion balance of [14C]-tolcapone in healthy volunteers (n = 6).
On the basis of the h.p.l.c. measurements in urine, the cumulative excretion of unchanged tolcapone in the urine was on average 0.5% (range 0.2–0.9%); no 3-OMT was detected in urine.
Metabolites in urine
The metabolite pattern obtained by h.p.l.c. of the pooled urine collected over the first 72 h after administration of tolcapone (88% of total urinary radioactivity) is shown in Figure 4a.
Figure 4.
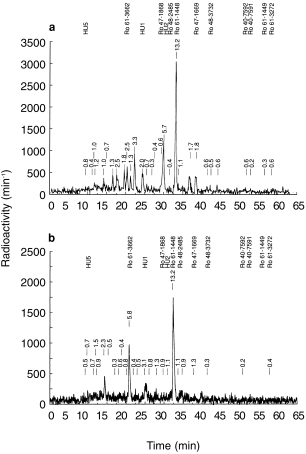
Excretion of tolcapone metabolites a) in urine collected 0–72 h and b) in pooled faecal samples collected from six participants after administration of 200 mg [14C]-tolcapone. HU5, amine sulphate; Ro 61–3662, amine derivative; HU1, amine glucuronide; Ro 47–1868, alcohol derivative; HU2, N-acetylamine glucuronide; Ro 48–2485, N-acetylamino derivative; Ro 61–1448, 3-O-β,d-glucuronide; Ro 47–1669, acid metabolite; Ro 48–3732, 3-O-methyltolcapone acid derivative; Ro 40–7592, unchanged tolcapone; Ro 40–7591, 3-O-methyltolcapone; Ro 61–1449, 4-O-methyltolcapone; Ro 61–3272, 3,4-O-dimethyltolcapone.
The principal urinary metabolite was the 3-O-β,d-glucuronide (Ro 61–1448, 13% of dose, 27% of urinary radioactivity). The second most abundant metabolite was the N-acetylamino glucuronide (HU2, 6% and 11%, respectively). Other metabolites identified in the urine included the amine derivative of tolcapone (Ro 61–3662, 5% of urinary radioactivity), and its glucuronide and sulphate conjugates (4% and 2% of urinary radioactivity, respectively). Furthermore, 3.7% and 1.2% of the radioactivity in urine were identified as the acid metabolite (Ro 47–1669) and the alcohol metabolite (Ro 47–1868), respectively. Tolcapone (0.6% of dose) and 3-OMT (0.2% of dose) occurred in urine only to a minor extent. Together, all of these metabolites accounted for 28% of the original dose (56% of urinary radioactivity); the remainder was accounted for by 25 minor metabolites. None of these minor metabolite fractions surpassed 5% of the dose and most of them accounted for less than 1%.
Metabolites in faeces
The metabolite pattern obtained by h.p.l.c. of the pooled faecal samples is shown in Figure 4b.
Approximately 24% of the dose (60% of the radioactivity in faeces) was accounted for by four metabolites: the 3-O-β,d-glucuronide Ro 61–1448 (13% of dose; 33% of radioactivity in faeces), the amine derivative Ro 61–3662 (6% and 14%, respectively), the amine glucuronide HU1 (3% and 8%, respectively) and an unknown metabolite (2% and 6%, respectively). Other metabolites did not account for more than 2% of the administered dose (alcohol metabolite 0.9%, acid metabolite 1.3%, N-acetylamino glucuronide 1.1%, amine sulphate 0.7%). Virtually no unchanged tolcapone was excreted in the faeces.
Discussion
The plasma concentration–time profile of tolcapone after oral administration is determined by rapid absorption from the gastrointestinal tract (tmax 2 h) and a relatively short t1/2,z of approximately 3 h. These characteristics were described in earlier pharmacokinetic studies [3–5, 10] and are confirmed here. Radioactivity measurements in plasma showed that, for the first 10 h after drug administration, tolcapone accounts for most of the drug-related material, whereas later, metabolites with slower elimination are responsible for the sustained excretion of total radioactivity. At 24 h after drug administration, on average 40% of drug-related material was excreted and, after 9 days, more than 95% of the radioactivity was recovered from all volunteers. Tolcapone is not eliminated intact but is extensively metabolized before excretion in the urine (60%) and faeces (40%).
The principal metabolic pathways for tolcapone are summarized in Figure 5. The biotransformation of tolcapone is confined exclusively to the periphery of the molecule with the benzophenone nucleus remaining unaltered. The primary metabolic pathways undergone by tolcapone in plasma are conjugative reactions involving glucuronidation and (to a lesser extent) methylation and oxidative hydroxylation and reduction reactions.
Figure 5.
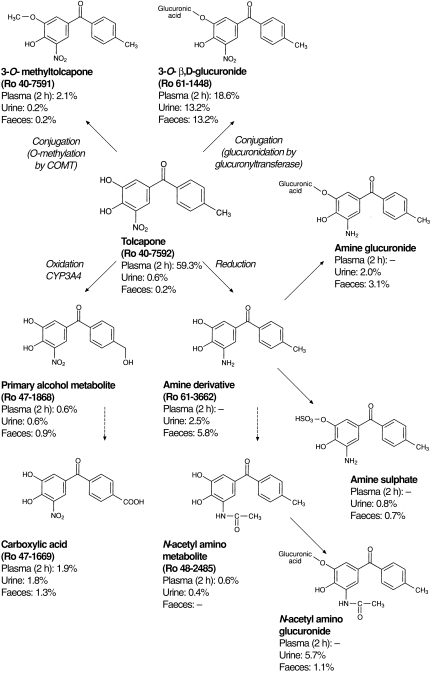
Potential metabolic pathways for tolcapone in humans. Tolcapone can undergo metabolism via conjugation catalysed by glucuronyltransferase or catechol-O-methyltransferase (COMT) or via oxidative or reductive metabolism and further conjugation of oxidized or reduced products.
The major pathway for tolcapone metabolism is direct glucuronidation by glucuronyltransferase at the hydroxyl group in position 3; more than 26% of the tolcapone dose is excreted in this form. The glucuronide is rapidly formed, as reflected by the early occurrence of this metabolite in plasma. At 2 h after drug administration, this metabolite accounted for the second largest fraction of drug-related material (19%), after tolcapone itself. Thereafter, the tolcapone glucuronide is quickly eliminated and excreted (approximately 50% via the kidney and 50% via the bile). The rapid appearance of the glucuronide in plasma may result from conjugation of tolcapone already in the intestinal wall; excretion of this conjugate into the bile and subsequent reabsorption from the gastrointestinal tract could explain the increase in plasma radioactivity seen approximately 12 h after administration of tolcapone. The alternative 4-O-glucuronidation of tolcapone has not been observed in our study, supporting the hypothesis that glucuronidation of nitrocatechols is highly regioselective in humans [13].
Several metabolites are derived from the amine derivative of tolcapone, which is produced from the reduction of the 5-nitro group. The amine can be excreted directly into urine (3% of dose) and faeces (6% of dose) or it can be metabolized further via glucuronidation (5% of dose), sulphation (1.5% of dose) or N-acetylation. Even though the N-acetylamino metabolite occurs in only very small amounts in urine, the evidence for this metabolic pathway is given by the occurrence of the N-acetylamino glucuronide in urine (6% of dose) and faeces (1% of dose). Neither the amine itself nor its sulphate or glucuronide conjugates have been identified in plasma; however, the N-acetylamino metabolite was measurable relatively late (12 h) in the general circulation.
The oxidative hydroxylation of the methyl group of tolcapone to a primary alcohol and subsequent oxidation to the carboxylic acid is a minor metabolic pathway for tolcapone. Neither metabolite contributes greatly to the radioactivity in plasma and less than 5% of the dose is excreted in either of these forms.
The last metabolic pathway, which should be mentioned even though it contributes to only a very small extent to the overall metabolism of tolcapone, is 3-O-methylation via COMT. This reaction reduces the polarity of the molecule and yields the long-lived metabolite 3-OMT (Ro 40–7591). This methylated derivative appeared in plasma within 4 h after administration of tolcapone and had become the principal plasma metabolite by 12 h after administration. Pharmacokinetic analysis showed that 3-OMT has a long t1/2,z, in agreement with previous studies [3–5, 10]. This t1/2,z determines the overall duration of excretion after a single administration. However, pharmacokinetic studies have shown that, 3-OMT does not accumulate after repeated administration, despite its long t1/2,z, because its formation is inhibited by tolcapone [4, 5]. Consequently, its long t1/2,z should not be of any clinical importance.
The present study gives an appropriate insight into the metabolism of the new COMT inhibitor tolcapone. Metabolites accounting for approximately 60% of the dose were identified in urine and faeces and more than 80% of the metabolites in plasma could be described. The remaining radioactivity was contained in a large number of small fractions, none exceeding 5% of the dose. We assume therefore that all principal metabolic pathways have been identified. The diversity of metabolites is based essentially on only a few phase I reactions, the products of which are multiplied by a large number of phase II reactions responsible for many of the minor peaks of radioactivity detected in urine and faeces. Moreover, the amine derivative is relatively unstable and may undergo spontaneous degradation, which could also account for some of these peaks.
On the basis of the pharmacokinetic–pharmacodynamic relationships derived for tolcapone, it has already been proposed that none of the metabolites contributes substantially to the pharmacological activity of tolcapone [4, 10]. This theory is further substantiated with the results from the present study. The two major plasma metabolites of tolcapone, the glucuronide and 3-OMT, are both conjugated at the catechol structure. Because this functional group is important in the binding of a compound to COMT, none of these metabolites is pharmacodynamically active. The metabolites resulting from oxidation or reduction can be active as inhibitors of COMT. However, these pathways are of minor importance, and the concentrations of the resulting metabolites remain too low for any substantial contribution to the action of tolcapone.
Since tolcapone is essentially completely metabolized prior to excretion it is reasonable to suspect that liver disease influences the elimination of the drug. As a consequence of the present results we undertook a study to investigate the effect of liver impairment on the pharmacokinetics of tolcapone and have shown that moderate liver cirrhosis results in a change of clearance of unbound drug leading to a recommendation of lower doses for patients suffering from this condition [14]. In contrast, based on the information obtained so far, renal impairment is not expected to have a major influence on tolcapone pharmacokinetics. Virtually no unchanged drug is excreted into urine and the major metabolite in urine is the inactive glucuronide. Renal impairment should not change the concentrations of tolcapone in the body, but could lead to a reduced renal elimination of the glucuronide. Since our study showed that the glucuronide could alternatively be excreted via the bile it appears unlikely that this metabolite will accumulate in patients with reduced renal function.
The metabolic profile of tolcapone as assessed in the current study predicts little potential for drug–drug interactions based on the metabolism. Oxidation contributes only to a small extent to the elimination of tolcapone and any inhibition or induction of the metabolizing enzymes should not have any effect on tolcapone pharmacokinetics. Whether or not tolcapone itself is an inhibitor or inducer of any cytochrome P450 isoenzyme needs to be addressed in a separate investigation. Clinically relevant competition for tolcapone’s major metabolic pathway with other drugs that are mainly glucuronidated appears unlikely, since the human body has a very high capacity for this conjugation reaction. It remains to be investigated, whether the inhibition of COMT by tolcapone leads to relevant pharmacokinetic interactions with drugs that are O-methylated by this enzyme such as apomorphine, α-methyldopa or dobutamine.
References
- 1.Reilly DK, Rivera-Calimlim L, van Dyke D. Catechol-O-methyltransferase activity: a determinant of levodopa response. Clin Pharmacol Ther. 1980;28:278–286. doi: 10.1038/clpt.1980.161. [DOI] [PubMed] [Google Scholar]
- 2.Fahn S. Adverse effects of levodopa in Parkinson’s disease. In: Calne DB, editor. Drugs for the Treatment of Parkinson’s Disease. Berlin, Germany: Springer-Verlag; 1989. pp. 385–409. [Google Scholar]
- 3.Dingemanse J, Jorga K, Zürcher G, et al. Pharmacokinetic–pharmacodynamic interaction between the COMT inhibitor tolcapone and single-dose levodopa. Br J Clin Pharmacol. 1995;40:253–262. doi: 10.1111/j.1365-2125.1995.tb05781.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dingemanse J, Jorga K, Zürcher G, et al. Multiple-dose clinical pharmacology of the catechol-O-methyl-transferase inhibitor tolcapone in elderly subjects. Eur J Clin Pharmacol. 1996;50:47–55. doi: 10.1007/s002280050068. [DOI] [PubMed] [Google Scholar]
- 5.Jorga KM, Sedek G, Fotteler B, Zürcher G, Nielsen T, Aitken JW. Optimizing levodopa pharmacokinetics with multiple tolcapone doses in the elderly. Clin Pharmacol Ther. 1997;62:300–310. doi: 10.1016/S0009-9236(97)90033-3. [DOI] [PubMed] [Google Scholar]
- 6.Jorga KM, Fotteler B, Schmitt M, Nielsen T, Zürcher G, Aitken J. The effect of COMT inhibition by tolcapone on tolerability and pharmacokinetics of different levodopa/benserazide formulations. Eur Neurol. 1997;38:59–67. doi: 10.1159/000112904. [DOI] [PubMed] [Google Scholar]
- 7.Sedek G, Jorga KM, Burns R, Schmitt M, Leese P. Effect of tolcapone on plasma levodopa levels after coadministration with levodopa/carbidopa to healthy volunteers. Clin Neuropharmacol. 1997;10:531–541. doi: 10.1097/00002826-199712000-00005. [DOI] [PubMed] [Google Scholar]
- 8.Borgulya J, Da Prada M, Dingemanse J, Scherschlicht R, Schläppi B, Zürcher G. Ro 40–7592: catecholamine-O-methyltransferase (COMT) inhibitor. Drugs Future. 1991;16:719–721. [Google Scholar]
- 9.Kurth MC, Adler CH, Saint Hilaire M, et al. Tolcapone improves motor function and reduces levodopa requirement in patients with Parkinson’s disease experiencing motor fluctuations: a multicenter, double-blind, randomized, placebo-controlled trial. Neurology. 1997;48:81–87. doi: 10.1212/wnl.48.1.81. [DOI] [PubMed] [Google Scholar]
- 10.Dingemanse J, Jorga KM, Schmitt M, et al. Integrated pharmacokinetics and pharmacodynamics of the novel catechol-O-methyltransferase inhibitor tolcapone during first administration to humans. Clin Pharmacol Ther. 1995;57:508–517. doi: 10.1016/0009-9236(95)90035-7. [DOI] [PubMed] [Google Scholar]
- 11.Heizmann P, Schmitt M, Leube J, Martin H, Saner A. Determination of the COMT-inhibitor tolcapone and three of its metabolites in animal and human plasma and urine by RP-HPLC with UV-detection. J Chromatogr. doi: 10.1016/s0378-4347(99)00146-2. submitted for publication. [DOI] [PubMed] [Google Scholar]
- 12.Gibaldi M, Perrier D. Pharmacokinetics. 2. New York: Marcel Dekker; 1982. [Google Scholar]
- 13.Wikberg T, Vuorela A, Ottoila P, Taskinen J. Identification of major metabolites of the catechol-O-methyltransferase inhibitor entacapone in rats and humans. Drug Metab Dispos. 1993;21:81–92. [PubMed] [Google Scholar]
- 14.Jorga KM, Kroodsma JM, Fotteler B, et al. The effect of liver impairment on the pharmacokinetics of tolcapone and its metabolites. Clin Pharmacol Ther. 1998;63:646–654. doi: 10.1016/S0009-9236(98)90088-1. [DOI] [PubMed] [Google Scholar]