

Abstract
Aims
The aims of this study were to describe paracetamol pharmacokinetics in neonates and infants.
Methods
Infants in their first 3 months of life (n = 30) were randomised to sequentially receive one of three paracetamol formulations (dose 30–40 mg kg−1) over a 2 day period. The formulations were (a) elixir, (b) glycogelatin capsule suppository and (c) triglyceride base suppository. Approximately six blood samples were taken after each dose over the subsequent 10–16 h. Data were analysed using a nonlinear mixed effect model. These neonatal and infant data were then included with data from four published studies of paracetamol pharmacokinetics (n = 221) and age-related pharmacokinetic changes investigated.
Results
Population pharmacokinetic parameter estimates and their coefficients of variation (CV%) for a one compartment model with first order input, lag time and first order elimination were volume of distribution 69.9 (18%) l and clearance 13.0 (41%) l h−1 (standardized to a 70 kg person). The volume of distribution decreased exponentially with a half-life of 1.9 days from 120 l 70 kg−1 at birth to 69.9 l 70 kg−1 by 14 days. Clearance increased from birth (4.9 l h−1 70 kg−1) with a half-life of 3.25 months to reach 12.4 l h−1 70 kg−1 by 12 months. The absorption half-life (tabs) for the oral preparation was 0.13 (154%) h with a lag time (tlag) of 0.39 h (31%). Absorption parameters for the triglyceride base and capsule suppositories were tabs 1.34 (90%) h, tlag 0.14 h (31%) and tabs 0.65 (63%) h, tlag 0.54 h (31%), respectively. The tabs for elixir and capsule suppository in children under 3 months were 3.68 and 1.51 times greater than children over 3 months. The relative bioavailability of rectal formulations compared with elixir were 0.67 (30%) and 0.61 (23%) for the triglyceride base and capsule suppositories, respectively.
Conclusions
Total body clearance of paracetamol at birth is 62% and volume of distribution 174% that of older children. A target concentration above 10 mg l−1 in approximately 50% subjects can be achieved by a dose from 45 mg kg−1 day−1 at birth and up to 90 mg kg−1 day−1 in 5-year-old children. A reduced dose of 75 mg kg−1 day−1 in an 8-year-old child is sufficient because clearance is a nonlinear function of weight.
Keywords: allometry, comparative bioavailability, NONMEM, paediatric, paracetamol, population pharmacokinetics
Introduction
Paracetamol (acetaminophen) elimination in neonates is all too frequently described only in terms of half-life [1–3], which is therefore confounded by volume of distribution. Levy et al. [3], in 1975 reported a half-life of 3.5 h based on serial urine collections analysed for paracetamol, paracetamol glucuronide, paracetamol sulphate and d-glucaric acid in 12 neonates aged 1 to 3 days old. More recently, Autret et al. [4] determined a similar half-life by studying an intravenous prodrug of paracetamol in five neonates less than 10 days old, as did Hopkins et al. [2], who studied neonates, infants and children given enteral paracetamol after cardiac surgery. There are no pharmaco-kinetic parameter estimates for enteral paracetamol in neonates in the literature. This paucity of information is reflected in clinical practice. A recent survey of paracetamol prescribing habits in our children's hospital [5] revealed 50% of practitioners either did not know safe doses or did not use paracetamol in the first 2 weeks of life.
We studied time-concentration profiles of paracetamol in neonates and young children in order to describe pharmacokinetics in this age group and to propose dosing regimens that achieve a target concentration [6] of 10 mg l−1.
Methods
The study protocol was approved by the Regional Health Authority Ethics Committee. Parental consent was obtained for each child. Neonates and infants in their first 3 months of life, admitted to a paediatric intensive care and requiring an arterial cannula for haemodynamic monitoring or frequent blood sampling, but without hepatic or renal disease, were eligible for entry into the study. Patients with a plasma bilirubin concentration above 150 µmol l−1 at the start of the study were excluded. Additional medications given during the study period included morphine, pancuronium, antibiotics, and diazepam. These drugs are not known to interfere with the paracetamol assay.
Subjects (n = 30) were randomised to sequentially receive one of three paracetamol formulations (dose 30–40 mg kg−1) over a 2 day period. The formulations were (a) oral elixir administered through a nasogastric feeding tube, (b) triglyceride base suppository or (c) glycogelatin capsule suppository. The oral elixir was supplied as a sugar free, alcohol free preparation with a standard strength of 250 mg 5 ml−1 (Wellcome, NZ). The suppositories were a paracetamol slurry (125 mg) contained in a glycogelatin capsule (Winthrop, Sterling Pharmaceuticals NZ Ltd) and the triglyceride base contained suspended paracetamol in a bullet shaped mould (25 and 50 mg doses). Nasogastric paracetamol was given 1 h before enteral feeding in those neonates receiving food.
Arterial blood samples were taken from heparinized arterial cannulae. Approximately six samples over 10–16 h were taken after each dose. Blood samples were taken hourly for the first 4 h after paracetamol administration, and then 2–4 hourly. The total amount of blood drawn was limited to 5 ml.
There are no published studies describing paracetamol pharmacokinetic changes with age. We investigated these changes by performing a pooled analysis of data from this current study combined with data from four previous published studies [2, 7–9] investigating paracetamol pharmacokinetics in children (n = 221). These data included three of our own studies [7–9] as well as data from a study by Hopkins et al. [2]. Our own studies used both the elixir and glycogelatin capsule suppository formulations. Sampling times in these previous studies were similar to those in the current study and few samples were taken in the first hour after dosing. Hopkins et al. [2] have reported time-concentration profiles in 28 febrile neonates, infants and children after cardiac surgery (n = 41). The elixir and triglyceride base suppository formulations were used. Observations were taken from time-concentration profiles in Figures 1 - 6 of their paper. Sampling was performed at 30, 60, 90, 120 min, 3, 4, 5, 6 and 8 h. Mean ages, weights and doses were taken from that paper's Table 1.
Figure 1.

Quality of fit of pharmacokinetic pooled analysis for all children over the study time. The y-axis of each panel displays the ratio of measured concentrations to those predicted from pharmacokinetic analysis. The upper panel 1a displays values from the population parameters. The lower panel 1b displays values from NONMEMs post hoc step based on values of the parameters for the specific individual. Subject 16 is shown as filled triangles to show the quality of the post hoc fit compared to the population fit.
Figure 6.

Simulation of paracetamol elixir dosing in 1000 1-month-old children given 25 mg kg−1 and then 15 mg kg−1 6 hourly. Children were given an age range of 0.9–1.1 months and a weight range of 3.5–4.5 kg. Variability is demonstrated using box and whisker plots. The central box represents the 50th centile. Indentations in this box indicate the median. Values outside the 97.5% centile are shown individually.
Table 1.
Characteristics of studied neonates and infants (n = 30).
| Geometric mean | %CV | Range | |
|---|---|---|---|
| Gestation at birth (weeks) | 38 | 6 | 31–40 |
| Postnatal age (days) | 7.3 | 164 | 1–90 |
| Weight (kg) | 3.5 | 25 | 2.5–6.8 |
| Morphine infusion | 17 of 30 neonates | ||
| Pathology | Respiratory illness (n = 12) | ||
| Neonatal surgery (n = 18) |
Paracetamol assay
Serum from each of the 30 neonates and infants was separated by centrifugation and stored at −20 °C until analysis. The stored samples were analysed batchwise, 17 samples at a time with three controls. Paracetamol was determined by fluorescence polarization immunoassay using the Abbott TDx (Abbott Laboratories, Abbott Park, IL 60064, USA) according to the manufacturer's instructions. All results were generated using the same calibration which was obtained using the calibrators supplied by Abbott Laboratories. The controls were Dade TDM (Baxter Diagnostics, Deerfield, IL 60015, USA). The Dade TDM level 1 and Dade TDM level 2 controls were used as supplied. The level 3 control was diluted 20 fold with blank serum. The mean values obtained for these controls were 55.9 (s.d. 2.4) mg l−1 (level 2), 30.2 (s.d. 1.4) mg l−1 (level 1) and 9.9 (s.d. 0.6) mg l−1 (diluted level 3). These are within the expected ranges quoted by the manufacturers. The between batch precision at these levels are 4.3%, 5.2% and 6.1%, respectively. The lower limit of quantification was 1.5 mg l−1 (CV 51%).
Pharmacokinetic modelling
A first order input with a lag time, first order elimination, one compartment disposition model was used to describe the time course of serum concentrations. The model was parameterized in terms of the absorption half-life (tabs, h), an absorption lag time (tlag, h), V/Foral is the apparent volume of distribution (l) and CL/Foral is the total body clearance (l h−1) after oral administration.
Paracetamol was administered as an extravascular dose and both clearance and distribution volume are confounded by bioavailability. Frectal/oral is used to refer to the relative bioavailability of the suppository compared with the oral formulation.
Population parameter estimations
Population parameter estimates were made using a nonlinear mixed effects model (NONMEM) [10]. This model accounts for both between subject and residual variability (random effects) as well parameter differences predicted by covariates (fixed effects). The between subject variability in model parameters was modelled by a proportional variance model. An additive term characterized the residual unknown variability (Err). This error model assumes that the residual variability is the same order of magnitude over the whole range of measurements. The population mean parameters, between subject variance and residual variance were estimated using the first order conditional estimate method using ADVAN 2 TRANS 2 of NONMEM V. The covariance between clearance, distribution volume and absorption half-time was incorporated into the model.
The between subject variability in NONMEM is modelled in terms of ETA (η) variables. Each of these variables is assumed to have mean 0 and a variance denoted by ω2, which is estimated. A covariance between two elements in η (e.g. CL and V) is a measure of statistical association between these two variables. Their covariance is related to their correlation (r) i.e.
Between occasion variability for the structural parameters of clearance and apparent volume were added to the model because each neonate received paracetamol on three different occasions. The between occasion covariance for clearance and volume was also estimated. This covariance is effected by factors that alter both clearance and volume together (e.g. protein binding, total body water), but variability of Felixir is the major contributor to this estimate.
In addition there were five different sources of data for the pooled population analysis. This between study variability was accounted for by giving each study its own separate residual error.
Covariate analysis
The parameter values were standardized for a body weight of 70 kg using an allometric model [11].
where Pi is the parameter in the ith individual, Wi is the weight in the ith individual and Pstd is the parameter in an individual with a weight Wstd of 70 kg. The PWR exponent was 0.75 for clearance, 1 for distribution volumes and 0.25 for elimination half time (t1/2) [12–15]. Once the final parameters in the pharmacokinetic model were estimated, the analysis was repeated using a Wstd of 15 kg (the mean weight of the sample population), in order to check if there was improved robustness of the estimate that depends on the choice of Wstd in relation to the actual weights in the sample.
The quality of fit of the pharmacokinetic model to the data was sought by NONMEM's objective function and by visual examination of plots of observed vs predicted concentrations. Models were nested (i.e. a full reduced set) and an improvement in the objective function was referred to the Chi-squared distribution to assess significance, e.g. an objective function change (OBJ) of 3.84 is significant at α = 0.05.
Covariate analysis included a model investigating age-related changes for clearance and volume of distribution:
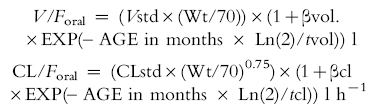 |
where Vstd and CLstd are the population estimates for V/Foral and CL/Foral, respectively, standardized to a 70 kg person using allometric models; βvol. and βcl are parameters estimating the fractional difference from Vstd and CLstd, respectively, at birth. tvol. and tcl describe the half-times of the age-related changes of V/Foral and CL/Foral.
The effect of age (above and below 3 months) was also investigated on relative bioavailability, absorption half-lives (tabs) and lag time (tlag) of the different formulations by applying a scaling factors (S elixir, S base, S cap) to each these parameters.
Simulation using parameter estimates
Pharmacokinetic simulations were performed in order to determine dosing regimens that would achieve trough concentrations of 10 mg l−1 in approximately 50% of simulated subjects. Paracetamol pharmacokinetic parameters and their coefficients of variation were used to predict time-concentration profiles (Pharsight Trial Designer, Version 1.2, Pharsight Corporation, Palo Alto, California) at ages 0, 1, 3, 6, 12 months, 5 and 8 years using a one compartment, first order input, first order elimination model. Pharmacokinetic parameter variability was assumed to have a log-normal distribution. Assumed average weights for these ages were 3.2, 4, 6, 7.5, 10, 18, and 30 kg, respectively. The weight for each child was sampled from a uniform distribution in the range (± 10%) associated with each group. An additive measurement error of 1.8 mg l−1 was incorporated into the model, with a minimal quantifiable concentration of 1.5 mg l−1.
Results
Thirty neonates were enrolled into the study. Sixteen were in their first 4 days of life and the remainders had postnatal ages that ranged from 2 weeks to 3 months. Characteristics of the studied neonates and infants are shown in Table 1. A total of 553 observations were collected for analysis. The mean infant and neonatal pharmacokinetic parameter estimates from NONMEM's first order conditional step (‘post hoc’ or ‘posterior individual’ estimates) for the final population model are shown in Table 2. These predictions from NONMEM's post hoc step are based on values of the pharmacokinetic parameters for the specific individual using the measured concentrations, rather than the typical (population) values of these parameters, which are based on covariate information.
Table 2.
Mean infant and neonatal parameter estimates taken from individual first order conditional estimates of the final population model (‘post hoc’ step of NONMEM).
| Parameter | Geometric Mean | %CV |
|---|---|---|
| V/Foral (l 70 kg−1) | 76.2 | 30 |
| CL/Foral (l h−1 70 kg−1) | 4.9 | 47 |
| Felixir | 1 fixed | |
| tabs elixir (h) | 0.54 | 132 |
| tlag elixir (h) | 0.39 | 20 |
| Frectal/oral triglyceride base | 0.73 | 18 |
| tabs triglyceride base (h) | 0.79 | 55 |
| tlag.triglyceride base (h) | 0.15 | 20 |
| Frectal/oral capsule | 0.63 | 13 |
| tabs.capsule (h) | 0.96 | 54 |
| tlag.capsule (h) | 0.55 | 20 |
CL/Foral = mean clearance after oral administration (l h−1 70 kg−1), V/Foral = mean volume of distribution l 70 kg−1), tabs = absorption half-life after nasogastric (elixir), triglyceride base suppository (base) and capsule suppository (cap) administration (h), tlag = absorption lag time after nasogastric (elixir), triglyceride base suppository (base) and capsule suppository (cap) administration (h), Frectal/oral = relative bioavailability of the rectal compared to the oral formulation.
The pooled analysis comprised 1653 observations from 221 children. These children had a mean age of 20 (CV% 236, range 1 day-15 years) months and weight of 15 (CV% 97, range 2.5–107) kg. Parameter estimates for this pooled analysis are shown in Table 3.Figure 1 demonstrates the quality of fit for pharmacokinetic data over the study time period – each subject's data is connected by a line. The weighted residuals (WRES) for each subject with values for each subject joined by vertical bars are shown in Figure 2. This plot gives a good indication of which patient's data deviated in a particular direction. The estimates from subject 16 have been highlighted in Figures 1 and 2. This subject deviates increasingly from the typical population estimate over time (Figure 1a); attributable to the low clearance (1.52 l h−1 70 kg−1) estimated in this individual. Figure 3 shows the time-concentration profile for subject 16. Post hoc estimates are in agreement with observations (Figures 1b and 3). The WRES for each predicted response are shown in Figure 4. In this graph responses are scaled to unit variance and within-subject correlations are minimized. The few outliers evident in this plot can be identified from Figure 2.
Table 3.
Standardized population pharmacokinetic parameter estimates (%CV is the coefficient of variation for the population parameter estimate, except for Vstd and CLstd, where between subject (BSV) and between occasion variability (BOV) were estimated; S.E. is the standard error of the structural parameter estimate, i.e. ‘stability' of the estimate).
| Parameter | Estimate | %CV | S.E |
|---|---|---|---|
| Vstd.(l 70 kg−1) | 69.9 | BSV 18 | 3.6 |
| BOV 24 | |||
| CLstd.(l h−1 70 kg−1) | 13.0 | BSV 41 | 0.8 |
| BOV 32 | |||
| Felixir | 1 fixed | ||
| tabs elixir.(h) | 0.13 | 154 | 0.02 |
| tlag elixir.(h) | 0.39 | 31 | 0.02 |
| Frectal/oral triglyceride base | 0.67 | 30 | 0.07 |
| tabs.triglyceride base (h) | 1.34 | 90 | 0.61 |
| tlag.triglyceride base (h) | 0.14 | 31 | 0.10 |
| Frectal/oral capsule | 0.61 | 23 | 0.04 |
| tabs.capsule (h) | 0.65 | 63 | 0.07 |
| tlag.capsule (h) | 0.54 | 31 | 0.03 |
CLstd = population estimate for CL/Foral (clearance after oral administration l h−1 70 kg−1), Vstd = population estimate for V/Foral (volume of distribution l 70 kg−1), tabs = absorption half-life after nasogastric (elixir), triglyceride base suppository and capsule suppository administration (h), tlag = absorption lag time after nasogastric (elixir), triglyceride base suppository and capsule suppository administration (h), Frectal/oral = relative bioavailability of the rectal compared with the oral formulation.
Figure 2.

The weighted residuals (WRES) for each subject with values for each subject joined by vertical bars. Subject 16 (filled triangles) can be seen to deviate in a positive direction.
Figure 3.

The time-concentration profile for subject 16. Post hoc estimates accurately mirror observations, but deviate from the population (typical) profile because of the low clearance in this individual.
Figure 4.

The weighted residuals (WRES) for the predicted population response.
The correlation of population parameter variability, introduced to increase the stability of the model, for CL/Foral and V/Foral and tabs was 0.17 and 0.04, respectively, while that between V/Foral and tabs was 0.03. There was little parameter interdependence. Changing the Wstd from 70 kg to the centred weight of 15 kg had no impact on parameter estimates. The CL/Foral, standardized to a 15 kg person, was 4.01 l h−1 while the V/Foral was 14.9 l. These estimates are proportional to those predicted using a Wstd of 70 kg.
Volume of distribution and clearance changes with age for the pooled analyses are shown in Figure 5. Addition of the covariate model for age-related change in V/Foral improved the OBJ by 271 (P < 0.001) while that for CL/Foral improved the OBJ by 370 (P < 0.001). The volume of distribution decreased exponentially (half-life 1.9 days) from 120 l 70 kg−1 at birth (1.74 times the eventual value) to 69.9 l 70 kg−1 by 14 days where it remained over the age span studied. Clearance increased exponentially (half-life 3.25 months) from birth (4.9 l h−1 70 kg−1) to reach 12.4 l h−1 70 kg−1 by 12 months. The covariate model for clearance (Table 4) predicts a mean clearance of 4.9, 6.4, 8.7, 10.7, 12.4 and 12.8 l h−1 70 kg−1 at birth, 1, 3, 6, 12 and 18 months, respectively. Clearance in an infant is 80% that of a 2-year-old by 6 months.
Figure 5.

Pharmacokinetic parameter changes with age for pooled analysis. Neonates and infants from current study are shown as open squares. The between occasion variability for each individual is demonstrated by linking estimates with a fine line. 5a Volume of distribution changes with age. Individual predicted volumes, standardized to a 70 kg person, from NONMEMs post hoc step are plotted against age. The solid line demonstrates the nonlinear relationship between volume of distribution and age. 5b Individual predicted clearances, standardized to a 70kg person, from NONMEMs post hoc step are plotted against age. The solid line demonstrates the nonlinear relationship between clearance and age.
Table 4.
Covariate models and estimates for pooled population parameters.
| (a) Volume of distribution | ||
| V/Foral = (Vstd × (Wt/70)) × (1 + βvol. × EXP(– AGE in months * Ln(2)/tvol)) l | ||
| (b) Clearance | ||
| CL/Foral = (CLstd × (Wt/70)0.75) × (1 + βcl × EXP(– AGE in months * Ln(2)/tcl)) l h−1 | ||
| (c) Absorption | ||
| Tabs elixir (infants) = S elixir × Tabs elixir (child) | ||
| Tabs base (infants) = S base × Tabs base (child) | ||
| Tabs cap (infants) = S cap × Tabs cap (child) | ||
| Parameter | Estimate | s.e. |
| βvol | 0.74 | 0.291 |
| Tvol | 1.9 days | 0.5 |
| βcl | −0.62 | 0.042 |
| tcl | 3.25 months | 1.4 |
| S elixir | 3.68 | 0.84 |
| S base | 0.40 | 0.14 |
| S cap | 1.51 | 0.31 |
βvol. and βcl are parameters estimating the fraction above or below V/Foral and CL/Foral, respectively, at birth; tvol. and tcl describe the half-times of the age-related changes of V/Foral and CL/Foral; S elixir, S base and S base are scaling factors applied to tabs for the elixir, triglyceride base and capsule suppository in infants 3 months and under.
The between occasion variability was 32% for clearance (ΔOBJ 53.36, P < 0.001) and 24% for the apparent volume (ΔOBJ 6.52, P < 0.02) and this variability can be seen in Figure 5a,b. Inclusion of this between occasion variability in the model had minimal effect on the clearance or apparent volume between subject variability (< 3% decrease in CV%). The residual errors for the different studies were 2.10 (current neonatal and infant study), 2.36 [8], 1.02 [7], 1.36 [9] and 0.92 mg l−1 [2]. Data from Hopkins et al. [2] were taken from the published figures and not tabulated results and consequently had potential for error in addition to the original error. However the residual error from Hopkin's data was, if anything, smaller than the residual error from our own data, suggesting that this was a negligible factor.
Scaling factors applied to the absorption half-lives in children 3 months and under improved the OBJ by 18.1 (P < 0.001). The absorption half-lives in children under 3 months were greater (elixir 368%, suppository 151%) than those of children over 3 months. We were unable to demonstrate any difference in the elixir tabs (t-test, P = 0.02) or tlag (t-test, P = 0.6) between those neonates given morphine and those not given morphine. The lag times and the relative bioavailability of suppositories in children under 3 months were similar to those of children over 3 months (OBJ 3.1 and 1.0, respectively, P = NS).
The final model incorporated the between occasion covariance for clearance and volume. This estimate of 25% reflects, to a large part, variability in the bioavailability of the elixir formulation. The addition of this parameter decreased the OBJ by 25 (P < 0.001). We were unable to estimate the standard errors of the structural model parameters in this final model. Final parameter estimates were, however, very close to those estimated in penultimate models and we have included the s.e. estimates from an earlier model in order to provide a measure of the stability of these structural parameter estimates.
Figure 6 shows the range of concentrations predicted in one month old infants given a loading dose of 25 mg kg−1 paracetamol elixir followed by 15 mg kg−1 6 hourly. There were 48.5%, 51.9% and 52.1% of simulated subjects with concentrations above 10 mg l−1 at the re-dosing times of 6 h, 12 h and 18 h. The dosing regimens which achieved similar time-concentration profiles for neonates, infants and children are shown in Table 5.
Table 5.
Dosing regimens that attain a target trough concentration of 10 mg l−1 in 50% of children.
| Loading dose (mg kg−1) | Maintenance dose (mg kg−1) | Dosing interval (h) | Total daily dose (mg kg−1 day−1) | |
|---|---|---|---|---|
| Oral dosing regimen | ||||
| New-born | 30 | 15 | 8 | 45 |
| 1 month | 25 | 15 | 6 | 60 |
| 3 months | 25 | 15 | 5 | 72 |
| 6 months | 25 | 15 | 4 | 90 |
| 12 months | 25 | 15 | 4 | 90 |
| 5 years | 25 | 15 | 4 | 90 |
| 8 years | 20 | 12.5 | 4 | 75 |
| Rectal dosing regimen | ||||
| New-born | 37.5 | 20 | 8 | 60 |
| 1 month | 30 | 20 | 6 | 80 |
| 3 months | 35 | 22.5 | 6 | 90 |
| 6 months | 35 | 17.5 | 4 | 105 |
| 12 months | 30 | 20 | 4 | 120 |
| 5 years | 30 | 20 | 4 | 120 |
| 8 years | 30 | 15 | 4 | 90 |
Discussion
Paracetamol prescribing practices in children have been determined not by pharmacokinetic-pharmacodynamic considerations, but by concerns about potential hepatotoxicity [16, 17]. Although the pharmacokinetics in children have been described [18, 19] there are few data concerning neonates and infants. This current paper describes paracetamol age-related pharmacokinetic changes from neonate through infant to child. A target concentration approach [6] using simulation was then used in order to rationalize dosing at different ages.
Size was the first covariate used in our analysis of the effects of age and weight. This deliberate choice was based on known biological principles. A great many physiological, structural and time related variables scale predictably within and between species with weight exponents of 0.75, 1 and 0.25, respectively [12]. We have used these ‘1/4 power models’ in this current study rather than centred weight, or some other function of weight, directly as a covariate in the model because the ‘1/4 power models’ have sound biological principles. West et al. [15] have used fractional geometry to mathematically explain this phenomenon. The 3/4 power law for metabolic rates was derived from a general model that describes how essential materials are transported through space-filled fractional networks of branching tubes [15]. These design principles are independent of detailed dynamics and explicit models and should apply to virtually all organisms [20]. By choosing weight as the primary covariate, the secondary effects of age could be investigated. We had no prior biological model for the effect of age on clearance or apparent volume, but assumed first order processes, which are common in biology.
Autret et al. [4], using an intravenous prodrug of paracetamol, under the assumption that 50% is converted to paracetamol, have reported a clearance of 0.149 l h−1 kg−1 in five neonates (weight 2.5 kg, s.d. 0.9) less than 10 days old but this does not help to predict clearances in older children because age and developmental changes are not known. We report a total body clearance (CL/Foral) in full term neonates of 4.9 (CV 38%) l h−1 70 kg−1 after enteral paracetamol. This clearance is 40% of those reported previously in children aged 2–15 years (CL/Foral 13.5, CV 46% L h−1 70 kg−1) [9] and in the current meta-analysis (13.0 l h−1 70 kg−1, CV 41%). Conversion of Autret et al.'s [4] estimates using allometric scaling reveals a clearance of 4.5 l h−1 70 kg−1 (0.149 l h−1 kg−1) in neonates and 14 l h−1 70 kg−1 (0.365 l h−1 kg−1) in infants (assuming a mean weight of 7 kg). This increase in size standardized clearance with age is similar in our results and those of Autret et al. [4]. We were unable to demonstrate an increase of clearance with age using our neonatal and infant data alone because the majority of neonates (16 of 30 subjects) were in their first 4 days of life. There were only 14 remaining neonates and infants younger than 3 months, where we might expect increased clearance. The pooled analysis showed an increase in clearance over the first year of an infant's life, reaching 80% of that seen in a child by 6 months.
These age-related clearance changes are consistent with immaturity of some of the metabolic pathways responsible for paracetamol clearance in neonates and infants. Sulphate conjugation is pronounced in neonates while glucuronide conjugation is deficit [1, 3]. The functional maturation profile of paracetamol clearance is similar to that of morphine, which is also metabolized by sulphate and glucuronide conjugation. Morphine clearance in infants at 6 months was approximately 80% of that described in adults when clearance was standardized for size [21, 22]. The relative contribution of sulphate and glucuronide conjugation change with age. The usual adult ratio of 2 : 1 glucuronide to sulphate conjugates of acetaminophen is reached by the age of 12 years [1, 3].
The population distribution volume of 69.9 (18%) l 70 kg−1 was similar to those reported in adults (56–70 l 70 kg−1) [23]. The volume of distribution for paracetamol in mammals [23], including humans, is 0.7–1.0 l kg−1, as we would expect from the allometric size model with a power function of 1. The volume of distribution was 1.7 times as large at birth, but decreased exponentially to 69.9 l 70 kg−1 by 2 weeks of age. This dramatic change in distribution volume probably reflects neonatal body composition and the rapid changes in body water distribution in early life. Paracetamol has a low molecular weight and reasonable lipid solubility [24]. Paracetamol crosses cell membranes easily distributes throughout all tissues and fluids [25], but does not bind to plasma proteins. Tissue:plasma concentration ratios are close to unity at equilibrium [23].
Absorption half-lives for both oral and capsule suppository formulations were prolonged in infants under three months compared to those seen in older children. The absorption half-life of nasogastric paracetamol has been reported in children given oral elixir to be as low as 0.04 h [9, 18]. The absorption half-life reported in adult series after oral administration ranges from 0.06 to 0. 6 h [23]. Paracetamol absorption depends on gastric emptying and gastric emptying is slow and erratic in the neonate [26]. Normal adult rates may not be reached until 6–8 months [27]. Gastric pH is near neutral in neonates. This increases the unionized form available for absorption for paracetamol, which has a pKa of 9.5 [28]. The stomach, however, is a poor route of entry to the circulation. The increased surface area, blood flow and permeability of the small intestine favour the intestinal route. Most of the neonates who presented with surgically correctable lesions received morphine infusions for analgesia. Morphine decreases gastric emptying [29], but we were unable to demonstrate a decreased elixir Tabs in those infants and neonates on morphine infusions when compared to those not receiving morphine. Absorption of the capsule suppository was also slow in neonates compared to older children. This may reflect the smaller surface area available for absorption. Although we have estimated that the tabs of the triglyceride suppository in infants under 3 months is lower than in older children (S base 0.40, s.e. 0.14), we have less confidence in this estimate. There were only nine children in the older age bracket given a triglyceride suppository and these subjects were all from the study of Hopkins et al. [2].
The relative bioavailability of the suppositories (Frectal/oral ≈ 0.64) compared with the elixir in infants under 3 months was similar to children. This relative bioavailability is higher than our previous estimate (Frectal/oral 0.52 for capsule suppository) [9] but lower than that reported by Coulthard et al. [30] (0.78, 95% CI 55 101) for triglyceride base and capsule suppositories in children. Paracetamol has low first pass metabolism and the hepatic extraction ratio is 0.11–0.37 in adults [31]. It has been suggested that the relative bioavailability of suppositories in the neonatal age group is higher than older children because suppository insertion depth may result in a different rectal venous drainage pattern [32]. The rectal route has the potential to partially reduce first pass hepatic loss by draining into the inferior and middle haemorrhoidal veins. Bioavailability strongly depends on site of absorption within the rectum [33, 34]. The impact of the rectal venous drainage pattern is questionable, given the degree of rectal venous anastomotic channels, slow absorption times and natural attrition from the rectum. We were unable to show an increased rectal bioavailability in infants 3 months and under.
The pharmacokinetic model used a first order input model with a lag time to describe absorption. The lag time for the triglyceride base was small (0.14 h, s.e 0.1) compared with the capsule suppository (0.54 h, s.e. 0.03). The standard error (‘stability’) of this parameter estimate for the base was high. Birmingham et al. [35] have reported a first order input model with a zero order dissolution time to describe absorption characteristics of a triglyceride base suppository in children. We were unable to explore this alternative absorption model in our study as we had fewer observations than in Birmingham's study for the first hour after rectal administration. The glycogelatin capsule suppository contains an paracetamol slurry that is released rapidly once the capsule wall is breached and consequently a lag time absorption model may have greater physiological relevance, although this was not investigated.
Autret et al. [4] separated infants in their study into those older and those younger than 10 days and suggested different dosing regimens for the two groups −30 mg kg−1 day−1 for those infants less than 10 days and 60 mg kg−1 day−1 for children aged over 10 days. This categorization fails to account for the gradual increase in clearance over the first year of life or the lower relative bioavailability of rectal formulations. In addition, the larger volume of distribution in the new-born means a larger initial dose is required to reach the target concentration of 10 mg l−1.
A target concentration of 10 mg l−1 is the steady-state concentration which is reported to reduce temperature to 50% of the maximum possible temperature reduction in febrile children [8] and is also the effect site concentration associated with pain scores less than 6/10 in 90% of children after tonsillectomy [9]. The dosing schedules proposed in Table 5 result in approximately 50% of simulated subjects having concentrations above 10 mg l−1 at trough. The daily elixir doses predicted using the target concentration approach in this current study are the same as those recommended by others who used potential hepatotoxicity as the primary determinant of dose [17, 36].
Penna & Buchanan [37] reported 7 deaths and 11 cases of hepatotoxicity associated with paracetamol. Survival was usually seen in those children suffering hepatotoxicity due to paracetamol greater than 150 mg day−1 kg−1 for 2 to 8 days [37]. However, hepatotoxicity has been reported with doses above 75 mg kg−1 day−1 in children and 90 mg kg−1 day−1 in infants [17, 36, 38, 39]. Hepatotoxicity from prolonged use of paracetamol in neonates is also possible. Neonates are also capable of forming hepatotoxic metabolites, despite a relatively low level of activity of the cytochrome P-450 CYP2E1 enzyme system [40, 41]. It is currently impossible to predict which individuals have an enhanced susceptibility to cellular injury from paracetamol. The toxic metabolite of paracetamol, N-acetyl-p-benzoquinone imine (NAPQI) is formed by the cytochrome P450s CYP2E1, 1A2 and 3A4 [42]. The coingestion of therapeutic drugs, foodstuffs or other xenobiotics has potential to induce these enzymes [17]. The influence of disease on paracetamol toxicity is unknown. Consequently even the proposed regimens may cause hepatotoxicity in some individuals if used for longer than 2–3 days [17].
Acknowledgments
This work was supported in part by grants from ‘The Children’s Pain Trust' and ‘SmithKline Beecham Pharmaceuticals’. Dr Holford is a member of the Scientific Advisory Board for Pharsight and is a stockholder in the company.
References
- 1.Miller RP, Roberts RJ, Fischer LT. Acetaminophen elimination kinetics in neonates, children and adults. Clin Pharmacol Ther. 1976;19:284–294. doi: 10.1002/cpt1976193284. [DOI] [PubMed] [Google Scholar]
- 2.Hopkins CS, Underhill S, Booker PD. Pharmacokinetics of paracetamol after cardiac surgery. Arch Dis Child. 1990;65:971–976. doi: 10.1136/adc.65.9.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levy G, Khanna NN, Soda DM, Tsuzuki O, Stern L. Pharmacokinetics of acetaminophen in the human neonate; formation of acetaminophin glucuronide and sulfate in relation to plasma bilirubin concentration and d-glucoric acid excretion. Pediatrics. 1975;55:818–825. [PubMed] [Google Scholar]
- 4.Autret E, Dutertre JP, Breteau M, Jonville AP, Furet Y, Laugier J. Pharmacokinetics of paracetamol in the neonate and infant after administration of propacetamol chlorhydrate. Dev Pharmacol Ther. 1993;20:129–134. doi: 10.1159/000457553. [DOI] [PubMed] [Google Scholar]
- 5.Anderson B, Anderson M, Hastie B. Paracetamol prescribing habits in a children's hospital. N Z Med J. 1996;109:376–378. [PubMed] [Google Scholar]
- 6.Holford NH. The target concentration approach to clinical drug development. Clin Pharmacokin. 1995;29:287–291. doi: 10.2165/00003088-199529050-00001. [DOI] [PubMed] [Google Scholar]
- 7.Anderson BJ, Woolard GA, Holford NH. Pharmacokinetics of rectal paracetamol after major surgery in children. Paediatr Anaesth. 1995;5:237–242. doi: 10.1111/j.1460-9592.1995.tb00291.x. [DOI] [PubMed] [Google Scholar]
- 8.Anderson BJ, Holford NH, Woollard GA, Chan PL. Paracetamol plasma and cerebrospinal fluid pharmacokinetics in children. Br J Clin Pharmacol. 1998;46:237–243. doi: 10.1046/j.1365-2125.1998.00780.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anderson BJ, Holford NH, Woollard GA, Kanagasundaram S, Mahadevan M. Perioperative pharmacodynamics of acetaminophen analgesia in children. Anesthesiology. 1999;90:411–421. doi: 10.1097/00000542-199902000-00014. [DOI] [PubMed] [Google Scholar]
- 10.Sheiner LB, Beal SL. NONMEM Users Guide. San Francisco: Division of Pharmacology: University of California; 1979. [Google Scholar]
- 11.Holford NHG. A size standard for pharmacokinetics. Clin Pharmacokin. 1996;30:329–332. doi: 10.2165/00003088-199630050-00001. [DOI] [PubMed] [Google Scholar]
- 12.Peters HP. Physiological correlates of size. In: Beck E, Birks HJB, Conner EF, editors. The ecological implications of body size. Cambridge: Cambridge University Press; 1983. pp. 48–53. Chapter 4. [Google Scholar]
- 13.Prothero JW. Scaling of blood parameters in animals. Comp Biochem Physiol. 1980;A67:649–657. [Google Scholar]
- 14.Gabrielsson J, Weiner D. Interspecies scaling. In: Gabrielsson J, Weiner D, editors. Pharmacokinetic and pharmacodynamic data analysis. Stockholm: Swedish Pharmaceutical Press Ltd; 1994. pp. 153–171. [Google Scholar]
- 15.West GB, Brown JH, Enquist BJ. A general model for the origin of allometric scaling laws in biology. Science. 1997;276:122–126. doi: 10.1126/science.276.5309.122. [DOI] [PubMed] [Google Scholar]
- 16.Shann F. Paracetamol: when, why and how much. J Paediatr Child Health. 1993;29:84–85. doi: 10.1111/j.1440-1754.1993.tb00454.x. [DOI] [PubMed] [Google Scholar]
- 17.Kearns GL, Leeder JS, Wasserman GS. Acetaminophen overdose with therapeutic intent. J Pediatr. 1998;132:5–8. doi: 10.1016/s0022-3476(98)70476-7. [DOI] [PubMed] [Google Scholar]
- 18.Brown RD, Wilson JT, Kearns GL, Eichler VF, Johnson VA, Bertrand KM. Single-dose pharmacokinetics of ibuprofen and acetaminophen in febrile children. J Clin Pharmacol. 1992;32:231–241. doi: 10.1002/j.1552-4604.1992.tb03831.x. [DOI] [PubMed] [Google Scholar]
- 19.Wilson JT, Brown RD, Kearns GL, et al. Single-dose, placebo-controlled comparative study of ibuprofen and acetaminophen antipyresis in children. J Pediatr. 1991;119:803–811. doi: 10.1016/s0022-3476(05)80307-5. [DOI] [PubMed] [Google Scholar]
- 20.West GB, Brown JH, Enquist BJ. The fourth dimension of life: fractal geometry and allometric scaling of organisms. Science. 1999;284:1677–1679. doi: 10.1126/science.284.5420.1677. [DOI] [PubMed] [Google Scholar]
- 21.Pokela ML, Olkkola KT, Seppala T, Koivisto M. Age-related morphine kinetics in infants. Dev Pharmacol Ther. 1993;20:26–34. doi: 10.1159/000457538. [DOI] [PubMed] [Google Scholar]
- 22.Anderson BJ, McKee AD, Holford NH. Size, myths and the clinical pharmacokinetics of analgesia in paediatric patients. Clin Pharmacokin. 1997;33:313–327. doi: 10.2165/00003088-199733050-00001. [DOI] [PubMed] [Google Scholar]
- 23.Prescott LF. Paracetamol (Acetaminophen) A Critical Bibliographic Review. 1. London: Taylor & Francis Publishers; 1996. [Google Scholar]
- 24.van Bree JB, de Boer AG, Danhof M, Ginsel LA, Breimer DD. Characterization of an ‘in vitro’ blood–brain barrier: effects of molecular size and lipophilicity on cerebrovascular endothelial transport rates of drugs. J Pharmacol Exp Ther. 1988;247:1233–1239. [PubMed] [Google Scholar]
- 25.Ochs HR, Greenblat DJ, Abernathy DR, et al. Cerebrospinal fluid uptake and peripheral distribution of centrally acting drugs: relation to lipid solubility. J Pharm Pharmacol. 1985;37:428–431. doi: 10.1111/j.2042-7158.1985.tb03030.x. [DOI] [PubMed] [Google Scholar]
- 26.Gupta M, Brans Y. Gastric retention in neonates. Pediatrics. 1978;62:26–29. [PubMed] [Google Scholar]
- 27.Grand RJ, Watkins JB, Torti FM. Development of the human intestinal tract: a review. Gastroenterology. 1976;70:790–810. [PubMed] [Google Scholar]
- 28.Forrest JA, Clements JA, Prescott LF. Clinical pharmacokinetics of paracetamol. Clin Pharmacokin. 1982;7:93–107. doi: 10.2165/00003088-198207020-00001. [DOI] [PubMed] [Google Scholar]
- 29.Nimmo WS, Heading RC, Wilson J, Tothill P, Prescott LF. Inhibition of gastric emptying and drug absorption by narcotic analgesics. Br J Clin Pharmacol. 1975;2:509–513. doi: 10.1111/j.1365-2125.1975.tb00568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coulthard KP, Nielson HW, Schroder M, et al. Relative bioavailability and plasma paracetamol profiles of Panadol suppositories in children. J Paediatr Child Health. 1998;34:425–431. doi: 10.1046/j.1440-1754.1998.00267.x. [DOI] [PubMed] [Google Scholar]
- 31.Rawlins MD, Henderson BD, Hijab AR. Pharmacokinetics of paracetamol (acetaminophen) after intravenous and oral administration. Eur J Clin Pharmacol. 1977;11:283–286. doi: 10.1007/BF00607678. [DOI] [PubMed] [Google Scholar]
- 32.van Hoogdalem EJ, de Boer AG, Breimer DD. Pharmacokinetics of rectal drug administration, Part II. Clinical applications of peripherally acting drugs, and conclusions. Clin Pharmacokin. 1991;21:110–128. doi: 10.2165/00003088-199121020-00003. [DOI] [PubMed] [Google Scholar]
- 33.Moolenar F, Schoonen AJM, Everts A, Huizinga T. Absorption rate and bioavailability from fatty suppositories. Pharmaceutisch Weekblod Sci Edition. 1979;112:89–94. [Google Scholar]
- 34.Seideman P, Alvan G, Andrews RS, Labross A. Relative bioavailability of a paracetamol suppository. Eur J Clin Pharmacol. 1980;17:465–468. doi: 10.1007/BF00570165. [DOI] [PubMed] [Google Scholar]
- 35.Birmingham PK, Tobin MJ, Henthorn TK, et al. Twenty-four-hour pharmacokinetics of rectal acetaminophen in children: an old drug with new recommendations. Anesthesiology. 1997;87:244–252. doi: 10.1097/00000542-199708000-00010. [DOI] [PubMed] [Google Scholar]
- 36.Heubi JE, Bien JP. Acetaminophen use in children: more is not better. J Pediatr. 1997;130:175–177. [PubMed] [Google Scholar]
- 37.Penna A, Buchanan N. Paracetamol poisoning in children and hepatotoxicity. Br J Clin Pharmacol. 1991;32:143–149. doi: 10.1111/j.1365-2125.1991.tb03873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heubi JE, Barbacci MB, Zimmerman HJ. Therapeutic misadventures with acetaminophen: hepatoxicity after multiple doses in children. J Pediatr. 1998;132:22–27. doi: 10.1016/s0022-3476(98)70479-2. [DOI] [PubMed] [Google Scholar]
- 39.Rivera Penera T, Gugig R, Davis J, et al. Outcome of acetaminophen overdose in pediatric patients and factors contributing to hepatotoxicity. J Pediatr. 1997;130:300–304. doi: 10.1016/s0022-3476(97)70359-7. [DOI] [PubMed] [Google Scholar]
- 40.Warner A. Drug use in the neonate: interrelationships of pharmacokinetics, toxicity, and biochemical maturity. Clin Chem. 1986;32:721–727. [PubMed] [Google Scholar]
- 41.Greene JW, Craft L, Ghishan F. Acetaminophen poisoning in infancy. Am J Dis Child. 1983;137:386–387. doi: 10.1001/archpedi.1983.02140300064018. [DOI] [PubMed] [Google Scholar]
- 42.Slattery JT, Nelson SD, Thummel KE. The complex interaction between ethanol and acetaminophen. Clin Pharmacol Ther. 1996;60:241–246. doi: 10.1016/S0009-9236(96)90050-8. [DOI] [PubMed] [Google Scholar]