

Abstract
Aims
To study the population pharmacokinetics of thioTEPA and its main metabolite TEPA in patients receiving high-dose chemotherapy consisting of thioTEPA (80–120 mg m−2 day−1), cyclophosphamide (1000–1500 mg m−2 day−1) and carboplatin (265–400 mg m−2 day−1) for 4 days.
Methods
ThioTEPA and TEPA kinetic data were processed with a two-compartment model using the nonlinear mixed effect modelling program NONMEM. Interindividual variability (IIV), interoccasion variability (IOV) and residual variability in the pharmacokinetics were estimated. The influence of patient characteristics on the pharmacokinetics was also determined.
Results
A total number of 40 patients receiving 65 courses of chemotherapy was included. Clearance of thioTEPA (CL) was 34 l h−1 with an IIV and IOV of 18 and 11%, respectively. The volume of distribution of thioTEPA was 47 l (IIV = 7.5%; IOV = 19%). The fraction of thioTEPA converted to TEPA divided by the volume of distribution of TEPA was 0.030 l−1 (IIV = 39%; IOV = 32%) and the elimination rate constant of TEPA was 0.64 h−1 (IIV = 27%; IOV = 32%). CL of thioTEPA was correlated with alkaline phosphatase and serum albumin. The volume of distribution of thioTEPA and the elimination rate constant of TEPA were correlated with total protein levels and body weight, respectively.
Conclusions
A model for the description of the pharmacokinetics of thioTEPA and TEPA was developed. Factors involved in the interpatient variability of thioTEPA and TEPA pharmacokinetics were identified. Since, IOV of both thioTEPA and TEPA was equal to or smaller than IIV, therapeutic drug monitoring based on data of previous courses may be meaningful using this population model.
Keywords: high-dose chemotherapy, pharmacokinetics, thioTEPA
Introduction
ThioTEPA (Figure 1) is an alkylating agent developed in the 1950s [1]. Interest in the drug was renewed with the finding that the dose can be increased dramatically when combined with peripheral blood progenitor cell or bone marrow transplantation. ThioTEPA is currently being used in many high-dose chemotherapy combination regimens [2], one of the most frequently used being thioTEPA, cyclophosphamide and carboplatin for solid tumours [2].
Figure 1.

Chemical structures of thioTEPA and its main metabolite TEPA.
ThioTEPA is rapidly metabolized by cytochrome P450 to its main metabolite TEPA (Figure 1), which shows a comparable alkylating activity with the parent drug [3–6]. This conversion is mediated by CYP2B and 2C [7, 8]. Very recently, two other alkylating metabolites of thioTEPA have been identified in the urine of patients treated with thioTEPA. Conversion of the aziridinyl function of TEPA into a β-chloroethyl group results in the formation of monochloroTEPA and glutathione conjugation of thioTEPA with subsequent loss of two amino acid residues results in the formation of thioTEPA-mercapturate. The clinical importance of these metabolites needs to be assessed in future studies [9]. It is likely that other metabolites are also formed, since the alkylating activity of urine of patients treated with thioTEPA is higher than can be explained from concentrations of thioTEPA and its metabolites alone [9, 10].
In contrast to TEPA, which is extensively protein bound, less than 15% of thioTEPA is bound to plasma proteins [11, 12]. The pharmacokinetics of thioTEPA and/or TEPA in conventional dose regimens both in adult and paediatric patients have been investigated in many studies [10, 13–21]. However the pharmacokinetics of thioTEPA in high-dose regimens in combination with haemato-logical support has been the subject of only a few studies [22–26] with only one presenting data on the pharmacokinetics of both thioTEPA and TEPA in such a regimen [27]. Relationships between pharmacokinetics of the drug and its toxicity have been described. Przepiorka et al. found a relationship between the sum of the area under the curves (AUC) of thioTEPA and TEPA and TEPA peak concentrations > 10 µm (1.75 µg ml−1) with the occurrence of major nonhaematopoietic toxicity in a high-dose regimen [27]. In conventional dose regimens correlations between thioTEPA AUC and haematological toxicity have also been described [16, 18, 19]. Furthermore, a relationship between thioTEPA AUC and mucositis was observed during treatment with high-dose thioTEPA, cisplatin and cyclophosphamide [26]. The large interpatient variability in the pharmacokinetics of thioTEPA found and the established relationships between its pharmacokinetics and pharmacodynamics justify pharmacokinetic guided dosing of thioTEPA. Dose individualization can either be based on pharmacokinetic parameters determined during previous courses or treatment days, on relevant patient characteristics or on a combination of both.
Accordingly the significant relationships between patient demographics and the pharmacokinetics of both thioTEPA and TEPA may thus facilitate pharmacokinetic guided dosing. Przepiorka et al. demonstrated that the volume of distribution (V) of thioTEPA was correlated with body weight and that dosing based on adjusted body weight resulted in less toxicity in obese patients [27]. No other relationships between patient characteristics and pharmacokinetics have been described thus far.
The aim of this study was to develop population pharmacokinetic models for thioTEPA and TEPA and to identify the relationships between their pharmacokinetics and patient characteristics. Moreover, interpatient and interoccasion variability in the pharmacokinetics of thioTEPA and TEPA were estimated.
Methods
Patients and treatment
Patients were entered in high-dose chemotherapy protocols with peripheral blood progenitor cell transplantation. They either had high-risk primary breast cancer and received high-dose chemotherapy as part of their adjuvant treatment, or had advanced breast, germ-cell or ovarian cancer. Recruited patients were below 60 years of age and had a good performance status (WHO 0 or 1) [28]. They had undergone no previous chemotherapy, unless it had been limited to nonanthracyclin-based adjuvant therapy at least 1 year before relapse. Before the first course of high dose chemotherapy all patients had adequate renal (creatinine clearance > 60 ml min−1) and hepatic (bilirubin < 20 µmol l−1, ALAT and ASAT < 1.5 times the upper limit of normal) functions. Each patient received induction and stem cell mobilization chemotherapy supported with G-CSF (filgrastim) prior to high-dose chemotherapy as described in detail previously [29].
Three weeks after the last mobilization course, the first high-dose course was started. Two regimens, CTC and tCTC, were used. The CTC regimen consisted of cyclophosphamide 1500 mg m−2 as a daily 1 h infusion immediately followed by 400 mg m−2 carboplatin as a daily 1 h infusion and 60 mg m−2 thioTEPA as twice daily 30 min infusions on 4 consecutive days. The tCTC regimen was identical to the CTC regimen except that the dose of each drug was two thirds of those of the latter. Patients received either one or two courses of CTC or multiple courses tCTC, every 4 weeks when possible. Treatment delays and dose adaptations were implemented as described previously [29]. Before a second or third course of tCTC was started, it was ensured that creatinine clearance was greater than 40 ml min−1 and loss of renal function as determined by the 24 h creatinine clearance was less than 20% of the baseline estimate. Furthermore, the hepatic function had to be demonstrated (bilirubin < 20 µmol l−1, ALAT and ASAT < 2 times the upper limit of normal).
MESNA (500 mg) was administered 6 times daily for a total of 36 doses, beginning 1 h prior to the first cyclophosphamide infusion. All patients received anti-emetics both prophylactically and as indicated, which usually included dexamethasone and granisetron. Patients received prophylactic antibiotics, including ciprofloxacin, itraconazole and acyclovir orally, starting 4 days before chemotherapy. Approximately 60 h after the last thioTEPA infusion, the peripheral blood progenitor cells were reinfused.
This study was approved by the Committee on Medical Ethics of the Netherlands Cancer Institute and written informed consent was obtained from all patients.
Sampling procedure and analytical methods
Blood samples were collected from a double lumen Hickman or a normal double lumen catheter inserted in a subclavian vein. The lumen ending most downstream was used for administration of the drugs, and the proximal lumen was used for the collection of the blood samples [30].
Samples were collected prior to the start of the infusions on all days of chemotherapy, and on day 1, day 2, 3, or 4, at 30 min after the start of the cyclophosphamide infusion (t = 30) and at t = 60 (the end of the cyclophosphamide infusion), 90, 120 (end of carboplatin infusion), 150 (the end of the thioTEPA infusion), 165, 180, 210, 285, 390 and 660 min. On day 5 an additional sample was collected approximately 22 h after the last cyclophosphamide infusion.
Samples were immediately placed on ice, plasma was separated by centrifuging the sample at 3000 g for 3 min at 4 °C. The plasma layer was collected and the sample was stored at −70 °C until analysis. ThioTEPA and TEPA were quantified using a validated gas chromatographic assay after liquid-liquid extraction with chloroform [31]. Accuracy, within-day and between-day precision were less than 10% for this assay. For thioTEPA the limit of detection (LOD) and the lower limit of quantification (LLQ) were 0.011 and 0.026 µm, respectively. For TEPA these values were 0.017 and 0.029 µm, respectively.
Pharmacokinetics
Data were analysed using the nonlinear mixed effect modelling software program NONMEM (version V, level 1.0, double precision) [32]. The first-order (FO) estimation procedure was used throughout. Both thioTEPA and TEPA data were described with an open two-compartment model with first-order elimination from the central compartment. The pharmacokinetic model used is schematically depicted in Figure 2. ThioTEPA and TEPA concentration data were expressed as µm, doses were expressed in µmol and time in h. ThioTEPA and TEPA data were fitted simultaneously using the NONMEM subroutine ADVAN6, which makes use of differential equations to describe the rate of mass transport between the different compartments. The differential equations used are described in equations 1–4.
Figure 2.

Schematic representation of the basic pharmacokinetic model.
| (1) |
| (2) |
| (3) |
| (4) |
In which A(n) and Vn describe the amount in and the volume of the nth compartment, respectively, kij represents the microconstant describing the mass transport between the ith and jth compartment, CL is the clearance of thioTEPA, fm is the fraction of the thioTEPA dose converted to TEPA and k is the elimination rate constant of TEPA.
Since the volume of distribution of TEPA is unknown, calculating with the amount of TEPA is impossible. Therefore, TEPA data were modelled with V3 = 1, whereafter fm, k34 and k43 can be calculated. The estimated fraction fm is, however, interpreted as the ratio of the fraction thioTEPA converted to TEPA and V3 and this ratio will be denoted fm*.
The AUC of thioTEPA and TEPA can be calculated from the empirical Bayesian estimates using equation 5 and 6.
| (5) |
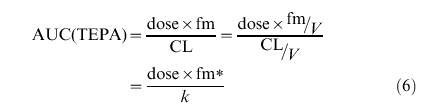 |
(6) |
The distribution and elimination half-life of thioTEPA were calculated from CL, V1, k12 and k21 using standard pharmacokinetic formulas [33].
Interindividual variability in the different pharmaco-kinetic parameters was estimated using a proportional error model. Variability in the pharmacokinetics of an individual during various courses (interoccasion variability) was also estimated using a proportional model as suggested by Karlsson & Sheiner [31]. For instance, variability in CL was estimated using CLij = θ1*exp(ηi + κj), in which CLij represents the CL of the ith individual on the jth occasion, θ1 is the population value of CL, η is the interindividual random effect with mean 0 and variance ω2 and κ is the interoccasion random effect with mean 0 and variance π2. Interindividual variability was estimated on all parameters and interoccasion variability was estimated for CL, V1, fm* and k. Residual variability was estimated with a combined proportional and additive error model assuming a constant coefficient of variation over the complete concentration range superimposed on a constant absolute error. Residual variability was calculated for both thioTEPA and TEPA. The performance of the model tested was judged using both statistical and graphical methods. The minimal value of the objective function as calculated by NONMEM was one of the parameters used to assess the goodness-of-fit. The minimal value of objective function is equal to minus twice the log likelihood of the data. An increase in goodness-of-fit is accompanied by a decrease of the objective function and this decrease is asymptotically distributed as a χ2-distribution. Significance of increase in goodness-of-fit was tested using the likelihood ratio test. Furthermore, standard errors for all parameters were calculated using the COVARIANCE option of NONMEM. For graphical model diagnostics the S-plus (MathSoft Inc, Seattle, USA) based model building aid Xpose 2.0 [34] was used.
Development population pharmacokinetic model including covariables
In order to establish possible relationships between the pharmacokinetics and patient characteristics, a population pharmacokinetic analysis was performed on the complete dataset. The following covariables were tested: age (years), body surface area (BSA, m2), body weight (WGT, kg), height (HGT, cm), serum creatinine (Scr, µm), alanine amino transferase (ALAT, U l−1), aspartate amino transferase (ASAT, U l−1), alkaline phosphatase (AP, U l−1), lactate dehydrogenase (LD, U l−1), serum total bilirubine (BILT, µm), serum total protein (TP, g l−1), serum albumin (ALB, g l−1), gamma glutamyl transferase (GGT, U l−1) and creatinine clearance (CLcr, ml min−1). Serum creatinine was determined using the Jaffé method and CLcr was calculated from serum creatinine, age, weight and gender using the Cockcroft-Gault formula without adjustment [35]. The different covariables were recorded before the start of a course.
A time efficient two-stage approach was chosen for model development. In the first step the different covariables were introduced separately into a pharmacokinetic model for thioTEPA using a standard two compartment model with first-order elimination from the central compartment using the NONMEM subroutine ADVAN3, which is equal to the model described in equation 1 and 2. The covariables were introduced on CL and V using linear relationships. For instance, CL was modelled using CL = θ1 + θ2*(covariable − median), in which θ1 represents the baseline CL and θ2 the relationship between the covariable and CL. Each model incorporating a covariable was tested against a model without this covariable. A covariable was considered potentially significant when the decrease in objective function was > 3.8 (P < 0.05). All potentially significant covariables were introduced in an intermediate model and sub-sequently the significant covariables were selected using a stepwise backward elimination procedure. In this step, a covariable was considered significant when elimination of this covariable resulted in an increase in objective function of> 7.9 (P < 0.005). Finally, individual empirical Bayesian pharmacokinetic parameters were estimated using the POSTHOC option of NONMEM. These individual parameters were used in the second step, where a population pharmacokinetic model was developed for TEPA using the model as described in equation 1,2,3 and 4. The dataset in this step consisted of the thioTEPA dose records and TEPA data. Covariables were introduced on fm* and k using the same procedure as applied for thioTEPA.
Finally, the two models were combined and a final backward elimination procedure was carried out in order to identify possible correlated covariables.
Results
ThioTEPA data were available from a total number of 40 patients (65 courses). TEPA data were also available from 30 of these patients (50 courses). Patient characteristics of the dataset are summarized in Table 1. The thioTEPA concentration was below the LLQ in a few samples immediately before the next thioTEPA infusion. Only samples above the LLQ were included in the analysis.
Table 1.
Patient characteristics.
| Number | |
|---|---|
| Patients | 40 |
| Male | 3 |
| Female | 37 |
| Site of disease and stage | |
| Breast cancer stage II or III | 14 |
| Breast cancer stage IV | 19 |
| Ovarian cancer | 4 |
| Germ cell cancer | 3 |
| Courses with pharmacokinetic data available | 65 |
| 1st course CTC | 20 |
| 2nd course CTC | 1 |
| 1st course tCTC | 20 |
| 2nd course tCTC | 15 |
| 3rd course tCTC | 91 |
| Total thioTEPA dose, number of courses | |
| 480 mg m−2 (CTC) | 21 |
| 320 mg m−2 (tCTC) | 43 |
| 240 mg m−2 (3rd course tCTC with dose reduction) | 1 |
| Median age (years) (range) | 46 (16–58) |
| Median body surface area (m2) (range) | 1.79 (1.55–1.98) |
| Median weight (kg) (range) | 68 (52–90) |
| Medium height (cm) (range) | 170 (155–185) |
| Median serum creatinine (µm) (range) | 66 (53–89) |
| Median creatinine clearance Cockcroft-Gault (ml min−1) (range) | 102 (64–162) |
| Median alanine amino transferase (U l−1) (range) | 20 (4–100) |
| Median aspartate amino transferase (U l−1) (range) | 12 (7–59) |
| Median alkaline phosphatase (U l−1) (range) | 57 (25–103) |
| Median lactate dehydrogenase (U l−1) (range) | 209 (154–504) |
| Median serum bilirubine (µm) (range) | 7 (5–21) |
| Median serum total protein (g l−1) (range) | 61 (54–77) |
| Median serum albumin (g l−1) (range) | 42 (38–48) |
| Median gamma glutamyl transferase (U l−1) (range) | 27 (7–254) |
| Number of thioTEPA concentrations | 1115 |
| Number of TEPA concentrations | 915 |
One third course of tCTC given without cyclophosphamide.
The basic pharmacokinetic model was applied to the dataset. The interpatient variability in k12 and k34 and the additive residual variability in TEPA pharmacokinetics could not be estimated. The results of the basic pharmacokinetic model are summarized in Table 2. For both thioTEPA and TEPA one-compartment models were tested but resulted in a significant worse fit of the data. Non-linear pharmacokinetic behaviour of thioTEPA and TEPA was not observed as judged by various diagnostic plots. Figure 3 shows the concentration-time data and the fitted curves of thioTEPA and TEPA for a representative patient. Figure 4 shows the plots of the weighted residuals vs the predicted concentrations of thioTEPA and TEPA. The population ratio of the AUC of TEPA and the AUC of thioTEPA was 1.76. The half-life of the distribution and elimination phase of thioTEPA were, 0.6 and 2.4 h, respectively.
Table 2.
Population pharmacokinetics of thioTEPA and TEPA (basic model).
| Parameter | Population value (RSD)1 | Interindividual variability (%) (RSD)1 | Interoccasion variability (%) (RSD)1 |
|---|---|---|---|
| CL (l h−1) | 34.5 (0.098) | 16 (0.45) | 15 (0.50) |
| V1 (l) | 45.8 (0.072) | 7.4 (2.8) | 19 (0.64) |
| k12 (h−1) | 0.273 (0.20) | ||
| k21 (h−1) | 0.459 (0.41) | 47 (1.1) | |
| fm* (l−1) | 0.0298 (0.20) | 36 (0.34) | 26 (0.41) |
| k (h−1) | 0.584 (0.18) | 21 (0.44) | 26 (0.50) |
| k34 (h−1) | 2.79 (0.22) | ||
| k43 (h−1) | 0.855 (0.088) | 24 (0.51) | |
| Proportional error thioTEPA (%) | 27.7 | ||
| Additive error thioTEPA (µm) | 0.0631 | ||
| Proportional error TEPA (%) | 20.9 |
RSD = relative standard error of estimate.
Figure 3.

Concentration-time data and fitted curves for thioTEPA (solid line and •) and TEPA (dashed line and □) in a representative patient.
Figure 4.

Weighted residuals vs the predicted concentrations of the basic pharmacokinetic model for thioTEPA (a) and TEPA (b).
The different covariables were introduced separately into the model for CL and V1 of the thioTEPA part of the model. Introduction of ALB, BILT, AP and ALAT on CL and weight, HGT, BSA, ALAT, ASAT, TP, BILT, AP and ALB on V1 provided an increase of goodness-of-fit associated with a P value < 0.05. After stepwise backward elimination only AP and ALB had a significant relation with CL and HGT and TP with V1.
Using the Bayesian estimates of the final thioTEPA model, a population pharmacokinetic model of TEPA was developed. Introduction of age, BSA, WGT, HGT, Scr, ALAT, ASAT, LD, TP, ALB, GGT and CLcr on fm* and BSA, WGT, HGT, Scr, ALAT, ASAT, TP, ALB, GGT and CLcr on k improved the goodness-of-fit with a P value < 0.05. Subsequent stepwise backward elimination of the different covariables resulted in a model incorporating only ALAT, WGT and TP on k. Finally, the different significant covariables were introduced into the combined model and a final backward elimination procedure was carried out. After this step only AP and ALB on CL, TP on V1 and WGT on k proved significant The results of this backward elimination procedure are shown in Table 3, and the results from this final pharmacokinetic model are summarized in Table 4.
Table 3.
Results of final backward elimination procedure.
Model: CL = θ1 + θ9*(AP-57) + θ10*(ALB-43); V1 = θ2 + θ11*>(TP-61); k12 = θ3; k21 = θ4, fm*= θ5; k = θ6 + θ12*(WGT-68); k34 = θ7; k43 = θ8.
| Eliminated parameter | Covariable | Objective function | ΔObjective function1 | P value2 |
|---|---|---|---|---|
| Basic population model | −2784.966 | |||
| Full population model | −2876.405 | |||
| θ9 CL | Alkaline phosphatase | −2853.050 | + 23.355 | < 0.0005 |
| θ10 CL | Serum albumin | −2864.130 | + 12.275 | 0.0005 |
| θ11V1 | Total protein | −2856.818 | + 19.227 | < 0.0005 |
| θ12k | Body weight | −2840.364 | + 36.041 | < 0.0005 |
Difference in objective function between indicated model and full population model.
P value calculated using the likelihood ratio test indicating the difference between the indicated (reduced) model and the full population model.
Table 4.
Population pharmacokinetic model of thioTEPA.
Model: CL = θ1 + θ9*(AP-57) + θ10*(ALB-43); V1 = θ2 + θ11*(TP-61); k12 = θ3; k21 = θ4, fm*= θ5; k = θ6 + θ12*(WGT-68); k34 = θ7; k43 = θ8.
| Parameter | Population value (RSD)1 | Interindividual variability (%) (RSD)1 | Interoccasion variability (%) (RSD)1 |
|---|---|---|---|
| θ1 CL (l*h–1) | 34.0 (0.067) | 18 (0.33) | 11 (0.35) |
| θ2V1 (l) | 46.9 (0.062) | 7.5 (2.1) | 19 (0.54) |
| θ3k12 (h–1) | 0.304 (0.17) | ||
| θ4k21 (h–1) | 0.408 (0.20) | 51 (0.60) | |
| θ5 fm* (l–1) | 0.0295 (0.17) | 39 (0.37) | 32 (0.59) |
| θ6k (h–1) | 0.636 (0.23) | 27 (0.67) | 32 (0.32) |
| θ7 k34 (h–1) | 2.75 (0.23) | ||
| θ8 k43 (h–1) | 0.851 (0.094) | 25 (058) | |
| θ9 CL Alkaline phosphatase | −0.0727 (0.53) | ||
| θ10 CL Serum albumin | 0.449 (0.49) | ||
| θ11V1 Total protein | 0.751 (0.45) | ||
| θ12k Body weight | 0.00736 (0.46) | ||
| Proportional error thioTEPA (%) | 28.7 | ||
| Additive error thioTEPA (µm) | 0.0209 | ||
| Proportional error TEPA (%) | 22.4 |
RSD = relative standard error of estimate.
Discussion
Application of the basic pharmacokinetic model to the complete dataset resulted in typical values of CL and V for thioTEPA of 35 l h−1 and 46 l, respectively. For thioTEPA a biphasic kinetic curve with a rapid distribution phase of approximately 10 min has been described in most studies [10, 15, 16, 18, 20, 22, 24, 27]. A terminal elimination half-life of thioTEPA between 1.5 and 4 h has been reported [10, 13–20, 22, 24, 25, 27]. In our study the half-life of the distribution phase and elimination phase were 0.6 and 2.4 h, respectively.
Some studies have shown nonlinear pharmacokinetics for thioTEPA which was explained by saturation of the elimination pathways [18, 20, 23, 26]. Hussein et al. showed a substantially lower clearance of thioTEPA at doses of 750–900 mg m−2 day−1 compared with 300–450 mg m−2 day−1 in a short infusion [26]. Henner et al. found an inverse relation between dose and clearance with doses between 180 and 900 mg m−2 in a 96 h infusion [23]. The doses employed in these studies were considerably higher than the doses used in our study. O'Dwyer et al. and Heideman et al. both showed that the formation of TEPA is saturated at conventional doses (30–75 mg m−2) causing nonlinear pharmaco-kinetics for thioTEPA and TEPA [18, 20]. Others demonstrated linear pharmacokinetics for thioTEPA at conventional doses [14, 19] as well as at high-doses [22, 25]. One study showed a linear increase in TEPA AUC when the dose increased from 60 mg to 80 mg [19]. We found no indication for nonlinear pharmacokinetics of thioTEPA and TEPA in the dose range of 80–120 mg m−2 day−1 although nonlinearity can not be excluded with only two dose levels. Values for CL of thioTEPA were in agreement with earlier studies both in high-dose regimens [22–25, 27] and conventional dose regimen [10, 14–19].
In most studies the interpatient variability in the pharmacokinetics of thioTEPA was modest with 10–40% variability in CL [14, 15, 17, 18, 23, 25, 27], although Lazarus et al. found an interpatient variability of up to 70% in a high-dose regimen [22]. Interpatient variability in TEPA pharmacokinetics was much higher compared with thioTEPA with up to three-fold difference in the AUC in patients receiving the same dose [16]. In our study, data from multiple courses were available enabling the estimation of interoccasion variability and interpatient variability. In previous studies the interoccasion variability was not taken into account. This is the main reason why the interpatient variability as observed in our study was smaller than previously reported. Interpatient variability in CL and V1 was small (16 and 7.4%, respectively) while the interpatient variability in k21 was 47%. Interoccasion variability in CL was 15%, which was comparable with the interpatient variability encountered. Interpatient and interoccasion variability in both the formation and elimination of TEPA exceeded the variability in thioTEPA pharmacokinetics. Relatively low interoccasion variability in thioTEPA and TEPA pharmacokinetics was found and the interoccasion variability of both thioTEPA and TEPA was equal to or smaller than the interpatient variability. Therefore, therapeutic drug monitoring based on data from previous courses may be meaningful using the developed model.
The population value of fm* in this study was 0.03 l−1. Since the fraction thioTEPA converted to TEPA cannot be greater than 1, the volume of distribution of TEPA must be considerably smaller than that of thioTEPA (46 l).
The AUC of TEPA generally exceeded that of thioTEPA due to its longer terminal half-life and smaller volume of distribution. The ratio of the TEPA and thioTEPA AUC varied between 1.1 and 4.9 [17–20, 27], whereas O'Dwyer et al. found a ratio of 0.79 in a conventional dose regimen [15]. The ratio of the TEPA and thioTEPA AUC found in this study (1.76), was in accordance with previously published data [17–20, 27].
In this study thioTEPA was administered as part of a high-dose combination regimen followed by peripheral blood progenitor cell transplantation. Besides the anti-cancer agents cyclophosphamide and carboplatin, patients received antimycotics as itraconazole, antibiotics such as ciprofloxacin, antiemetics such as granisetron and dexamethasone and the uroprotective agent MESNA. These comedicated drugs may introduce pharmacokinetic interactions, in particular through induction or inhibition of cytochrome P450 activity. ThioTEPA is metabolized by CYP2B and 2C families to TEPA [7, 8]. The CYP2C family is also involved in the 4-hydroxylation of cyclophosphamide [36, 37]. However, an inhibitory effect of cyclophosphamide on the formation of TEPA has not been demonstrated in vivo and in vitro [23, 25, 26, 38, 39]. Henner et al. have described an enhancement of thioTEPA elimination after a 96 h continuous infusion of cyclophosphamide and thioTEPA, which was possibly caused by an inductive effect of cyclophosphamide [23]. In our study, however, no induction of thioTEPA elimination was observed. Although some of the other comedicated drugs may have large effects on cytochrome P450 enzymes, van Maanen et al. showed that these drugs did not have any effects on the thioTEPA and TEPA pharmacokinetics [39].
The patient population in this study was highly selected. Only patients eligible for high-dose chemo-therapy protocols were included, and all patients had a good clinical condition with normal renal and hepatic function. Moreover, the population consisted of mainly young women. Nowadays, thioTEPA is almost exclusively used in these high-dose regimens for solid tumours and therefore, the study population may be representative of the current population treated with thioTEPA.
The effects of the various covariables were tested in a model including estimation of interoccasion variability. Failure to model interoccasion variability may lead to the introduction of nonsignificant covariables and model overparameterization as described by Karlsson et al. [40]. In the final model, CL of thioTEPA and thus formation of TEPA was significantly related to serum AP and serum albumin concentrations. This indicates that CL is positively related to liver function, which may be explained by the fact that less than 2% of the total thioTEPA dose is excreted unchanged in the urine [9, 10]. The volume of distribution of thioTEPA was related to total protein concentration and the elimination of TEPA was related to body weight. Although renal clearance of TEPA may play a considerable role in its elimination no significant relationships between TEPA elimination and creatinine clearance or serum creatinine were identified.
In conclusion, a model for the pharmacokinetics of thioTEPA and TEPA was developed. Some demographic and biochemical factors involved in the interpatient variability of thioTEPA and TEPA pharmacokinetics were identified. The model which enables the estimation of individual pharmacokinetic parameters using a Bayesian approach with limited individual data, will be validated prospectively and used for pharmacokinetic guided dosing of thioTEPA in future studies.
Acknowledgments
This research was supported with a grant fromt the Dutch Cancer Society (project NKI 97–1439).
References
- 1.Sykes M, Karnofsky D, Phillips F, Burchenal J. Clinical studies of triethylenethiophosphoramide and diethylenephosphoramide compounds with nitrogen mustard-like activity. Cancer. 1953;6:61–70. [Google Scholar]
- 2.van der Wall E, Beijnen JH, Rodenhuis S. High-dose chemotherapy for solid tumors. Cancer Treat Rev. 1995;21:105–132. doi: 10.1016/0305-7372(95)90023-3. [DOI] [PubMed] [Google Scholar]
- 3.Cole DE, Johnson G, Tartaglia RL, O'Dwyer P, Balis F, Poplack DG. Correlation between plasma pharmacokinetics and in vitro cytotoxicity of thiotepa (tt) and tepa (tp) Proc Am Ass Clin Oncol. 1989;8:72. [Google Scholar]
- 4.Breau AP, Field L, Mitchell WM. Thiono compounds. 4. in vitro mutagenic and antineoplastic activity of tepa and thio-tepa. Cell Biol Toxicol. 1984;1:21–30. doi: 10.1007/BF00125562. [DOI] [PubMed] [Google Scholar]
- 5.Teicher BA, Waxman DJ, Holden SA, et al. Evidence for enzymatic activation and oxygen involvement in cytotoxicity and antitumor activity of N,N',N′′-triethylenethiophosphoramide. Cancer Res. 1989;49:4996–5001. [PubMed] [Google Scholar]
- 6.Miller B, Tenenholz T, Egorin MJ, Sosnovsky G, Maheswara Rao NU, Gutierrez PL. Cellular pharmacology of N,N',N′′-triethylenethiophosphoramide. Cancer Lett. 1988;41:157–168. doi: 10.1016/0304-3835(88)90112-7. [DOI] [PubMed] [Google Scholar]
- 7.Ng S, Waxman DJ. Biotransformation of N.N',N′′-triethylenethiophosphoramide: oxidative desulfuration to yield N.N',N′′-triethylenephosphoramide associated with suicide inactivation of a phenobarbital- inducible hepatic P-450 monooxygenase. Cancer Res. 1990;50:464–471. [PubMed] [Google Scholar]
- 8.Ng S, Waxman DJN. N',N′′-triethylenethiophosphoramide (thio-TEPA) oxygenation by constitutive hepatic P450 enzymes and modulation of drug metabolism and clearance in vivo by P450-inducing agents. Cancer Res. 1991;51:2340–2345. [PubMed] [Google Scholar]
- 9.Van Maanen MJ, Tijhof IM, Damen MA, et al. A search for new metabolites of N,N',N′′-triethylenethiophosphoramide. Cancer Res. 1999;59:4720–4724. [PubMed] [Google Scholar]
- 10.Cohen BE, Egorin MJ, Kohlhepp EA, Aisner J, Gutierrez PL. Human plasma pharmacokinetics and urinary excretion of thiotepa and its metabolites. Cancer Treat Rep. 1986;70:859–864. [PubMed] [Google Scholar]
- 11.Jones RB, Matthes S. Pharmacokinetics. In: Armitage JO, Antman KH, editors. High-Dose Cancer Therapy: Pharmacology, Hematopoietins, Stem Cells. Baltimore: Williams & Wilkins; 1992. pp. 55–56. [Google Scholar]
- 12.Hagen B, Nilsen OG. The binding of thio-TEPA in human serum and to isolated serum protein fractions. Cancer Chemother Pharmacol. 1987;20:319–323. doi: 10.1007/BF00262584. [DOI] [PubMed] [Google Scholar]
- 13.Hagen B, Walseth F, Walstad RA, Iversen T, Nilsen OG. Single and repeated dose pharmacokinetics of thio-TEPA in patients treated for ovarian cancer. Cancer Chemother Pharmacol. 1987;19:143–148. doi: 10.1007/BF00254567. [DOI] [PubMed] [Google Scholar]
- 14.Hagen B, Walstad RA, Nilsen OG. Pharmacokinetics of thio-TEPA at two different doses. Cancer Chemother Pharmacol. 1988;22:356–358. doi: 10.1007/BF00254246. [DOI] [PubMed] [Google Scholar]
- 15.O'Dwyer PJ, LaCreta FP, Schilder R, et al. Phase I trial of thiotepa in combination with recombinant human granulocyte-macrophage colony-stimulating factor. J Clin Oncol. 1992;10:1352–1358. doi: 10.1200/JCO.1992.10.8.1352. [DOI] [PubMed] [Google Scholar]
- 16.O'Dwyer PJ, LaCreta D, Nash S, et al. Phase I study of thiotepa in combination with the glutathione transferase inhibitor ethacrynic aced. Cancer Res. 1991;51:6059–6065. [PubMed] [Google Scholar]
- 17.Hagen B, Neverdal G, Walstad RA, Nilsen OG. Long-term pharmacokinetics of thio-TEPA, TEPA and total alkylating activity following i.v. bolus administration of thio-TEPA in ovarian cancer patients. Cancer Chemother Pharmacol. 1990;25:257–262. doi: 10.1007/BF00684882. [DOI] [PubMed] [Google Scholar]
- 18.O'Dwyer PJ, LaCreta F, Engstrom PF, et al. Phase I/pharmacokinetic reevaluation of thioTEPA. Cancer Res. 1991;51:3171–3176. [PubMed] [Google Scholar]
- 19.Hagen B. Pharmacokinetics of thio-TEPA and TEPA in the conventional dose-range and its correlation to myelosppressive effects. Cancer Chemother Pharmacol. 1991;27:373–378. doi: 10.1007/BF00688860. [DOI] [PubMed] [Google Scholar]
- 20.Heideman RL, Cole DE, Balis F, et al. Phase I and pharmacokinetic evaluation of thiotepa in the cerebrospinal fluid and plasma of pediatric patients: evidence for dose-dependent plasma clearance of thiotepa. Cancer Res. 1989;49:736–741. [PubMed] [Google Scholar]
- 21.Shapiro CL, Dezube BJ, Tretyakov O, et al. Dose escalation of N,N',N′′-triethylenethiophosphoramide combined with pentoxifylline for advanced breast cancer. Clin Cancer Res. 1995;1:791–796. [PubMed] [Google Scholar]
- 22.Lazarus HM, Reed MD, Spitzer TR, Rabaa MS, Blumer JL. High-dose iv thiotepa and cryoperserved autologous bone marrow transplantation for therapy of refractory cancer. Cancer Treat Rep. 1987;71:689–695. [PubMed] [Google Scholar]
- 23.Henner WD, Shea TC, Furlong EA, et al. Pharmacokinetics of continuous-infusion high-dose thiotepa. Cancer Treat Rep. 1987;71:1043–1047. [PubMed] [Google Scholar]
- 24.Kletzel M, Kearns GL, Wells TG, Thompson HC. Pharmacokinetics of high dose thiotepa in children undergoing autologous bone marrow transplantation. Bone Marrow Transplant. 1992;10:171–175. [PubMed] [Google Scholar]
- 25.Ackland SP, Choi KE, Ratain MJ, et al. Human plasma pharmacokinetics of thiotepa following administration of high-dose thiotepa and cyclophosphamide. J Clin Oncol. 1988;6:1192–1196. doi: 10.1200/JCO.1988.6.7.1192. [DOI] [PubMed] [Google Scholar]
- 26.Hussein AM, Petros WP, Ross M, et al. A phase I/II study of high-dose cyclophosphamide, cisplatin, and thioTEPA followed by autologous bone marrow and granulocyte colony-stimulating factor-primed peripheral-blood progenitor cells in patients with advanced malignancies. Cancer Chemother Pharamcol. 1996;37:561–568. doi: 10.1007/s002800050429. [DOI] [PubMed] [Google Scholar]
- 27.Przepiorka D, Madden T, Ippoliti C, Estrov Z, Dimopoulos M. Dosing of thioTEPA for myeloablative therapy. Cancer Chemother Pharmacol. 1995;37:155–160. 10.1007/s002800050381. [PubMed] [Google Scholar]
- 28.WHO Handbook for Reporting Results of Cancer Treatment. Geneva. Switzerland: World Health Organization; 1979. [Google Scholar]
- 29.Rodenhuis S, Westermann A, Holtkamp MJ, et al. Feasibility of multiple courses of high-dose cyclophosphamide, thioTEPA and carboplatin for breast cancer or germ cell cancer. J Clin Oncol. 1996;14:1473–1483. doi: 10.1200/JCO.1996.14.5.1473. [DOI] [PubMed] [Google Scholar]
- 30.Huitema ADR, Holtkamp M, Tibben MM, Rodenhuis S, Beijnen JH. Sampling technique from central venous catheters proves to be critical for pharmacokinetic studies. Ther Drug Monit. 1999;21:102–104. doi: 10.1097/00007691-199902000-00016. [DOI] [PubMed] [Google Scholar]
- 31.Huitema ADR, Tibben MM, Kerbusch T, Zwikker JW, Rodenhuis S, Beijnen JH. Simultaneous determination of N,N',N′′-triethylenethiophosphoramide, cyclophosphamide and some of their metabolites in plasma using capillary gas chromatography. J Chromatogr B. 1998;716:177–186. doi: 10.1016/s0378-4347(98)00300-4. [DOI] [PubMed] [Google Scholar]
- 32.Beal SL, Sheiner LB. San Fransisco: University of California at San Fransisco; 1998. NONMEM Users'Guides, NONMEM. Project Group. [Google Scholar]
- 33.Rowland M, Tozer TN. Clinical pharmacokinetics. Concepts and Applications. 2. Philadelphia: Lea & Febiger; 1989. p. 304. [Google Scholar]
- 34.Jonsson EN, Karlsson MO. Xpose-an S-PLUS based model building aid for population analysis with NONMEM. In: Aarons L, Balant LP, Danhof M, editors. The population approach: measuring and managing variability in response, concentration and dose. Brussels: European Commission; 1997. [Google Scholar]
- 35.Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16:31–41. doi: 10.1159/000180580. [DOI] [PubMed] [Google Scholar]
- 36.Ren S, Yang JS, Kalhorn TF, Slattery JT. Oxidation of cyclophosphamide to 4-hydroxycyclophosphamide and deschoroethylcyclophosphamide in human liver microsomes. Cancer Res. 1997;47:4229–4235. [PubMed] [Google Scholar]
- 37.Chang TKH, Yu L, Goldstein JA, Waxman DJ. Identification of the polymorphically expressed CYP 2C19 and the wild-type CYP 2C9-ILE359 allele as low-Km catalysts of cyclophosphamide and ifosfamide activation. Pharmacogenetics. 1997;7:211–221. doi: 10.1097/00008571-199706000-00006. [DOI] [PubMed] [Google Scholar]
- 38.Dobrovolskaja A, Nitz U, Frick M, Bender HG, Burk M. High-dose chemotherapy with stem cell support in breast cancer: does cyclophosphamide alter high-dose thiotepa pharmacokinetics? Eur J Cancer. 1998;34(Suppl 1):S37. (Abstract) 10.1016/s0959-8049(98)80136-6. [Google Scholar]
- 39.van Maanen MJ, Huitema ADR, Beijnen JH. Influence of co-medicated drugs on the biotransformation of thioTEPA to TEPA and thioTEPA-mercapturate. Anticancer Res. in press. [PubMed]
- 40.Karlsson MO, Sheiner LB. The importance of modeling interoccasion variability in population pharmacokinetic analyses. J Pharmacokin Biopharm. 1993;21:735–750. doi: 10.1007/BF01113502. [DOI] [PubMed] [Google Scholar]