

Abstract
Aims
To investigate the effects of various anticancer drugs on the major metabolic pathways (glucuronidation and 6-methylhydroxylation) of DMXAA in human liver microsomes.
Methods
The effects of various anticancer drugs at 100 and 500 µm on the formation of DMXAA acyl glucuronide (DMXAA-G) and 6-hydroxymethyl-5-methylxanthenone-4-acetic acid (6-OH-MXAA) in human liver microsomes were determined by high performance liquid chromatography (h.p.l.c.). For those anticancer drugs showing significant inhibition of DMXAA metabolism, the inhibition constants (Ki) were determined. The resulting in vitro data were extrapolated to predict in vivo changes in DMXAA pharmacokinetics.
Results
Vinblastine, vincristine and amsacrine at 500 µm significantly (P < 0.05) inhibited DMXAA glucuronidation (Ki = 319, 350 and 230 µm, respectively), but not 6-methylhydroxylation in human liver microsomes. Daunorubicin and N-[2-(dimethylamino)-ethyl]acridine-4-carboxamide (DACA) at 100 and 500 µm showed significant (P < 0.05) inhibition of DMXAA 6-methylhydroxylation (Ki = 131 and 0.59 µm, respectively), but not glucuronidation. Other drugs such as 5-fluoroucacil, paclitaxel, tirapazamine and methotrexate exhibited little or negligible inhibition of the metabolism of DMXAA. Pre-incubation of microsomes with the anticancer drugs (100 and 500 µm) did not enhance their inhibitory effects on DMXAA metabolism. Prediction of DMXAA–drug interactions in vivo based on these in vitro data indicated that all the anticancer drugs investigated except DACA appear unlikely to alter the pharmacokinetics of DMXAA, whereas DACA may increase the plasma AUC of DMXAA by 6%.
Conclusions
These results indicate that alteration of the pharmacokinetics of DMXAA appears unlikely when used in combination with other common anticancer drugs. However, this does not rule out the possibility of pharmacokinetic interactions with other drugs used concurrently with this combination of anticancer drugs.
Keywords: anti-cancer, DMXAA, drug interaction, human liver microsomes
Introduction
5,6-Dimethylxanthenone-4-acetic acid (DMXAA) (Figure 1) is an anticancer drug with an unusual mechanism of action compared with conventional cytotoxic anticancer drugs. DMXAA induces rapid vascular collapse and necrosis in transplantable murine tumours, thought to be due to immune modulation and the induction of cytokines, in particular tumour necrosis factor-α, interferons, serotonin and nitric oxide [1–4]. Co-administration of DMXAA with other drugs has been shown to result in enhanced antitumour activity and alterations in pharmacokinetics, as reported for the combination of DMXAA with melphalan, thalidomide, and the bioreductive agent tirapazamine, in mouse models [5–8]. These results suggest that coadministration of DMXAA with other anticancer drugs may be a useful strategy for enhancing their antitumour activity. DMXAA is extensively metabolized, mainly by glucuronidation of its acetic acid side chain and 6-methylhydroxylation [9–11], giving rise to DMXAA acyl glucuronide (DMXAA-G), and 6-hydroxymethyl-5-methylxanthenone-4-acetic acid (6-OH-MXAA), which are excreted into bile and urine. Studies have indicated that DMXAA glucuronidation is catalysed by uridine diphosphate glucuronosyltransferases (UGT1A9 and UGT2B7) [10], and 6-methylhydroxylation by cytochrome P450 (CYP1A2) [12].
Figure 1.

The structure of DMXAA.
The aim of this study was to investigate the effects of various anticancer drugs on the metabolism of DMXAA in human liver microsomes, and if significant inhibition is observed, to predict in vivo DMXAA–drug pharmacokinetic interactions.
Methods
Chemicals and reagents
DMXAA, 2,5-dimethylxanthenone-4-acetic acid (as internal standard), N-[2-(dimethylamino)-ethyl]acridine-4-carboxamide (DACA), and amsacrine were synthesized in the Auckland Cancer Society Research Centre (ACSRC) as described [13, 14]. DMXAA was protected from light exposure to avoid degradation [15]. Authentic DMXAA-G and 6-OH-MXAA were isolated and purified by a solid phase extraction method from the bile and urine of rats treated with DMXAA. Both metabolites had a purity of 99% as determined by h.p.l.c, and their structure was confirmed by mass spectrometry and [1H]-nuclear magnetic resonance [11]. Daunorubicin, 5-fluorouracil, paclitaxel, cisplatin, tirapazamine, irinotecan, methotrexate, melphalan, 6-thioguanine, 6-mercaptopurine, cyclophosphamide, folic acid, vinblastine, vincristine and 6-methylguanine were purchased from Sigma-Aldrich Chemical Co. (Auckland, NZ). Uridine diphosphate glucuronic acid (UDPGA) and NADPH were purchased from Roche Diagnostics NZ Ltd. (Auckland, NZ). All other reagents were of analytical or h.p.l.c grade as appropriate.
Preparation of human liver microsomes
Human liver samples were obtained under strict ethical conditions from donors, and were stored at −80 °C prior to use. Histological examination of the resected livers ensured the use of healthy liver tissue. Relevant details of the donors have been described elsewhere [12]. Ethical approval from the Northern New Zealand Research Ethics Committee and written informed consent for liver tissue to be used for research was obtained. Liver microsomes were prepared by differential centrifugation as described [16] and microsomes were stored at −80 °C until used. Microsomal fractions used in this study were from HL6, HL7, HL8, HL12, HL13 and HL14 from our human liver bank. Microsomal protein concentration was determined by the bicinchoninic acid method [17]. The CYP content was determined as described [18].
In vitro metabolic inhibition studies
The effects of a number of anticancer drugs (100 and 500 µm) on DMXAA glucuronidation and 6-methylhydroxylation in human liver microsomes were investigated using optimized incubation conditions [12]. Typical incubations (total volume=200 µl, in triplicate) for DMXAA glucuronidation contained liver microsomal protein (0.1 mg ml−1, pooled from HL6, HL7 and HL8), 10 mm UDPGA, 5 mm MgCl2, 0.1 mg ml−1 d-saccharic acid 1,4-lactone, Brij 58 (0.1–0.25 : 1, ratio of Brij 58 over microsome, w/w), inhibitor (100 and 500 µm), and DMXAA in 0.1 m phosphate buffer (pH 6.8). d-Saccharic acid 1,4-lactone was used to inhibit the activity of β-glucuronidase in microsomes. Typical incubations (total volume=200 µl, in triplicate) for 6-methylhydroxylation contained 1 mg ml−1 liver microsomal protein (from three human livers, HL12, HL13, and HL14), 5 mm MgCl2, 0.5 mm NADPH, inhibitor (100 and 500 µm), and DMXAA in 0.1 m phosphate buffer (pH 7.4). The concentrations of DMXAA were 100 µm for glucuronidation, and 25 µm for 6-methylhydroxylation (the corresponding apparent Km values for each metabolic pathway). Pooled human liver microsomes were used for DMXAA glucuronidation as this pathway is catalysed by multiple UGT enzymes (UGT1A9/UGT2B7) [10], and the glucuronidation activity for DMXAA was similar between human livers; whereas CYP1A2 was responsible for DMXAA 6-methylhydroxylation, and significant interindividual variation in the activity was observed [12]. The reactions were initiated by the addition of NADPH or UDPGA as appropriate. Pre-incubations were also performed in duplicate in the presence of inhibitor and cofactor (NADPH or UDPGA) for 0 or 15 min prior to the addition of DMXAA at 37 °C in a shaking water-bath. Incubations were stopped by cooling on ice and adding 2 volumes of an ice-cold acetonitrile: methanol mixture (3 : 1, v/v) containing 2 µm internal standard, and vortexing vigorously. Mixtures were centrifuged (3000 g for 10 min) to remove the precipitated microsomal protein. The supernatant was removed, evaporated under nitrogen, and the residue reconstituted with mobile phase for injection onto the h.p.l.c. All anticancer drugs were dissolved in dimethyl sulphoxide (DMSO), which was used at a final concentration of 1% (v/v) in incubations. DMSO reduced the rate of DMXAA hydroxylation by 22%, but had no significant effect on DMXAA glucuronidation. Each drug was also incubated with microsomes and UDPGA or NADPH in the absence of DMXAA to identify any chromatographic peaks which might interfere with the measurement of DMXAA-G or 6-OH-MXAA.
For those drugs showing significant inhibitory effects, further inhibition kinetic studies were performed to determine the mechanism of inhibition and the apparent Ki values. To construct Dixon plots, DMXAA (25–100 µm for glucuronidation; 6.25–25 µm for 6-methylhydroxylation) was incubated at 37 °C with human liver microsomes in the presence of inhibitors. The inhibitor concentrations used were 50–400 µm for amsacrine, for 62.5–500 µm for vinblastine and vincristine, for 37.5–300 µm daunorubicin, and 0.625–5 µm for DACA.
High performance liquid chromatography (h.p.l.c)
The determination of DMXAA-G and 6-OH-MXAA has been described previously [19]. Briefly, the h.p.l.c system consisted of a solvent delivery system, a Model SF250 fluorescence detector (excitation and emission wavelength, 345 nm and 409 nm, respectively), a Model 460 autosampler, and a Model D450 data processing system (All from Kontron Instrument Co., Milan, Italy). A Luna C18 guard column and a 5-µm Spherex C18 analytical column (150 × 4.6 mm; Phenomenex) were used. The mobile phase was acetonitrile: 10 mm ammonium acetate buffer (24 : 76, v/v, pH 5.0) at a flow rate of 2.5 ml min−1. The difference between the theoretical and measured concentration, and the coefficient variation, were less than 15% at the low quality control (QC) concentration (0.5 µm), and less than 10% at the medium (2.5 µm) and high (10 µm) QC concentrations. The limit for the determination of DMXAA-G and 6-OH-MXAA was 0.25 µm for a 75 µl injection volume. Assay specificity was indicated by the absence of interfering chromatographic peaks in microsomal samples and in incubations with anticancer drugs.
Prediction of drug interaction based on in vitro data
For the inhibition of a metabolic pathway by a drug, the degree of inhibition (R) can be calculated by the following equation 1 [20]:
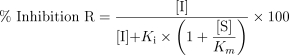 |
1 |
where [I] is the unbound concentration of the inhibitor; Ki is the inhibition constant; and [S] is the unbound therapeutic substrate concentration.
As DMXAA has a low plasma clearance (2–5 ml min−1 kg−1) in cancer patients [21], the degree of change (Rc) in the plasma area under the concentration-time curve (AUC) by the inhibiting drug can be calculated as follows [22]:
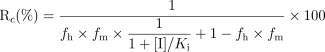 |
2 |
where fh is the fraction of hepatic clearance in total clearance, and fm the fraction of metabolic pathway in hepatic clearance. Approximated fh and fm for DMXAA were estimated from the urine of a patient treated with DMXAA, where 2.4%, 35.9% and 5.5% was excreted as unchanged DMXAA, DMXAA-G and 6-OH-MXAA, respectively [23]. Thus the approximate fh, fm (glucuronidation) and fm (6-methylhydroxylation) were 97.6%, 86.7% and 13.3%, respectively, assuming that biliary excretion of DMXAA in humans is similar to that in the urine, as was found in the rat and rabbit [9, 11]. The unbound fraction of DMXAA (2000 µm) in human plasma was 0.123 as determined by ultrafiltration followed by h.p.l.c [24].
Data analysis
Data were expressed as mean ± s.d. The initial estimate of the apparent Ki values and the nature of inhibition were obtained from Dixon plots, where the apparent Ki was given by the intersection point of the linear regression lines for data sets of 1/v against the concentration of inhibitor. Several inhibition models (competitive, uncompetitive, and mixed inhibition), represented by the following equations, were fitted to the data and compared using Prism 3.0 program (Graphpad Software Co., CA).
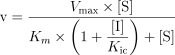 |
3 |
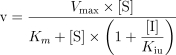 |
4 |
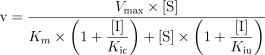 |
5 |
where v is the rate of metabolism, Vmax, is the maximum velocity, Km, the Michaelis-Menten constant, [S], the substrate concentration, [I], the inhibitor concentration, Ki; the apparent inhibition constant and subscripts c and u represent competitive and uncompetitive inhibition. The appropriate model was chosen by comparing and reviewing the relative residuals and the standard error of the parameter estimates. The significance of differences in the formation of DMXAA metabolites was assessed by Student's unpaired t-test. Differences were considered statistically significant when P values were < 0.05.
Results
Effects on anticancer drugs on DMXAA metabolism in vitro
The effects of various anticancer drugs on DMXAA glucuronidation and 6-methylhydroxylation by human liver microsomes are reported in Table 1. Vinblastine, vincristine and amsacrine at 500 µm significantly (P < 0.05) inhibited DMXAA glucuronidation (Ki = 319, 350 and 230 µm, respectively), but not 6-methylhydroxylation in human liver microsomes. Daunorubicin and DACA at 100 and 500 µm showed significant (P < 0.05) inhibition of DMXAA 6-methylhydroxylation (Ki = 131 and 0.59 µm, respectively), but not glucuronidation. Figure 2 illustrates the Dixon plots and the estimated apparent Ki values for inhibition of DMXAA metabolism by various anticancer drugs. The mechanism of inhibition of either DMXAA glucuronidation or 6-methyl-hydroxylation by these anticancer drugs was competitive, as indicated by the smallest mean sum of squares compared with noncompetitive and mixed models. Other drugs such as 5-fluorouracil, paclitaxel, cisplatin, irinotecan, tirapazamine and methotrexate exhibited little or negligible inhibition of the metabolism of DMXAA. Pre-incubation of microsomes with the anticancer drugs (100 and 500 µm) did not enhance their inhibitory effects on DMXAA metabolism.
Table 1.
Effects of various anticancer drugs on DMXAA glucuronidation and 6-methylhydroxylation by human liver microsomes1
| Glucuronidation (% enzyme activity remaining) | 6-Methylhydroxylation (% enzyme activity remaining) | |||
|---|---|---|---|---|
| Drugs | at 100 µm inhibitor | at 500 µm inhibitor | 100 µm inhibitor | 500 µm inhibitor |
| Folic acid | 73 ± 8 | 70 ± 2 | 102 ± 5 | 111 ± 12 |
| Vincristine | 74 ± 14 | 46 ± 8 | 101 ± 0 | 67 ± 14 |
| Amsacrine | 82 ± 12 | 27 ± 1 | 102 ± 19 | 91 ± 5 |
| Daunorubicin | 84 ± 19 | 63 ± 7 | 59 ± 2 | 15 ± 2 |
| Paclitaxel | 88 ± 7 | 78 ± 5 | 98 ± 8 | 95 ± 6 |
| Cisplatin | 91 ± 5 | 67 ± 2 | 100 ± 10 | 86 ± 7 |
| DACA | 96 ± 1 | 71 ± 16 | 17 ± 2 | 6 ± 1 |
| Irinotecan | 97 ± 7 | 71 ± 4 | 95 ± 9 | 92 ± 5 |
| Tirapazamine | 97 ± 7 | 95 ± 7 | 93 ± 3 | 86 ± 4 |
| Vinblastine | 99 ± 4 | 40 ± 0 | 104 ± 9 | 113 ± 1 |
| 6-Thioguanine | 100 ± 2 | 98 ± 7 | 89 ± 2 | 64 ± 1 |
| Cyclophosphamide | 101 ± 29 | 74 ± 12 | 95 ± 2 | 85 ± 4 |
| 6-Mercaptopurine | 102 ± 5 | 96 ± 10 | 97 ± 8 | 94 ± 6 |
| 5-Fluorouracil | 104 ± 11 | 103 ± 7 | 97 ± 7 | 96 ± 10 |
| Methotrexate | 108 ± 8 | 88 ± 2 | 109 ± 8 | 89 ± 14 |
| Melphalan | 113 ± 15 | 104 ± 15 | 99 ± 7 | 101 ± 5 |
For glucuronidation, DMXAA (100 µm, ≈ Km) was incubated for 20 min at 37 °C with 0.1 mg ml−1 Brij 58-activated liver microsomes from pooled HL6, HL7 and HL8 in the presence of various anticancer drugs. For 6-methylhydroxylation, DMXAA (25 µm, ≈ Km) was incubated for 40 min at 37 °C with 1.0 mg ml−1 liver microsomes from HL12, HL13 and HL14 in the presence of various anticancer drugs. Results are the mean ± s.d. of at least three determinations from pooled human liver microsomes for glucuronidation and mean ± s.d. of data from three individual livers for 6-methylhydroxylation.
Figure 2.

Dixon plots for the inhibition of DMXAA glucuronidation by amsacrine (a), vinblastine (b) and vincristine (c), and 6-methylhydroxylation by daunorubicin (d) and DACA (e) in human liver microsomes. Concentrations of DMXAA are shown alongside individual plots. Each data point is the mean of at least three determinations.
Quantitative prediction of DMXAA–drug interaction in vivo
The predicted percentage inhibition (R) and percentage increase in AUC of DMXAA (Rc) that might be caused by coadministration of various anticancer drugs based on our in vitro studies are shown in Table 2. An increase of 6% in the plasma AUC was predicted with DACA, which is of insignificant magnitude to be of clinical relevance. With amsacrine, daunorubicin, vinblastine and vincristine, there was no predicted in vivo inhibition of DMXAA metabolism and change in plasma AUC of DMXAA. Predictions of in vivo DMXAA–drug interactions in patients based on these in vitro data indicated that all the anticancer drugs tested except DACA appear unlikely to alter the pharmacokinetics of DMXAA.
Table 2.
Predicted DMXAA–anticancer drug interactions in vivo.
| Anti-cancer drugs | Apparent Ki value (µm) | Metabolic pathway of inhibition | [I] (µm) | Predicted R (%) | Rc (%) |
|---|---|---|---|---|---|
| Daunorubicin | 131 | Hydroxylation | 0.01–1.001 | 0 | 0 |
| DACA | 0.59 | Hydroxylation | 0.21–18.52 | 0.1–6.4 | 0.1–6.6 |
| Vinblastine | 319 | Glucuronidation | 0.01–0.173 | 0 | 0 |
| Vincristine | 350 | Glucuronidation | 0.01–0.083 | 0 | 0 |
| Amsacrine | 230 | Glucuronidation | 0.71–3.704 | 0 | 0 |
Discussion
There is an increasing interest in the drug–drug interaction studies using human liver microsomes or recombinant human drug-metabolizing isozymes (mainly CYPs) in the early stages of drug development, as this may help identify possible drug interactions and avoid drug toxicity in vivo [22]. Our results indicate that of the anticancer drugs investigated, only vinblastine, vincristine, amsacrine, DACA and daunorubicin at supratherapeutic concentrations (≥ 100 µm) caused a significant inhibition of either glucuronidation or 6-methylhydroxylation of DMXAA with an apparent Ki of 0.59–350 µm. The inhibition by these anticancer agents appears to be competitive, with no involvement of mechanism-based inhibition. When interpreting the clinical relevance of these inhibition studies, the unbound concentration of both the substrate and inhibitors utilized in vitro, and those that exist in vivo, must be taken into consideration. The concentrations for DMXAA (25–140 µm) used in this study have been observed as unbound plasma concentrations in patients in a Phase I trial [21].
Numerous models have been used to correlate in vitro with in vivo drug interactions, with some success [22]. A model for low hepatic clearance drugs administered by intravenous injection was used to extrapolate our in vitro drug interaction data to the in vivo state. In this model, it is assumed that both inhibitor and DMXAA are metabolized only in the liver, with possible competitive inhibition, and that there are only two metabolic pathways for DMXAA. Our results indicate that none of the anticancer drugs investigated would cause a clinically significant reduction in DMXAA plasma clearance. However, these conclusions must be regarded with caution due to the inherent assumptions within the model. In addition, nonspecific microsomal binding may be another important factor influencing the accuracy of the extrapolation from the in vitro to in vivo state [25–27]. Non-specific microsomal binding of substrates will result in a higher apparent Km value determined from the total (added) concentration, rather than the unbound concentration. However, DMXAA as a weak acid is predicted to have insignificant nonspecific microsomal binding, since the microsomal membrane has a net negative charge, and acidic drugs such as caffeine, tolbutamide and naproxen do not bind significantly to it [27]. Furthermore, the model used in this study does not take into account inhibition of metabolism by indirect mechanisms such as alterations in cytokine and/or nitric oxide concentrations, or changes in other drug disposition processes, such as absorption, renal metabolism, or active transport mechanisms.
According to equation 2, the contribution to overall clearance of a particular metabolic pathway subject to inhibition, is an important determining factor in the prediction of in vivo drug interactions [22]. Our in vitro and in vivo studies have indicated that glucuronidation is the major metabolic pathway for DMXAA, whereas 6-methylhydroxylation is a minor metabolic pathway [11, 23]. The intrinsic clearance ratio of glucuronidation: 6-methylhydroxylation was 2.6 from our in vitro human studies [Paxton et al. personal communication]. Previously Miners et al. [10] screened various drugs, mainly substrates of UGTs, for their potential to interact with DMXAA using human liver microsomes and cDNA-expressed UGTs. Significant inhibition of DMXAA glucuronidation was observed with diclofenac, epirubicin, indomethacin, R,S-ketoprofen, lorazepam, S-naproxen, oxazepam, and temazepam with apparent Ki values ranging from 10 to 318 µm. Recently, our mouse studies indicated that diclofenac at 100 mg kg−1 administered by intraperitoneal injection increased the plasma AUC of DMXAA by 24% [8]. This was considered to be mainly due to inhibition of DMXAA glucuronidation. DMXAA is 6-methylhydroxylated by CYP1A2 [12]. However, alterations in this relatively minor pathway of DMXAA metabolism would not be expected to have a clinically significant effect on DMXAA disposition. As none of the anticancer drugs investigated are known substrates of the UGTs, it is perhaps of no surprise that there was little inhibition of DMXAA glucuronidation by these drugs, with the exception of amsacrine, vinblastine and vincristine at high concentration.
In conclusion, despite the occurrence in vitro of inhibition of DMXAA metabolism by several anticancer drugs, such as daunorubicin, DACA and amsacrine, it appears unlikely that combinations of these drugs will result in clinically significant drug interactions. However, this does not rule out the possibility of pharmacokinetic interactions with other drugs used concurrently with this combination of anticancer drugs.
Acknowledgments
The authors gratefully appreciate the support by the Maurice and Phyllis Paykel Trust and the University of Auckland Research Fund. SF Zhou is supported by an Auckland Medical Research Foundation Senior Scholarship.
References
- 1.Thomsen LL, Ching L-M, Baguley BC. Evidence for the production of nitric oxide by activated macrophages treated with the antitumor agents flavone-8-acetic acid and xanthenone-4-acetic acid. Cancer Res. 1990;50:6966–6970. [PubMed] [Google Scholar]
- 2.Thomsen LL, Ching L-M, Zhuang L, Gavin JB, Baguley BC. Tumor-dependent increased plasma nitrate concentrations as an indication of the antitumor effect of flavone-8-acetic acid and analogues in mice. Cancer Res. 1991;51:77–81. [PubMed] [Google Scholar]
- 3.Baguley BC, Cole G, Thomsen LL, Li Z. Serotonin involvement in the antitumour and host effects of flavone-8-acetic acid and 5,6-dimethylxanthenone-4-acetic acid. Cancer Chemother Pharmacol. 1993;33:77–81. doi: 10.1007/BF00686027. [DOI] [PubMed] [Google Scholar]
- 4.Philpott M, Baguley BC, Ching L-M. Induction of tumour necrosis factor-alpha by single and repeated doses of the antitumour agent 5,6-dimethylxanthenone-4-acetic acid. Cancer Chemother Pharmacol. 1995;36:143–148. doi: 10.1007/BF00689199. 10.1007/s002800050299. [DOI] [PubMed] [Google Scholar]
- 5.Pruijn FB, Vandaalen M, Holford NHG, Wilson WR. Mechanisms of enhancement of the antitumour activity of melphalan by the tumour-blood-flow inhibitor 5,6-dimethylxanthenone-4-acetic acid. Cancer Chemother Pharmacol. 1997;39:541–546. doi: 10.1007/s002800050611. 10.1007/s002800050611. [DOI] [PubMed] [Google Scholar]
- 6.Ching L-M, Browne WL, Tchernegovski R, Gregory T, Baguley BC, Palmer BD. Interaction of thalidomide, phthalimide analogues of thalidomide and pentoxifylline with the anti-tumour agent 5,6-dimethylxanthenone-4-acetic acid: concomitant reduction of serum tumour necrosis factor-alpha and enhancement of anti-tumour activity. Br J Cancer. 1998;78:336–343. doi: 10.1038/bjc.1998.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lash CJ, Li AE, Rutland M, Baguley BC, Zwi LJ, Wilson WR. Enhancement of the anti-tumour effects of the antivascular agent 5,6-dimethylxanthenone-4-acetic acid (DMXAA) by combination with 5-hydroxytryptamine and bioreductive drugs. Br J Cancer. 1998;78:439–445. doi: 10.1038/bjc.1998.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou SF, Paxton JW, Tingle MD, Kestell P, Ching L-M. In vitro and in vivo kinetic interactions of the anti-tumour agent 5,6-dimethylxanthenone-4-acetic acid with thalidomide and diclofenac. Cancer Chemother Pharmacol. 2001 doi: 10.1007/s002800000249. in press. [DOI] [PubMed] [Google Scholar]
- 9.Webster LK, Ellis AG, Kestell P, Rewcastle GW. Metabolism and elimination of 5,6-dimethylxanthenone-4-acetic acid in the isolated perfused rat liver. Drug Metab Dispos. 1995;23:363–368. [PubMed] [Google Scholar]
- 10.Miners JO, Valente L, Lillywhite KJ, et al. Preclinical prediction of factors influencing the elimination of 5,6-dimethylxanthenone-4-acetic acid, a new anticancer drug. Cancer Res. 1997;57:284–289. [PubMed] [Google Scholar]
- 11.Kestell P, Paxton JW, Rewcastle GW, Dunlop I, Baguley BC. Plasma disposition, metabolism and excretion of the experimental antitumour agent 5,6-dimethylxanthenone-4-acetic acid in the mouse, rat and rabbit. Cancer Chemother Pharmacol. 1999;43:323–330. doi: 10.1007/s002800050902. 10.1007/s002800050902. [DOI] [PubMed] [Google Scholar]
- 12.Zhou SF, Paxton JW, Tingle MD, Kestell P. Identification of the human liver cytochrome P450 isozyme responsible for the 6-methylhydroxylation of the novel anticancer drug 5,6-dimethylxanthenone-4-acetic acid. Drug Metab Dispos. 2000;28:1449–1456. [PubMed] [Google Scholar]
- 13.Rewcastle GW, Atwell GJ, Baguley BC, Calveley SB, Denny WA. Potential antitumour agents. 58. Synthesis and structure-activity relationships of substituted xanthenone-4-acetic acids active against the colon 38 tumour in vivo. J Med Chem. 1989;32:793–799. doi: 10.1021/jm00124a012. [DOI] [PubMed] [Google Scholar]
- 14.Kestell P, Dunlop IC, McCrystal MR, et al. Plasma pharmacokinetics of N-[2-(dimethylamino) ethyl]acridine-4-carboxamide in a phase I trial. Cancer Chemother Pharmacol. 1999;44:45–50. doi: 10.1007/s002800050943. 10.1007/s002800050943. [DOI] [PubMed] [Google Scholar]
- 15.Rewcastle GW, Kestell P, Baguley BC, Denny WA. Light-induced breakdown of flavone acetic acid and xanthenone analogues in solution. J Natl Cancer Inst. 1990;82:528–529. doi: 10.1093/jnci/82.6.528. [DOI] [PubMed] [Google Scholar]
- 16.Robson RA, Matthews AP, Miners JO, et al. Characterisation of theophylline metabolism by human liver microsomes. Br J Clin Pharmacol. 1987;24:293–300. doi: 10.1111/j.1365-2125.1987.tb03172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith PK, Krohn RI, Hermanson GT, et al. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 18.Omura T, Sato R. The carbon monoxide binding pigment of liver microsomes. I. Evidence for its hemoprotein nature. J Biol Chem. 1964;23:2370–2378. [PubMed] [Google Scholar]
- 19.Zhou SF, Paxton JW, Tingle MD, McCall J, Kestell P. Determinaton of two major metabolites of the novel anti-tumour agent 5,6-dimethylxanthenone-4-acetic acid in hepatic microsomal incubations by high-performance liquid chromatography with fluorescence detection. J Chromatogr (B) 1999;734:129–136. doi: 10.1016/s0378-4347(99)00341-2. [DOI] [PubMed] [Google Scholar]
- 20.von Moltke LL, Greenblatt DJ, Schmider J, Wright CE, Harmatz JS, Shader RI. In vitro approaches to predicting drug interactions in vivo. Biochem Pharmacol. 1998;55:113–122. doi: 10.1016/s0006-2952(97)00239-6. 10.1016/s0006-2952(97)00239-6. [DOI] [PubMed] [Google Scholar]
- 21.Jameson MB, Thomson PI, Baguley BC, et al. Phase I pharmacokinetic and pharmacodynamic study of 5,6-dimethylxanthenone-4-acetic acid (DMXAA), a novel antivascular agent. Proc Annu Meet Am Soc Clin Oncol. 2000;19:705. [Google Scholar]
- 22.Ito K, Iwatsubo T, Kanamitsu S, Ueda K, Suzuki H, Sugiyama Y. Prediction of pharmacokinetic alterations caused by drug–drug interactions: metabolic interaction in the liver. Pharmacol Rev. 1998;50:387–411. [PubMed] [Google Scholar]
- 23.Zhou SF, Paxton JW, Tingle MD, et al. Identification and reactivity of the major metabolite (b-1-glucuronide) of the anti-tumour agent 5,6-dimethylxanthenone-4-acetic acid (DMXAA) in humans. Xenobiotica. 2001 doi: 10.1080/00498250110043544. in press. [DOI] [PubMed] [Google Scholar]
- 24.Zhou SF, Paxton JW, Kestell P, Tingle MD. Reversible binding of the novel anti-tumour agent 5,6-dimethylxanthenone-4-acetic acid to plasma proteins and blood cells in various species. J Pharm Pharmacol. 2001 doi: 10.1211/0022357011775758. in press. [DOI] [PubMed] [Google Scholar]
- 25.Tucker GT. The rational selection of drug interaction studies: implications of recent advances in drug metabolism. Int J Clin Pharmacol Ther Toxicol. 1992;30:550–553. [PubMed] [Google Scholar]
- 26.Obach RS. Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: An examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab Dispos. 1999;27:1350–1359. [PubMed] [Google Scholar]
- 27.McLure JA, Miners JO, Birkett DJ. Nonspecific binding of drugs to human liver microsomes. Br J Clin Pharmacol. 2000;49:453–461. doi: 10.1046/j.1365-2125.2000.00193.x. 10.1046/j.1365-2125.2000.00193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paul C, Liliemark J, Tidefelt U, Gahrton G, Peterson C. Pharmacokinetics of daunorubicin and doxorubicin in plasma and leukemic cells from patients with acute nonlymphoblastic leukemia. Ther Drug Monitor. 1989;11:140–148. doi: 10.1097/00007691-198903000-00004. [DOI] [PubMed] [Google Scholar]
- 29.Galettis P, Boutagy J, Ma DD. Daunorubicin pharmacokinetics and the correlation with P-glycoprotein and response in patients with acute leukaemia. Br J Cancer. 1994;70:324–329. doi: 10.1038/bjc.1994.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Evans SM, Robertson IG, Paxton JW. Plasma protein binding of the experimental antitumour agent acridine-4-carboxamide in man, dog, rat and rabbit. J Pharm Pharmacol. 1994;46:63–67. doi: 10.1111/j.2042-7158.1994.tb03722.x. [DOI] [PubMed] [Google Scholar]
- 31.Nelson RL, Dyke RW, Root MA. Comparative pharmacokinetics of vindesine, vincristine and vinblastine in patients with cancer. Cancer Treat Rev. 1980;7(Suppl 1):17–24. doi: 10.1016/s0305-7372(80)80003-x. [DOI] [PubMed] [Google Scholar]
- 32.Donigian DW, Owellen RJ. Interaction of vinblastine, vincristine and colchicine with serum proteins. Biochem Pharmacol. 1973;22:2113–2119. doi: 10.1016/0006-2952(73)90110-x. [DOI] [PubMed] [Google Scholar]
- 33.Petros WP, Rodman JH, Mirro JJ, Evans WE. Pharmacokinetics of continuous-infusion amsacrine and teniposide for the treatment of relapsed childhood acute nonlymphocytic leukemia. Cancer Chemother Pharmacol. 1991;27:397–400. doi: 10.1007/BF00688865. [DOI] [PubMed] [Google Scholar]
- 34.Paxton JW, Jurlina JL, Foote SE. The binding of amsacrine to human plasma proteins. J Pharm Pharmacol. 1986;38:432–438. doi: 10.1111/j.2042-7158.1986.tb04606.x. [DOI] [PubMed] [Google Scholar]