

Abstract
Background & Purpose:
Drug-resistant cancer cells frequently display efflux pumps such as P-glycoprotein (P-gp), the multidrug resistance associated protein (MRP1) or the transporter ABCG2. These transporters are each capable of mediating the active efflux of numerous anticancer drugs and display relatively distinct substrate preferences. The last, most recently discovered member, ABCG2, plays a major role in resistance in several types of cancer and the precise pharmacology of this multidrug transporter remain unresolved as does the nature of substrate binding.
Experimental Approach:
Plasma membranes from insect cells expressing ABCG2 were used to characterise binding of [3H]daunomycin to the multidrug transporter. The kinetics of association and dissociation for this substrate and several other compounds were also determined in this experimental system.
Key Results:
The dissociation constant for [3H]daunomycin binding was 564±57 nM and a Hill slope of 1.4 suggested cooperative binding. Doxorubicin, prazosin and daunomycin completely displaced the binding of radioligand, while mitoxantrone and Hoechst 33342 produced only a partial displacement. Analysis of the dissociation rates revealed that [3H]daunomycin and doxorubicin bind to multiple sites on the transporter.
Conclusions:
Both kinetic and equilibrium data support the presence of at least two symmetric drug binding sites on ABCG2, which is distinct from the asymmetry observed for P-gp. The data provide the first molecular details underlying the mechanism by which this transporter is capable of interacting with multiple substrates.
Keywords: multidrug resistance, cancer chemotherapy, breast cancer resistance protein, ABCG2, ABC transporter, drug binding, daunomycin, drug efflux
Introduction
Reduction of anticancer drug accumulation is a primary or initial mechanism in the development of a resistant phenotype in cancer (Gottesman et al., 2002; Leonard et al., 2003). Efflux pumps mediate the reduced accumulation via active drug extrusion and their expression is correlated with poor prognosis, reduced rates of remission and low efficacy of conventional chemotherapy (Chan et al., 1991; Hornicek et al., 2000; van den Heuvel-Eibrink et al., 2000; Burger et al., 2004). The most widely investigated ‘multidrug' efflux pumps are P-glycoprotein (P-gp or ABCB1) and MRP1 (ABCC1) with the former being associated with the extrusion of over 200 known compounds (Wang et al., 2003). The third ‘multidrug' transporter contributing to cancer resistance was discovered using a series of cell lines resistant to mitoxantrone, topotecan and anthracyclines, but not expressing ABCB1 or ABCC1 (Doyle et al., 1998; Miyake et al., 1999). The protein responsible for the resistance was ABCG2, also a member of the ABC transporter family. Unlike ABCB1 and ABCC1, ABCG2 is a ‘half-transporter' that is assumed to form a dimer in order to display a role in transport (Kage et al., 2002; Litman et al., 2002; Polgar et al., 2004; Xu et al., 2004). In addition, the ABCG2 transporter displays a ‘reverse topology' with an N-terminal nucleotide-binding domain and a C-terminal membrane spanning domain (Allikmets et al., 1998).
The function of ABCG2 is best reflected by the localization profile in the small and large intestine, placenta, renal proximal tubules and the bile canalicular membrane (Allikmets et al., 1998; Fetsch et al., 2005). This physiological expression profile suggests a protective role for ABCG2, by limiting the absorption and increasing the excretion of hydrophobic biological molecules (Chan et al., 2004). In contrast, the precise role of ABCG2 in specific tumour types remains somewhat controversial. Expression of the transporter has been demonstrated in resistant variants of several breast, colon, ovarian and small cell lung cancer cultured cell lines (Diestra et al., 2002; Ejendal and Hrycyna, 2002). There are, however, fewer reports of its presence in cancers in vivo. Its relevance in acute myelogenous leukaemia (AML) and myelodysplastic syndrome (MDS) is well established and expression of ABCG2 is observed in 30% of patients (Benderra et al., 2004; Ross, 2004). There is also a role in solid tumours since expression of ABCG2 is used as a prognostic marker in lung cancer (Bessho et al., 2006; Nagashima et al., 2006). A recent report has also provided evidence that ABCG2 can affect the bioavailability of topoisomerase inhibitors in patients with gastrointestinal cancer (Kuppens et al., 2004).
There are several contradictory reports on the substrate specificity of ABCG2. For example, ABCG2 was cloned from mitoxantrone resistant colon cancer cell lines and the protein product (initially termed MXR) conferred resistance to mitoxantrone, methotrexate and camptothecins (Miyake et al., 1999). A concurrent investigation cloned ABCG2 from drug-resistant breast cancer cells and the transporter (initially termed BCRP) conferred resistance to mitoxantrone, doxorubicin, daunomycin and camptothecins (Doyle et al., 1998; Miyake et al., 1999). While there was overlap with substrates such as mitoxantrone, there were defined differences (methotrexate versus anthracyclines), yet it was clear that the MXR and BCRP proteins were similar. A systematic study (Honjo et al., 2001) explored the efflux and cytotoxicity of rhodamine123 and doxorubicin in 11 cell lines expressing MXR/BCRP. The data was correlated with protein sequence and it was apparent that the proteins varied at position 482. By comparison with the gene obtained from healthy placental tissue (Allikmets et al., 1998), the protein with an arginine at position 482 was designated as the wild-type and this protein could not transport rhodamine123 or doxorubicin (Honjo et al., 2001), but was capable of mediating methotrexate efflux (Mitomo et al., 2003). In contrast, a protein with glycine or threonine at position 482 was capable of transporting rhodamine123 and doxorubicin, but provided less resistance to SN-38 and topotecan (Honjo et al., 2001). Position 482 is proximal to transmembrane segment three; however, there is no biochemical data on the role of this residue in the translocation pathway.
The precise molecular mechanisms underlying ‘multidrug' transport remain a biological enigma despite considerable progress with ABCB1 and ABCC1. It is clear that while the multidrug transporters share broad substrate specificity and their substrate profiles overlap, there are important distinctions in the mode of drug–protein interaction and subsequent mechanism of translocation. The information on ABCG2-mediated transport lags considerably behind that available for ABCB1 and ABCC1. The majority of the available data report cytotoxicity profiles or steady-state rhodamine123 accumulation, neither of which provide molecular mechanistic information (Honjo et al., 2001; Robey et al., 2001; Mitomo et al., 2003; Bessho et al., 2006; Nagashima et al., 2006). ABCG2 expressed in insect cells demonstrated that prazosin and to a lesser extent, mitoxantrone, were able to stimulate the hydrolysis of ATP by the protein (Ozvegy et al., 2001, 2002). However, the majority of substrates did not significantly alter ATPase activity, suggesting that the protein displays weak or inefficient coupling.
The present manuscript explores the initial interaction of drugs with the transporter using radioligand-binding studies. The objective is to provide the mechanism underlying multidrug recognition by the R482G isoform (ABCG2R482G) by examination of the number of drug-binding sites and whether competitive or allosteric interactions exist between substrates on the transporter. The R482G isoform was chosen for these investigations because of its wider pharmacological spectrum, thereby enabling a more exhaustive characterization of drug binding to the transporter. Detailed characterization will facilitate future efforts to map the drug-binding site of the protein and examine the molecular impact of the numerous polymorphic variants of ABCG2 observed (Ishikawa et al., 2005).
Methods
Generation of recombinant baculovirus encoding N-His6- ABCG2R482G
An ABCG2 cDNA containing plasmid, encoding glycine at position 482 (ABCG2R482G), was obtained from Dr T Litman (University of Copenhagen). Oligonucleotide primers (5′ CTAGACTCGAGCGGCGCCATGTCTTCCAGTAATG, & 3′ CT TGGTACCGAGCGCGGCCGCCTAAGAATATTTTTTAAG) were employed to amplify the cDNA and add a 5′ KasI and 3′ NotI restriction site (underlined). The resultant PCR product was digested and subcloned into the pFastBac-HTC vector (Invitrogen, Paisley, UK) generating a cDNA that encodes an N-terminal extension to ABCG2R482G of MSYYHHHHHH DYDIPTTENLYFQGAMSSSN, where the affinity tag is italicized, the TEV-protease cleavage site is in bold and the first five amino acids of the open reading of ABCG2R482G are underlined. The fidelity of PCR was confirmed by DNA sequencing of this resultant construct. This vector was transformed into chemically competent DH10Bac Escherichia coli. Putative recombinant bacterial colonies were restreaked to ensure no contamination by nonrecombinant bacteria and were further screened by PCR, following isolation of high molecular weight bacmid DNA. Insect cells (Sf9, 9 × 105 cells per 30 mm dish) were transfected with recombinant bacmid DNA (3 μg DNA; 6 μg lipofectin) to produce an initial viral titre suitable for further amplification as previously described (Taylor et al., 2001).
Protein expression
The Trichoplusia ni (high five) cell line was routinely used for the expression of ABCG2 and maintained in shaking suspension cultures as previously described (Rothnie et al., 2004). High Five cells at a density of ∼3 × 106 cells ml−1 were infected with recombinant baculovirus (∼5 × 107 plaque-forming units ml−1) at a multiplicity of infection of 10. After 2 h of incubation with virus, the cells were diluted to a density of 1.5 × 106 cells ml−1 and grown for a further 3 days before harvesting by centrifugation (2000 g, 10 min).
Membrane preparation
Crude membrane preparations were isolated as previously described (Taylor et al., 2001), with the exception that buffers contained 20 mM MOPS, pH 7.4, 200 mM NaCl, 0.25 M sucrose. Briefly, cells were ruptured with four rounds of nitrogen cavitation using 1500 p.s.i. at 4°C with 20 min incubation in each round. Cellular debris was removed by centrifugation at 2000 g for 10 min. The crude membranes were isolated by ultracentrifugation at 100 000 g for 60 min at 4°C. Membranes were resuspended at protein concentrations of approximately 50 mg/ml in isolation buffer (0.25 M sucrose, 20 mM MOPS pH 7.4 1 mM benzamidine, 0.1 mg/ml pepstatin and 0.1 mg/ml leupeptin) and stored at −80°C.
Drug-binding assay
Radio-labelled drug-binding assays were based on a previously published procedure used to investigate ABCB1 (Martin et al., 1999). Membranes (10 μg) were incubated with the radiolabel, [3H]daunomycin, in a total volume of 100 μl in polypropylene test tubes. The membranes, [3H]daunomycin and any other drugs were incubated in hypotonic-binding buffer, which comprised 50 mM Tris-HCl pH 7.4. Times and temperatures of incubation varied depending on the nature of the specific experiment as outlined below. Nonspecific binding (NSB) was defined as the amount of [3H]daunomycin bound in the presence of a large molar excess (30 μM) of unlabelled daunomycin or doxorubicin. Drugs were added from concentrated stocks in DMSO and the solvent concentration was maintained at less than 1% (v/v). Actual concentrations of [3H]daunomycin added to the tubes were determined by liquid scintillation counting. Unbound ligand was separated from bound using rapid vacuum filtration through porous glass-fibre filters (GF/F) on a 48-well manifold. Samples on the filters were rinsed twice with 10 ml ice-cold wash buffer (50 mM Tris-HCl pH 7.4, 20 mM MgSO4). The GF/F filters were presoaked in wash buffer supplemented with 0.1% (w/v) BSA for 10 min. Bound [3H]daunomycin on filters was measured using liquid scintillation counting using Ready Protein+ Fluid.
Equilibrium drug-binding assays
Saturation isotherm analysis involved examination of the amount of [3H]daunomycin bound as a function of concentration added to the tubes. ABCG2R482G containing membranes were incubated at 20°C for 2 h in the presence of 1–1000 nM [3H]daunomycin as described above. Each radiolabel concentration was assayed in triplicate; two tubes were used to measure total binding (Btot) and third tube was used to determine the level of NSB. Specific binding (i.e. bound to ABCG2R482G) was defined as Btot – NSB and was converted to nmol/mg membrane protein. The amount bound was plotted as a function of [3H]daunomycin concentration and nonlinear regression of the Langmuir Adsorption Isotherm (Equation 1) was used to ascertain the affinity (Kd) and maximal binding capacity (Bmax).
Heterologous displacement assays were used to determine the ability and potency of drugs to interact with ABCG2R482G. The crude membranes were incubated with a single concentration of [3H]daunomycin (120–180 nM) at 20°C for 2 h. Unlabelled drugs were added over the concentration range 10 nM to 300 μM, obtained from serial dilution of a concentrated stock in DMSO. The only exceptions were Hoechst 33342, which is from a saline stock, and prazosin, which was stored as a 50:50 mixture of DMSO:methanol. The amount of [3H]daunomycin bound at each concentration of heterologous drug was expressed as a fraction of that obtained with radiolabel alone. The fraction bound was plotted as a function of added drug concentration and nonlinear regression of the general dose response relation (Equation 2) was used to ascertain the potency (IC50) and degree of displacement (FD).
Kinetic drug-binding assays
The rate of formation of the drug-ABCG2R482G complex was examined using a kinetic assay, which generated the association rate constant and permitted the analysis of drug–drug interactions with the transporter. Membranes were incubated with a fixed concentration of [3H]daunomycin for varying times (0, 1, 2, 3, 4, 5, 6, 8, 10, 12, 15, 17, 20, 30, 45 and 60 min) at 4, 10 or 20°C. Each time point sample comprised three tubes, one of which contained an excess of doxorubicin (30 μM) to enable determination of NSB, while the other two tubes indicated total binding. Association was initiated by the addition of [3H]daunomycin to tubes containing membranes. The specific binding was plotted as a function of time and nonlinear regression of Equation 3 to obtain the observed association rate constant (kobs). This parameter comprises both the association and dissociation rate constants, related by the expression kobs=k1·[L]+k−1. To obtain the association rate constant, kobs was measured at a range of [3H]daunomycin concentrations (50–400 nM) and plotted as a function of the amount of radiolabel added. The slope of this relationship provided the association rate constant.
The dissociation rate constant was also determined using a kinetic adaptation of the radioligand-binding assay. ABCG2R482G containing membranes were incubated with a fixed concentration of [3H]daunomycin (120–180 nM) for 2 h to attain equilibrium. This initial phase was always carried out at 20°C, regardless of the temperature used for the dissociation. At this point, the association reaction was effectively ‘blocked' by diluting the samples from 100 μl to 8 ml in binding buffer. The dilution was invoked for specific time intervals (40, 35, 30, 25, 20, 15, 10, 8, 6, 4, 2 and 0 min) and the rate of decay of the [3H]daunomycin- ABCG2R482G complex provided the dissociation rate constant. The total binding at each time point was expressed as a fraction of the amount bound at equilibrium. Fractional binding at each time point was plotted as a function of time and nonlinear regression of Equation 4 was used to measure the dissociation rate constant. To ascertain the effects of other compounds on the rate of dissociation, an excess of unlabelled drug was added to the dilution buffer.
Data analysis
Nonlinear regression analyses were generated using the GraphPad Prism4.0 program. Statistical analyses between data sets were carried out with ANOVA with the Tukey post hoc test and statistical significance was achieved where P<0.05. Comparisons of mean±s.e.m. were undertaken with at least three independent observations in each case. The following equations were used in nonlinear regressions:
 |
B=specific bound [3H]daunomycin (nmol/mg); Bmax=maximal binding capacity; Kd=dissociation constant (nM) and [L]=ligand concentration.
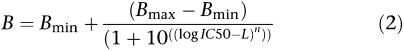 |
B=maximal [3H]daunomycin binding; Bmax=maximal binding; Bmin=minimum binding IC50=concentration of drug that leads to half maximal binding of radiolabel (nM); n=hill slope factor and L=log10(ligand concentration (M)). The binding capacities are expressed as either fraction of total or in absolute units of d.p.m.
B=fraction of maximal [3H]daunomycin binding; Bmax=maximal binding; t=time (min) and kobs=observed association rate (min−1).
B=fraction of maximal [3H]daunomycin binding; Bmax=maximal binding; Bmin=minimum binding; t=time (min) and koff=dissociation rate constant (nM min−1).
Materials
[3H]Daunomycin (8.2 Ci/mmol) was purchased from Perkin Elmer LAS (Beaconsfield, UK) and Ready Protein+ scintillation fluid was obtained from Beckman Coulter (High Wycombe, UK). Daunomycin, doxorubicin, mitoxantrone, methotrexate, rhodamine123, prazosin nicardipine and verapamil were purchased from Sigma (Poole, UK). Hoecsht 33342 was obtained from Molecular Probes (Leiden, Netherlands) and GF/F filters were purchased from VWR International (Lutterworth, UK). All consumables for DNA manipulation were obtained from New England Biolabs (Hitchin, UK) or from Qiagen (Crawley, UK). DNA sequencing was performed by the Biopolymer Synthesis and Analysis Unit, University of Nottingham. Insect Xpress media was obtained from Cambrex (Reading, UK) and Ex-cell 405 media from JRH Biosciences (Andover, UK).
Results
Equilibrium drug binding to ABCG2R482G
Daunomycin was used as the primary radioligand for investigation of binding by ABCG2R482G due to its commercial availability and established interaction with this isoform (Ozvegy et al., 2001; Allen et al., 2002). Membranes expressing the R482G isoform of ABCG2 containing an N-terminal hexahistidine tag were incubated in the presence of varying [3H]daunomycin concentrations. The amount of [3H]daunomycin bound as a function of concentration is shown in Figure 1. NSB was linear and accounted for < 15% of the total binding, while membranes not expressing ABCG2R482G did not display specific [3H]daunomycin binding (data not shown). Total binding accounted for < 10% of the amount of drug added to the tubes. Figure 1 demonstrates that the binding of [3H]daunomycin did not fully saturate due to the inability to attain sufficiently high concentrations using the commercially available preparation. As the binding was not fully saturated at concentrations up to 1 μM [3H]daunomycin, the maximal binding capacity (0.856±0.035 nmol/mg) and the dissociation constant (564±57 nM) can only be considered estimates of these parameters. The slope factor defining the relationship was 1.46, and the regression with this value was statistically more significant than using a value of 1.0 (P<0.05, F-test of slopes). This suggests that the binding of [3H]daunomycin cannot be described by a simple, single site process. In order to examine this issue and to generate a more accurate measure of the dissociation constant, kinetic analyses were applied to [3H]daunomycin binding.
Figure 1.

Saturation isotherm analysis for binding of [3H]daunomycin to ABCG2R482G. Insect cell membranes (10 μg) expressing ABCG2R482G were incubated with varying concentrations of [3H]daunomycin (1–1000 nM) for 2 h at 20°C. The total amount bound was collected by rapid filtration procedures and the NSB subtracted. The specific binding of drug to the membranes was plotted as a function of added [3H]daunomycin concentration and fitted to Equation 2 using nonlinear regression. The graph represents pooled data from six independent preparations.
Kinetics of drug-binding to ABCG2R482G
Figure 2a shows a representative data set of the specific binding of [3H]daunomycin to membranes containing ABCG2R482G, as a function of time. The time constant from this relationship is known as the observed association rate (kobs) since it encompasses both the formation and breakdown of the [3H]daunomycin-ABCG2R482G complex. The kobs was converted to the true association rate constant (kon) as described in the Methods. At a temperature of 20°C, the kon was estimated as 3.31±0.38 pM−1 min−1 (Table 1) from a number of independent data sets. At this temperature, the formation of the [3H]daunomycin-ABCG2R482G complex was rapid, thereby introducing systematic error in the estimation of kobs and kon. Consequently, the association rate constant was also evaluated at 4 and 10°C to ensure more reliable determination of the data, with the results shown in Table 1.
Figure 2.

Kinetics of [3H]daunomycin binding to ABCG2R482G. (a) A representative data set of the association kinetics for specific [3H]daunomycin (100 nM) binding to ABCG2R482G containing membranes (10 μg) at 10°C. NSB was calculated in the presence of 30 μM unlabelled doxorubicin and subtracted from total binding. Specific binding was measured at various intervals from 1 to 50 min. The observed rate constant (kobs) was determined by nonlinear regression of the single-phase association curve (Equation 3) to the data. (b) Dissociation kinetics for [3H]daunomycin from membranes containing ABCG2R482G in the absence or presence of 30 μM doxorubicin. Membranes (10 μg) and [3H]daunomycin (150 nM) were allowed to reach equilibrium for 2 h at 20°C. Dissociation was initiated by dilution of the samples (100 μl) with 8 ml buffer. Specific binding was measured at various intervals from 1 to 40 min. The dissociation rate constant (koff) was determined by nonlinear regression of the single-phase dissociation curve (Equation 4) to the data. Data represent the mean±s.e.m. of four independent observations.
Table 1.
Kinetic parameters for [3H]daunomycin binding to ABCG2R482G containing membranes
| Temperature T (°C) | Association rate constant kon (pM−1 min−1) | Dissociation rate constant koff (min−1) | Dissociation half-life t1/2 (min) | Kinetically derived Kd (nM) |
|---|---|---|---|---|
| 4 | 1.83±0.67 (3) | 0.181±0.010 (4) | 3.87±0.24 | 98.9 |
| 10 | 3.73±0.49 (8) | 0.265±0.013 (13) | 2.91±0.20 | 71.0 |
| 20 | 3.31±0.38 (9) | 0.325±0.054 (4) | 2.32±0.38 | 98.2 |
Membrane preparations (10 μg protein) from high five cells expressing ABCG2R482G were incubated with [3H]-daunomycin to measure the association and dissociation rate constants. Membranes were incubated with varying concentrations of [3H]-daunomycin for periods ranging from 1 to 120 min at 4, 10 or 20°C to determine the association rate constant. The observed rate constant for this reaction was determined by nonlinear regression of Equation 3 and used to estimate the true association rate constant as described in Methods. In order to determine dissociation kinetics, membranes and [3H]-daunomycin (150–200 nM) were incubated for 2 h at various temperatures to attain equilibrium binding. The membranes were diluted to 100 × volume to initiate dissociation of [3H]-daunomycin. The dissociation rate constant (koff) was determined by nonlinear regression of Equation 4. Values represent the mean±s.e.m and values in parentheses refer to the number of independent observations used to obtain the value.
Figure 2b (solid symbols) demonstrates the dissociation of the [3H]daunomycin- ABCG2R482G complex following the attainment of equilibrium, which is defined as a fraction bound (Bt/B0) of 1.0. The dissociation profile was best fitted with a single phase exponential decay. The dissociation rate constant derived from this equation at 20°C was rapid (Table 1) and equated to a decay half-life of approximately 2 min. The dissociation rate constant displayed a positive temperature dependence as demonstrated in Table 1, with the rate constant at 4°C about half that at 20°C.
The dissociation constant is defined as Kd=koff/kon and consequently the two rate constants were used to provide an estimate of the affinity of [3H]daunomycin binding to ABCG2R482G. The estimates obtained at each temperature (4, 10 and 20°C) are included in Table 1. The values obtained at 4°C are the most reliable due to the errors associated with assigning rate constants under the rapid conditions of binding at higher temperatures, although the three values obtained are in close agreement. The dissociation constant for the binding of [3H]daunomycin binding to ABCG2R482G was therefore defined as 99 nM and provides a more reliable measure than the higher value obtained using equilibrium binding. The rate of association of daunomycin to ABCG2R482G was more rapid than vinblastine binding to ABCB1 (Martin et al., 1999), which may be related to the relatively greater hydrophilicity of the former. In contrast, the rate of dissociation of daunomycin from ABCG2R482G was approximately 10-fold greater than observed for vinblastine or XR9576 interaction with ABCB1 (Martin et al., 1999).
Heterologous binding interactions on ABCG2R482G
Subsequent investigations focussed on further characterizing the pharmacology of drug interaction with ABCG2R482G, particularly examining possible drug–drug interactions on this multidrug transporter. This was achieved initially with heterologous displacement assays using compounds previously demonstrated to interact with the R482G isoform of ABCG2 (primarily through steady-state accumulation and ATPase assays). Figure 3a demonstrates the ability of unlabelled daunomycin and the related anthracycline anticancer drug doxorubicin to displace the binding of [3H]daunomycin to ABCG2R482G. Both compounds caused >90% reduction in the binding of [3H]daunomycin, however, the potency of daunomycin to displace binding was 3.3-fold lower than that observed for doxorubicin (Table 2). Other drugs, structurally unrelated to anthracyclines, were also investigated in this assay (Table 2, Figure 3b). Mitoxantrone is one of the few compounds demonstrated to interact with both the wild-type and R482G mutant isoforms of ABCG2 (Mitomo et al., 2003; Sarkadi et al., 2004). Mitoxantrone was also more potent than unlabelled daunomycin in displacing [3H]daunomycin, however, the fraction displaced (FD) was only approximately 60% of total binding. The DNA intercalating compound Hoechst33342 also produced a partial displacement of [3H]daunomycin binding to ABCG2R482G (Table 2). The fraction displaced was only about 30% of total binding, yet the potency of this compound remained similar to that of daunomycin. The partial displacement of [3H]daunomycin binding to ABCG2R482G by mitoxantrone and Hoechst33342 is suggestive of multiple drug-binding sites. Moreover, the data indicates that the radioligand itself may bind to two sites on ABCG2R482G and that mitoxantrone/Hoechst33342 displace from one, but not the other. Of the compounds tested, prazosin was the least potent in displacing bound [3H]daunomycin (Table 2 and Figure 3). This antihypertensive drug is a known stimulator of ATP hydrolysis by ABCG2R482G and a substrate for transport (Ozvegy et al., 2001; Robey et al., 2001). The fraction of [3H]daunomycin binding displaced by prazosin was about the same as that displaced by unlabelled daunomycin but prazosin had an IC50 value almost 100-fold higher than that of daunomycin. As demonstrated in Table 2, neither rhodamine123 nor methotrexate was able to displace more than 10% of the [3H]daunomycin binding.
Figure 3.

Heterologous displacement of [3H]daunomycin binding to ABCG2R482G. Membranes were incubated with radiolabel and varying concentrations (1 nM–50 μM) of displacing agent for 2 h at 20°C. The binding of [3H]daunomycin at each drug concentration was expressed as a fraction of that observed in the absence of displacing drug. The IC50 value for each drug was obtained from nonlinear regression of the general dose–response relationship (Equation 2). (a) Representative curve demonstrating the displacement of [3H]daunomycin (150 nM) binding to ABCG2R482G membranes by doxorubicin and daunomycin. (b) Representative curve demonstrating the displacement of [3H]daunomycin (150 nM) binding to ABCG2R482G membranes by mitoxantrone, Hoecsht33342 and prazosin.
Table 2.
Displacement of [3H]daunomycin from ABCG2R482G containing membranes
| Drug | N | FD | IC50 (μM) |
|---|---|---|---|
| Daunomycin | 6 | 0.921±0.024 | 2.49±0.32 |
| Doxorubicin | 6 | 0.915±0.014 | 0.75±0.09a |
| Mitoxantrone | 5 | 0.629±0.061b | 1.32±0.30a |
| Hoecsht33342 | 5 | 0.357±0.022b | 2.53±0.68 |
| Prazosin | 3 | 0.896±0.035 | 237±33a |
| Rhodamine 123 | 3 | <0.1 | ND |
| Methotrexate | 3 | <0.1 | ND |
Abbreviations: ND, not determined.
Membrane preparations (10 μg protein) of high five cells expressing ABCG2R482G were incubated with [3H]-daunomycin (150–200 nM) in the presence of several compounds (10−8–5 × 10−4 M) for 2 h at room temperature. The fraction of displaced [3H]-daunomycin (FD) and the potency (IC50) for each drug to displace the binding were determined by nonlinear regression of the general dose–response relationship. Values represent the mean±s.e.m. from n independent observations.
Indicates that the IC50 was different (P<0.05 ANOVA) from that produced by daunomycin.
Indicates that the FD was different (P<0.05 ANOVA) from that produced by daunomycin.
Allosteric interactions between drugs binding to ABCG2R482G
The kinetic parameters of drug binding were examined for ABCG2R482G to characterize the nature of drug interaction with the transporter. For example, according to the law of mass action, any alteration in the dissociation rate constant can only be attributed to an allosteric (i.e. noncompetitive) effect (Weiland and Molinoff, 1981; Motulsky and Mahan, 1983; Kenakin, 1997). Consequently, the dissociation of [3H]daunomycin binding was measured in the presence or absence of drugs to define whether their interaction was competitive.
In the presence of 30 μM prazosin, the dissociation rate of [3H]daunomycin (koff) was not significantly different from the control value (Table 3). The fraction displaced was >90% and indicates a competitive interaction between prazosin and daunomycin. Similarly, the dissociation of [3H]daunomycin was not significantly altered in the presence of Hoechst33342 (Table 3), also suggesting a purely competitive interaction. However, the fraction of radiolabel displaced in the presence of Hoechst33342 was 0.811±0.021 and therefore a proportion of the [3H]daunomycin (∼20% of total bound) remains bound to the protein – even with longer incubation. Increasing the radioligand concentration exacerbated this effect (data not shown) and suggests a proportion of the [3H]daunomycin bound is unable to dissociate from ABCG2R482G. Mitoxantrone, like prazosin and Hoechst33342, failed to cause any significant increase in the dissociation rate of [3H]daunomycin, which is suggestive of competitive displacement. Mitoxantrone also failed to completely dissociate [3H]daunomycin binding, with only about half displaced (Table 3) and this residual binding remained at this level for an extended period. As the dilution strategy employed in this investigation is the stimulus for [3H]daunomycin dissociation, a significant residual binding remaining was unexpected. This unusual finding further suggests that mitoxantrone and Hoechst33342 competitively displace [3H]daunomycin from one of its binding sites on ABCG2R482G, but lead to ‘stabilization' at the alternate site. These results indicate that ABCG2R482G contains multiple drug-binding sites with a communication network between them.
Table 3.
Dissociation kinetics of [3H]-daunomycin from ABCG2R482G containing membranes in the presence of drugs
| Drug | N | koff (min−1) | FD |
|---|---|---|---|
| Control | 13 | 0.248±0.013 | 0.935±0.010 |
| Daunomycin | 4 | 2.065±0.522a | 0.970±0.004 |
| Doxorubicin | 3 | 3.353±0.253a | 0.979±0.002 |
| Mitoxantrone | 9 | 0.602±0.065 | 0.563±0.031b |
| Prazosin | 4 | 0.313±0.024 | 0.935±0.006 |
| Hoechst33342 | 3 | 0.301±0.016 | 0.811±0.021 |
Membrane preparations (10 μg protein) of high five cells expressing ABCG2R482G were incubated with [3H]-daunomycin (150–200 nM) for 2 h at 20°C to attain equilibrium binding. The membranes were diluted to 100 × volume to initiate dissociation of [3H]-daunomycin. Dissociation measurements were undertaken in the presence or absence of various compounds (30 μM). The fraction of displaced [3H]-daunomycin (FD) and the dissociation rate constant (koff) were determined by nonlinear regression of the Equation 4. Values represent the mean±s.e.m. from n independent observations.
Indicates that the koff was different (P<0.05 ANOVA) from the control value.
Indicates that the FD was different (P<0.05 ANOVA) from the control value.
The anticancer drug doxorubicin, was also examined for its ability to displace [3H]daunomycin binding (Figure 2b); these two compounds are both anthracyclines and therefore share many structural and functional similarities. The decay of the [3H]daunomycin-ABCG2R482G complex in the presence of 30 μM doxorubicin was increased 13.5-fold (Table 3). This clearly indicates that despite their similarity, the two drugs may bind to distinct sites on the transporter, which are linked by a negative heterotropic allosteric communication.
Kinetic studies with unlabelled daunomycin added a degree of complexity to the interpretation of drug-ABCG2R482G interactions since it also accelerated the rate of [3H]daunomycin dissociation by 8.3-fold (Table 3). This indicates a negative cooperativity for the binding of daunomycin to ABCG2R482G.
Discussion
The ability of a single transporter to induce the efflux of a large number of structurally and functionally unrelated compounds remains a biological enigma. Understanding the mechanism is a key element to facilitate the rational design of pharmacological inhibitors of proteins such as ABCG2, ABCB1 and ABCC1 in order to restore efficacy of chemotherapy. Radioligand-binding studies with ABCB1 have demonstrated the presence of at least four distinct binding sites and similar investigations with ABCC1 defined a drug-binding site and an allotropic modulator site that interacts with glutathione. The present investigation provides the first comparable characterization of the ABCG2 transporter and enables assignment of three distinct pharmacological binding sites in the ABCG2R482G isoform. Two of the sites display considerable allosteric communication while there was no interaction observed with the third site. There is also a negative cooperativity for the binding of a single ligand, daunomycin, something not observed for ABCB1 or ABCC1. In addition, certain compounds elicit a stabilization of one daunomycin-binding site via a positive heterotrophic allostery. A further distinction between drug binding to ABCG2R482G and to ABCB1 is in the kinetics of association and dissociation. We observed that both processes occur significantly faster for daunomycin with ABCG2R482G, compared to the interaction of vinblastine or XR9576 with ABCB1 (Martin et al., 1999), although this may be related to the greater hydrophilicity of daunomycin compared to the other drugs.
How does our binding data compare to the existing data on transport of drugs by both wild-type and R482G ABCG2? The antifolate chemotherapeutic drug methotrexate was not able to displace drug binding to the ABCG2R482G isoform, which is in agreement with studies demonstrating that it is not transported by the ABCG2R482G isoform (Chen et al., 2003; Mitomo et al., 2003). In apparent contrast, rhodamine123 is a substrate capable of stimulating the ATPase activity of the ABCG2R482G isoform, but not the wild-type protein (R482) (Ozvegy et al., 2001), and is also exported from cells expressing ABCG2R482G. This illustrates that the difference in transport capabilities of isoforms differing at position 482 is at the level of initial drug interaction with the transporters, rather than an inability to progress through the translocation pathway. The existing sets of data (Ozvegy et al., 2001; Mitomo et al., 2003) demonstrate that rhodamine123 directly interacts with ABCG2R482G. The inability to displace [3H]daunomycin binding using concentrations up to 500 μM does not contradict this, but it indicates that the two compounds interact at pharmacologically distinct sites on the protein and that these two regions are not connected allosterically. The fact that rhodamine123, a known substrate of ABCG2R482G, could not displace [3H]daunomycin binding and that mitoxantrone and Hoechst33342 caused only partial displacement strongly suggests multiple sites for interaction on the protein. As a consequence, noncompetitive or allosteric drug interactions are likely to be a hallmark of this transporter.
Interestingly, despite their structural similarity, the two anthracycline chemotherapeutic agents, doxorubicin and daunomycin, exhibited a negative allosteric interaction on the ABCG2R482G transporter. This type of communication is similar to the types of drug–drug interactions reported for ABCB1 (Martin et al., 2000) and the L-type Ca2+ channel (Glossmann et al., 1984). Moreover, there was also a negative cooperativity and by definition, multiple sites for the binding of daunomycin to ABCG2R482G. The binding of [3H]vinblastine to the bacterial multidrug ‘half-transporter' LmrA also displayed cooperativity and this data was interpreted as proof of two binding sites for the radioligand (van Veen et al., 2000), which is in contrast to findings with the single polypeptide transporter ABCB1 (Martin et al., 2000). Consequently, daunomycin and doxorubicin may not bind to distinct sites on the transporter; instead the binding interactions reported here are explained by the presence of two allosterically linked sites for anthracyclines (conceivably one on each ABCG2R482G monomer). The presence of multiple interacting binding sites for [3H]daunomycin is also consistent with the Hill slope (1.46) obtained from analysis of the equilibrium binding data.
Figure 4 summarizes drug binding to the ABCG2R482G isoform, which contains two units each with two binding sites. In the simplest scenario each of these units would correspond to a monomer of ABCG2R482G (but see below for an alternative interpretation). Each unit contains distinct and noninteracting binding sites for rhodamine123 and [3H]daunomycin. The model also depicts that mitoxantrone, Hoechst33342 and daunomycin bind within the same pocket of ABCG2R482G. This is consistent with the partly competitive interactions provided above. Daunomycin and doxorubicin may bind to each of the units in the model and there is a negative allosteric interaction between the two sites to explain the interactions producing accelerated radioligand dissociation. However, this negative allosteric interaction between the ‘daunomycin-binding sites' is not observed for all ligands. Binding of mitoxantrone and Hoechst33342 to a site will displace daunomycin at that site in a competitive fashion and generate a positive allostery at the alternate site, leading to ‘stabilization' of binding.
Figure 4.

Schematic representation of drug-binding sites on ABCG2R482G. The model depicts two identical units, each capable of binding substrates. One site on each unit exclusively interacts with rhodamine123 while the other was characterized as the binding site for [3H]daunomycin. The latter site is also capable of binding doxorubicin, prazosin, mitoxantrone and Hoechst33342. Interactions also exist between the two protein units as demonstrated by the arrows; the ⊖ symbol indicates a negative allosteric interaction while the ⊕ symbol represents a positive cooperativity. The ‘protein units' may be monomers of ABCG2R482G, or alternatively each unit comprises a functional transporter (i.e. a dimer of ABCG2R482G). It is not possible from the present data to determine whether the two rhodamine123 sites interact, or whether a second site exists for the binding of mitoxantrone and Hoechst33342 and is responsible for the ‘stabilization' effect.
The presence of multiple binding sites for a single ligand on a protein, or the negative cooperativity between them is not an uncommon property for membrane proteins. For example, the rate of [3H]-bradykinin dissociation from the BK-receptor is also accelerated by the unlabelled compound and this negative cooperativity has been interpreted as a mechanism of desensitization (Pizard et al., 1998). It is unclear whether a single BK-receptor contained two ligand-binding sites, or whether the phenomenon occurred via interaction between proximal receptors. Cytokine binding to hetero- and homo-oligomers of the CCR2/CCR5 receptors also displayed negative allostery (Springael et al., 2006).
The binding site interactions reported here for the ABCG2R482G transporter may be explained by two daunomycin-binding sites on a functional dimeric assembly of the protein. A great body of biochemical and structural evidence with intact ABC transporters indicates that two nucleotide-binding domains are required to generate a functional transporter (al-Shawi and Senior, 1993; Urbatsch et al., 1995a, 1995b; Lacaille and Androlewicz, 1998). By implication, half-transporters including ABCG2 will dimerize in order to satisfy this functional criterion (Graf et al., 2003; Guimaraes et al., 2004). Consequently, a great deal of effort has been invested to demonstrate that ABCG2 exists as a functional homo-dimer, although there are suggestions that the protein forms higher order oligomeric species (Kage et al., 2002; Litman et al., 2002; Polgar et al., 2004; Xu et al., 2004). However, the possibility that multiple homodimeric assemblies of ABCG2 interact to generate the allosteric binding data presented here cannot be discounted. Recent structural analyses obtained using electron microscopy have demonstrated that the purified transporter may be found in a large complex consisting of a tetramer of dimers (McDevitt et al., 2006). This association of monomers into a complex would allow for functional interactions between multiple transporter units, which could influence many aspects of observed pharmacology; for example, ligand binding may stabilize subunit interactions or conversely the interactions may modulate substrate binding to regulate the ultimate functional outcome (Williams et al., 2004). Further studies will also be required to compare the binding of drugs to wild-type ABCG2 and the data obtained with the R482G isoform will be useful in eventually describing the location and molecular properties of drug interactions with this transporter.
Acknowledgments
This research was funded by a Cancer Research UK Program grant (SP1861/0401). IDK acknowledges the Royal Society and the University of Nottingham for support. RC would like to thank Drs George, Megzee, Sheyla and Smočin for assistance with the GMT program.
Abbreviations
- ABC
ATP binding cassette
- AML
acute myelogenous leukaemia
- BCRP
breast cancer resistance protein
- Btot
total binding
- DMSO
dimethyl sulphoxide
- FD
fraction displaced
- KD
dissociation constant
- kobs
observed rate constant
- koff
dissociation rate constant
- kon
association rate constant
- MDS
myelodysplastic syndrome
- MRP
multidrug resistance associated protein
- MXR
mitoxantrone resistance protein
- NSB
nonspecific binding
- P-gp
P-glycoprotein
Conflict of interest
The authors state no conflict of interest.
References
- al-Shawi MK, Senior AE. Characterization of the adenosine triphosphatase activity of Chinese hamster P-glycoprotein. J Biol Chem. 1993;268:4197–4206. [PubMed] [Google Scholar]
- Allen JD, Jackson SC, Schinkel AH. A mutation hot spot in the Bcrp1 (Abcg2) multidrug transporter in mouse cell lines selected for Doxorubicin resistance. Cancer Res. 2002;62:2294–2299. [PubMed] [Google Scholar]
- Allikmets R, Schriml LM, Hutchinson A, Romano-Spica V, Dean M. A human placenta-specific ATP-binding cassette gene (ABCP) on chromosome 4q22 that is involved in multidrug resistance. Cancer Res. 1998;58:5337–5339. [PubMed] [Google Scholar]
- Benderra Z, Faussat AM, Sayada L, Perrot JY, Chaoui D, Marie JP, et al. Breast cancer resistance protein and P-glycoprotein in 149 adult acute myeloid leukemias. Clin Cancer Res. 2004;10:7896–7902. doi: 10.1158/1078-0432.CCR-04-0795. [DOI] [PubMed] [Google Scholar]
- Bessho Y, Oguri T, Achiwa H, Muramatsu H, Maeda H, Niimi T, et al. Role of ABCG2 as a biomarker for predicting resistance to CPT-11/SN-38 in lung cancer. Cancer Sci. 2006;97:192–198. doi: 10.1111/j.1349-7006.2006.00164.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger H, van Tol H, Boersma AW, Brok M, Wiemer EA, Stoter G, et al. Imatinib mesylate (STI571) is a substrate for the breast cancer resistance protein (BCRP)/ABCG2 drug pump. Blood. 2004;104:2940–2942. doi: 10.1182/blood-2004-04-1398. [DOI] [PubMed] [Google Scholar]
- Chan HS, Haddad G, Thorner PS, DeBoer G, Lin YP, Ondrusek N, et al. P-glycoprotein expression as a predictor of the outcome of therapy for neuroblastoma. N Engl J Med. 1991;325:1608–1614. doi: 10.1056/NEJM199112053252304. [DOI] [PubMed] [Google Scholar]
- Chan LM, Lowes S, Hirst BH. The ABCs of drug transport in intestine and liver: efflux proteins limiting drug absorption and bioavailability. Eur J Pharm Sci. 2004;21:25–51. doi: 10.1016/j.ejps.2003.07.003. [DOI] [PubMed] [Google Scholar]
- Chen ZS, Robey RW, Belinsky MG, Shchaveleva I, Ren XQ, Sugimoto Y, et al. Transport of methotrexate, methotrexate polyglutamates, and 17beta-estradiol 17-(beta-D-glucuronide) by ABCG2: effects of acquired mutations at R482 on methotrexate transport. Cancer Res. 2003;63:4048–4054. [PubMed] [Google Scholar]
- Diestra JE, Scheffer GL, Catala I, Maliepaard M, Schellens JH, Scheper RJ, et al. Frequent expression of the multi-drug resistance-associated protein BCRP/MXR/ABCP/ABCG2 in human tumours detected by the BXP-21 monoclonal antibody in paraffin-embedded material. J Pathol. 2002;198:213–219. doi: 10.1002/path.1203. [DOI] [PubMed] [Google Scholar]
- Doyle LA, Yang W, Abruzzo LV, Krogmann T, Gao Y, Rishi AK, et al. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc Natl Acad Sci USA. 1998;95:15665–15670. doi: 10.1073/pnas.95.26.15665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ejendal KF, Hrycyna CA. Multidrug resistance and cancer: the role of the human ABC transporter ABCG2. Curr Protein Pept Sci. 2002;3:503–511. doi: 10.2174/1389203023380521. [DOI] [PubMed] [Google Scholar]
- Fetsch PA, Abati A, Litman T, Morisaki K, Honjo Y, Mittal K, et al. Localization of the ABCG2 mitoxantrone resistance-associated protein in normal tissues. Cancer Lett. 2005;235:84–92. doi: 10.1016/j.canlet.2005.04.024. [DOI] [PubMed] [Google Scholar]
- Glossmann H, Ferry DR, Goll A, Rombusch M. Molecular pharmacology of the calcium channel, evidence for sub-types, multiple drug receptor sites, channel sub-units, and the development of a radioiodinated 1,4-dihydropyridine calcium channel label, [125I]Iodipine. J Cardiovascular Pharmacol. 1984;6:S608–S621. [PubMed] [Google Scholar]
- Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- Graf GA, Yu L, Li WP, Gerard R, Tuma PL, Cohen JC, et al. ABCG5 and ABCG8 are obligate heterodimers for protein trafficking and biliary cholesterol excretion. J Biol Chem. 2003;278:48275–48282. doi: 10.1074/jbc.M310223200. [DOI] [PubMed] [Google Scholar]
- Guimaraes CP, Domingues P, Aubourg P, Fouquet F, Pujol A, Jimenez-Sanchez G, et al. Mouse liver PMP70 and ALDP: homomeric interactions prevail in vivo. Biochim Biophys Acta. 2004;1689:235–243. doi: 10.1016/j.bbadis.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Honjo Y, Hrycyna CA, Yan QW, Medina-Perez WY, Robey RW, van de Laar A, et al. Acquired mutations in the MXR/BCRP/ABCP gene alter substrate specificity in MXR/BCRP/ABCP-over expressing cells. Cancer Res. 2001;61:6635–6639. [PubMed] [Google Scholar]
- Hornicek FJ, Gebhardt MC, Wolfe MW, Kharrazi FD, Takeshita H, Parekh SG, et al. P-glycoprotein levels predict poor outcome in patients with osteosarcoma. Clin Orthop Relat Res. 2000;382:11–17. doi: 10.1097/00003086-200004000-00003. [DOI] [PubMed] [Google Scholar]
- Ishikawa T, Tamura A, Saito H, Wakabayashi K, Nakagawa H. Pharmacogenomics of the human ABC transporter ABCG2: from functional evaluation to drug molecular design. Naturwissenschaften. 2005;92:451–463. doi: 10.1007/s00114-005-0019-4. [DOI] [PubMed] [Google Scholar]
- Kage K, Tsukahara S, Sugiyama T, Asada S, Ishikawa E, Tsuruo T, et al. Dominant-negative inhibition of breast cancer resistance protein as drug efflux pump through the inhibition of S-S dependent homodimerization. Int J Cancer. 2002;97:626–630. doi: 10.1002/ijc.10100. [DOI] [PubMed] [Google Scholar]
- Kenakin T. Pharmacologic Analysis of Drug-Receptor Interaction. Lippincott-Raven; 1997. [Google Scholar]
- Kuppens IE, Beijnen J, Schellens JH. Topoisomerase I inhibitors in the treatment of gastrointestinal cancer: from intravenous to oral administration. Clin Colorectal Cancer. 2004;4:163–180. doi: 10.3816/ccc.2004.n.017. [DOI] [PubMed] [Google Scholar]
- Lacaille VG, Androlewicz MJ. Herpes simplex virus inhibitor ICP47 destabilizes the transporter associated with antigen processing (TAP) heterodimer. J Biol Chem. 1998;273:17386–17390. doi: 10.1074/jbc.273.28.17386. [DOI] [PubMed] [Google Scholar]
- Leonard GD, Fojo T, Bates SE. The role of ABC transporters in clinical practice. Oncologist. 2003;8:411–424. doi: 10.1634/theoncologist.8-5-411. [DOI] [PubMed] [Google Scholar]
- Litman T, Jensen U, Hansen A, Covitz KM, Zhan Z, Fetsch P, et al. Use of peptide antibodies to probe for the mitoxantrone resistance-associated protein MXR/BCRP/ABCP/ABCG2. Biochim Biophys Acta. 2002;1565:6–16. doi: 10.1016/s0005-2736(02)00492-3. [DOI] [PubMed] [Google Scholar]
- Martin C, Berridge G, Higgins CF, Mistry P, Charlton P, Callaghan R. Communication between multiple drug binding sites on P-glycoprotein. Mol Pharmacol. 2000;58:624–632. doi: 10.1124/mol.58.3.624. [DOI] [PubMed] [Google Scholar]
- Martin C, Berridge G, Mistry P, Higgins C, Charlton P, Callaghan R. The molecular interaction of the high affinity reversal agent XR9576 with P-glycoprotein. Br J Pharmacol. 1999;128:403–411. doi: 10.1038/sj.bjp.0702807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDevitt C, Collins RF, Conway M, Storm J, Kerr ID, Ford RC, et al. Purification and 3-D structural analysis of oligomeric human multidrug transporter ABCG2 Structure 2006(in press) [DOI] [PubMed]
- Mitomo H, Kato R, Ito A, Kasamatsu S, Ikegami Y, Kii I, et al. A functional study on polymorphism of the ATP-binding cassette transporter ABCG2: critical role of arginine-482 in methotrexate transport. Biochem J. 2003;373:767–774. doi: 10.1042/BJ20030150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake K, Mickley L, Litman T, Zhan Z, Robey R, Cristensen B, et al. Molecular cloning of cDNAs which are highly over expressed in mitoxantrone-resistant cells: demonstration of homology to ABC transport genes. Cancer Res. 1999;59:8–13. [PubMed] [Google Scholar]
- Motulsky HJ, Mahan LC. The kinetics of competitive radioligand binding predicted by the law of mass action. Mol Pharmacol. 1983;25:1–9. [PubMed] [Google Scholar]
- Nagashima S, Soda H, Oka M, Kitazaki T, Shiozawa K, Nakamura Y, et al. BCRP/ABCG2 levels account for the resistance to topoisomerase I inhibitors and reversal effects by gefitinib in non-small cell lung cancer. Cancer Chemother Pharmacol. 2006;58:594–600. doi: 10.1007/s00280-006-0212-y. [DOI] [PubMed] [Google Scholar]
- Ozvegy C, Litman T, Szakacs G, Nagy Z, Bates S, Varadi A, et al. Functional characterization of the human multidrug transporter, ABCG2, expressed in insect cells. Biochem Biophys Res Commun. 2001;285:111–117. doi: 10.1006/bbrc.2001.5130. [DOI] [PubMed] [Google Scholar]
- Ozvegy C, Varadi A, Sarkadi B. Characterization of drug transport, ATP hydrolysis, and nucleotide trapping by the human ABCG2 multidrug transporter. Modulation of substrate specificity by a point mutation. J Biol Chem. 2002;277:47980–47990. doi: 10.1074/jbc.M207857200. [DOI] [PubMed] [Google Scholar]
- Pizard A, Marchetti J, Allegrini J, Alhenc-Gelas F, Rajerison RM. Negative cooperativity in the human bradykinin B2 receptor. J Biol Chem. 1998;273:1309–1315. doi: 10.1074/jbc.273.3.1309. [DOI] [PubMed] [Google Scholar]
- Polgar O, Robey RW, Morisaki K, Dean M, Michejda C, Sauna ZE, et al. Mutational analysis of ABCG2: role of the GXXXG motif. Biochemistry. 2004;43:9448–9456. doi: 10.1021/bi0497953. [DOI] [PubMed] [Google Scholar]
- Robey RW, Honjo Y, van de Laar A, Miyake K, Regis JT, Litman T, et al. A functional assay for detection of the mitoxantrone resistance protein, MXR (ABCG2) Biochim Biophys Acta. 2001;1512:171–182. doi: 10.1016/s0005-2736(01)00308-x. [DOI] [PubMed] [Google Scholar]
- Ross DD. Modulation of drug resistance transporters as a strategy for treating myelodysplastic syndrome. Best Pract Res Clin Haematol. 2004;17:641–651. doi: 10.1016/j.beha.2004.08.014. [DOI] [PubMed] [Google Scholar]
- Rothnie A, Storm J, Campbell J, Linton KJ, Kerr ID, Callaghan R. The topography of transmembrane segment six is altered during the catalytic cycle of P-glycoprotein. J Biol Chem. 2004;279:34913–34921. doi: 10.1074/jbc.M405336200. [DOI] [PubMed] [Google Scholar]
- Sarkadi B, Ozvegy-Laczka C, Nemet K, Varadi A. ABCG2 – a transporter for all seasons. FEBS Lett. 2004;567:116–120. doi: 10.1016/j.febslet.2004.03.123. [DOI] [PubMed] [Google Scholar]
- Springael JY, Nguyen PL, Urizar E, Costagliola S, Vassart G, Parmentier M. Allosteric modulation of binding properties between units of chemokine receptor homo- and hetero-oligomers. Mol Pharmacol. 2006;69:1652–1661. doi: 10.1124/mol.105.019414. [DOI] [PubMed] [Google Scholar]
- Taylor AM, Storm J, Soceneantu L, Linton KJ, Gabriel M, Martin C, et al. Detailed characterization of cysteine-less P-glycoprotein reveals subtle pharmacological differences in function from wild-type protein. Br J Pharmacol. 2001;134:1609–1618. doi: 10.1038/sj.bjp.0704400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbatsch IL, Sankaran B, Bhagat S, Senior AE. Both P-glycoprotein nucleotide-binding sites are catalytically active. J Biol Chem. 1995a;270:26956–26961. doi: 10.1074/jbc.270.45.26956. [DOI] [PubMed] [Google Scholar]
- Urbatsch IL, Sankaran B, Weber J, Senior AE. P-glycoprotein is stably inhibited by vanadate-induced trapping of nucleotide at a single catalytic site. J Biol Chem. 1995b;270:19383–19390. doi: 10.1074/jbc.270.33.19383. [DOI] [PubMed] [Google Scholar]
- van den Heuvel-Eibrink MM, Sonneveld P, Pieters R. The prognostic significance of membrane transport-associated multidrug resistance (MDR) proteins in leukemia. Int J Clin Pharmacol Ther. 2000;38:94–110. doi: 10.5414/cpp38094. [DOI] [PubMed] [Google Scholar]
- van Veen HW, Margolles A, Muller M, Higgins CF, Konings WN. The homodimeric ATP-binding cassette transporter LmrA mediates multidrug transport by an alternating two-site (two-cylinder engine) mechanism. EMBO J. 2000;19:2503–2514. doi: 10.1093/emboj/19.11.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RB, Kuo CL, Lien LL, Lien EJ. Structure-activity relationship: analyses of p-glycoprotein substrates and inhibitors. J Clin Pharm Ther. 2003;28:203–228. doi: 10.1046/j.1365-2710.2003.00487.x. [DOI] [PubMed] [Google Scholar]
- Weiland GA, Molinoff PB. Quantitative analysis of drug-receptor interactions: I. Determination of kinetic and equilibrium properties. Life Sci. 1981;29:313–330. doi: 10.1016/0024-3205(81)90324-6. [DOI] [PubMed] [Google Scholar]
- Williams DH, O'Brien DP, Sandercock AM, Stephens E. Order changes within receptor systems upon ligand binding: receptor tightening/oligomerization and the interpretation of binding parameters. J Mol Biol. 2004;340:373–383. doi: 10.1016/j.jmb.2004.04.056. [DOI] [PubMed] [Google Scholar]
- Xu J, Liu Y, Yang Y, Bates S, Zhang JT. Characterization of oligomeric human half-ABC transporter ATP-binding cassette G2. J Biol Chem. 2004;279:19781–19789. doi: 10.1074/jbc.M310785200. [DOI] [PubMed] [Google Scholar]