

Abstract
Protein engineers use a variety of mutagenic strategies to adapt enzymes to novel substrates. Directed evolution techniques (random mutagenesis and high-throughput screening) offer a systematic approach to the management of protein complexity. This sub-discipline was galvanized by the invention of DNA shuffling, a procedure that randomly recombines point mutations in vitro. In one influential study, Escherichia coli β-galactosidase (BGAL) variants with enhanced β-fucosidase activity (tenfold increase in kcat/KM in reactions with the novel para-nitrophenyl-β-d-fucopyranoside substrate; 39-fold decrease in reactivity with the “native” para-nitrophenyl-β-d-galactopyranoside substrate) were evolved in seven rounds of DNA shuffling and screening. Here, we show that a single round of site-saturation mutagenesis and screening enabled the identification of β-fucosidases that are significantly more active (180-fold increase in kcat/KM in reactions with the novel substrate) and specific (700,000-fold inversion of specificity) than the best variants in the previous study. Site-saturation mutagenesis thus proved faster, less resource-intensive and more effective than DNA shuffling for this particular evolutionary pathway.
Keywords: random mutagenesis, molecular recognition, in vitro evolution, high-throughput screen, substrate specificity
Introduction
Protein engineers redesign proteins in order to improve their biomedical or industrial utility. We are particularly interested in re-engineering enzyme substrate specificity. Advances in directed evolution (also called in vitro evolution or laboratory evolution) techniques have enabled the direct reconfiguration of protein function without a complete understanding of protein structure. Diverse populations of molecules are generated either by random mutagenesis of a protein-coding gene,1–3 or chimeragenesis of two or more genes.4–6 The resulting libraries are expressed in recombinant microorganisms; clones exhibiting improvement in reactivity (e.g. with a novel substrate) are isolated in high-throughput screens or selections. The selected clones are generally further mutated and/or randomly recombined for the next round of expression and screening.
Experimental evolutionists have developed a variety of methods to generate molecular diversity. No consensus has emerged, however, about which method is most effective for the evolution of enzyme substrate specificity. It remains unclear, for example, whether it is more efficient to mutate the whole enzyme randomly or just its active site.7,8 The development of directed evolution techniques was stimulated by the advent of DNA shuffling, a PCR-like process that introduces and recombines random point mutations within a particular gene.9 DNA shuffling experiments sometimes showed that amino acid changes distant from the active site can affect substrate specificity.10,11 Such changes can work by altering the orientation of active-site residues or the conformational dynamics of the entire protein,12–14 and are therefore difficult to anticipate.
The more orthodox view is that amino acid changes near the substrate are more likely to alter substrate specificity.15,16 Structure-based, site-directed mutagenesis is usually applied to residues in or near the active site, and this approach has produced many enzyme variants with dramatically altered specificities.17–24 Some employ site-saturation mutagenesis, in which a small number of active site residues are “randomized” (mutated randomly).25–29 Experimental evolutionists generally base their methodological decisions upon intuition rather than any systematic understanding of adaptive protein evolution. Persuasive case studies all too often convince workers to employ techniques that fail to solve seemingly analogous problems. We therefore advocate the direct comparison of evolutionary methods using common model proteins.
The work described here was inspired by the directed evolution of β-galactosidase (BGAL) variants with β-fucosidase activity.11 In that influential study, a population of BGAL was evolved in seven iterated cycles of DNA shuffling and high-throughput screening. The enzyme derived from the fittest clone contained eight amino acid changes, including two in the active site. It exhibited approximately tenfold improvement (kcat/KM) in reactivity with para-nitrophenyl-β-d-fucopyranoside (pNP-fuc, “novel” substrate) and a 39-fold decrease in reactivity with para-nitrophenyl-β-d-galactopyranoside (pNP-gal, “native” substrate). The evolved enzyme retained a 2.7-fold preference for pNP-gal over pNP-fuc. This study provided proof-of-principle that substrate specificity could be altered even in the complete absence of structural information.
Here, we compare the efficiency of DNA shuffling with that of site-saturation mutagenesis. We repeated the directed evolution of BGAL, using the latter technique to randomize three selected active site residues. This semi-rational approach effected greater improvement in β-fucosidase activity and greater substrate specificity in a single round of mutagenesis and screening. We describe the conditions under which site-saturation muta-genesis is expected to outperform DNA shuffling, as well as conditions that justify the opposite approach.
Results
Our goal is to adapt enzymes to novel substrates using the most efficient design algorithms. An influential study showed that DNA shuffling of the Escherichia coli BGAL gene (lacZ) enabled the directed evolution of variants with increased β-fucosidase activity. The best β-fucosidase after seven rounds of DNA shuffling and screening exhibited approximately tenfold improvement in kcat/KM in reactions with pNP-fuc (novel substrate).11 The endpoint of the present study was to impart greater improvement in fewer rounds of directed evolution through the semi-rational site-saturation mutagenesis approach.25–27
BGAL is a good model for directed evolution studies because it is amenable to high-throughput screening, and because its structure and function are well understood. The structure of pNP-gal (native substrate) differs from that of pNP-fuc (novel substrate) by an oxygen atom on the C6 substituent (hydroxymethyl versus methyl). It is therefore reasonable to presume that both substrates bind the active site in the same way. We examined the published co-crystal structure of the E537Q BGAL/pNP-gal complex.30 The C6 hydroxyl group of pNP-gal (absent from pNP-fuc) forms hydrogen bonds with His540, Asn604, and with a fixed sodium ion (Figure 1). Asp201 helps hold this sodium ion in place, and is only ~3.1 Å away from the C6 hydroxyl group. We reasoned that site-saturation mutagenesis of codons 201, 540 and 604 would produce variants that no longer bind the sodium ion, but instead interact directly with the C6 methyl group of pNP-fuc. The randomization of three codons should produce a library containing 32,768 clones (32 × 32 × 32), which is comparable to the throughput of our screen.
Figure 1.
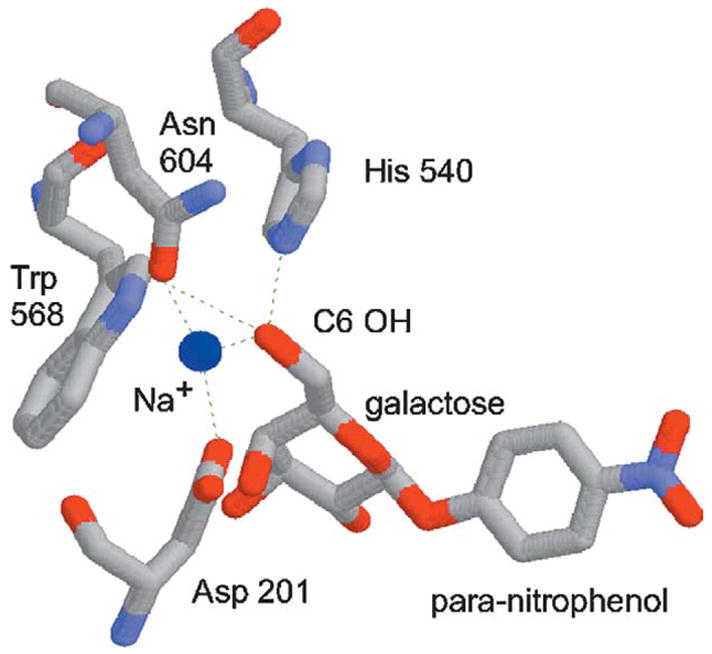
Structure of the Escherichia coli β-galactosidase active site.30 The para-nitrophenyl-β-d-fucopyranoside (“novel substrate”) is identical with para-nitrophenyl-β-d-galactopyranoside (“native substrate”, shown here), except that it lacks the C6 hydroxyl group. The dotted lines represent hydrogen bonds. The Asp201, His540, and Asn604 residues were “randomized” in this study. The sodium ion (blue sphere) is reduced in scale so as not to obscure these amino acid residues.
We designed three complementary primer pairs (top and bottom) encoding the sequence surrounding codons 201, 540 and 604 (Table 1). Each of these codons was replaced in the primers with degenerate NNK sequence, where K = G or T. The primer lacZ D201X, for example, has the sequence: 5′-ATCTGGAAGATCAGNNKATGTGGCGGATGA-3′, where codon 201 = NNK (bold) and the surrounding sequence (underlined) encodes wild-type codons 197–200, 201–206. We also designed two non-degenerate back-to-back primers complementary to vector sequence downstream of the lacZ gene (Table 1, 3′pET3 and 3′pETout2). We employed 6his-lacZ-pET28a+ as a template for four separate PCR reactions (Figure 2, PCR 1–4: 201–540, 540–604, 604-vector, vector-201). The four PCR products were combined in an overlap PCR,25,31 and the full-length recombinant insert-vector product was amplified, purified, self-ligated and used to transform E. coli.
Table 1.
Primers used in this study
| Primer name | Sequencea | Use |
|---|---|---|
| lacZ D201X | ATCTGGAAGATCAGNNKATGTGGCGGATGA | PCR 1: randomize D201 |
| lacZ D201X rev | TCATCCGCCACATMNNCTGATCTTCCAGATA | PCR 4: randomize D201 |
| lacZ H540X | TTGCGAATACGCCNNKGCGATGGGTAACA | PCR 2: randomize H540 |
| lacZ H540X rev | TGTTACCCATCGCMNNGGCGTATTCGCAAAG | PCR 1: randomize H540 |
| lacZ N604X | CCAGTTCTGTATGNNKGGTCTGGTCTTTG | PCR 3: randomize N604 |
| lacZ N604X rev | CAAAGACCAGACCMNNCATACAGAACAGGC | PCR 2: randomize N604, adds Q600L mutation |
| 3′ pET3 | Phosphate-GTCGACGTTGGAGTCCACGTTCTTTAATA | PCR 3 and 5: amplify whole plasmid |
| 3′ pETout2 | Phosphate-GCATGCCGTAAAGCACTAAATCGGAACC | PCR 4 and 5: amplify whole plasmid |
| lacZ 454 rev | CGACCCAGCGCCCGTTGCACCACAG | lacZ sequencing |
| lacZ 377 | AATCCGACGGGTTGTTACTCGCTCAC | lacZ sequencing |
| lacZ 770 | GAGTTGCGTGACTACCTACGGGTAAC | lacZ sequencing |
| lacZ 1154 | CTGAACGGCAAGCCGTTGCTGATTC | lacZ sequencing |
| lacZ 1536 | CGCGTGGATGAAGACCAGCCCTTC | lacZ sequencing |
| lacZ 1920 | CGGGCAAACCATCGAAGTGACCAGC | lacZ sequencing |
| lacZ 2269 | CATCGAGCTGGGTAATAAGCGTTGGC | lacZ sequencing |
| lacZ 2679 | TGCCAGCTGGCGCAGGTAGCAGAG | lacZ sequencing |
K = G or T; M = C or A.
Table 3.
Steady-state kinetic parameters of BGAL variants
| pNP-gal
|
pNP-fuc
|
|||||
|---|---|---|---|---|---|---|
| kcat (s−1) | KM (μM) | kcat/KM (s−1 M−1) | kcat (s−1)a | KM (μM) | kcat/KM | |
| Wild-type | 33.4 ± 3.7 | 48.0 ± 5.4 | 723,000 ± 143,000 | > 3000 | 47 ± 3 | |
| H540V | 1.0 ± 0.0 | 916.9 ± 75.3 | 1114 ± 111 | > 3000 | 10,610 ± 4516 | |
| N604T | 4.1 ± 0.1 | 64.5 ± 3.6 | 64,928 ± 6,084 | > 3000 | 110 ± 4 | |
| H540V/N604T | 0.1 ± 0.0 | 702.0 ± 22.9 | 179 ± 2 | 21.5 ± 2.4 | 2563.1 ± 78.6 | 8365 ± 735 |
The wild-type, H54-V and N604T BGAL variants could not be saturated with pNP-fuc, so neither the kcat nor KM value could be determined accurately.
Figure 2.
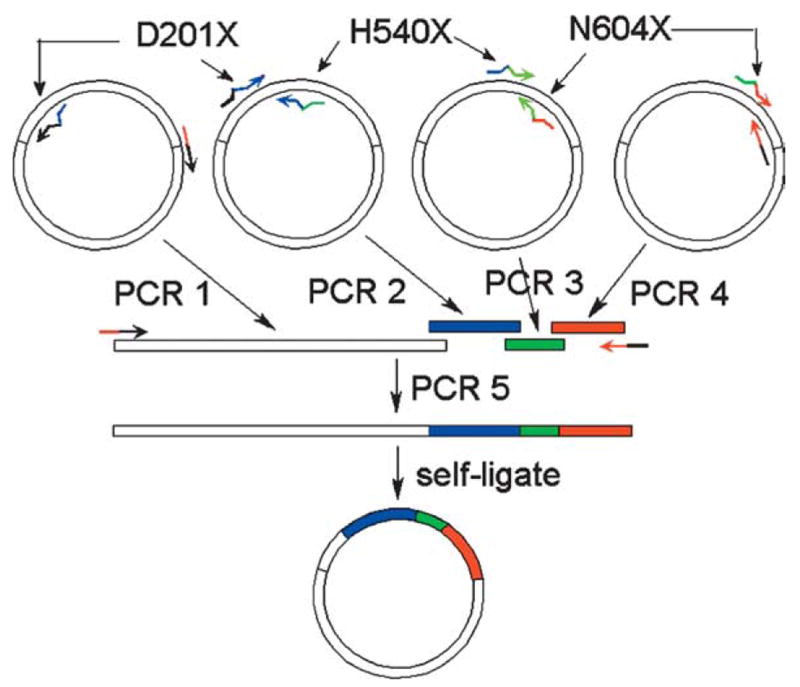
Randomization of β-galactosidase residues 201, 540 and 604. Complementary primer pairs (arrows) encoding vector sequence (black) or lacZ (colored) were synthesized. The lacZ-specific primers were degenerate (NNK) at codons 201, 540 and 604. The 6his-lacZ-pET28a+ (white circle) was employed as a template in four separate PCR reactions (white bar = vector–201, blue bar = 201–540, green bar = 540–604, red bar = 604–vector). Each PCR product overlapped one or two others in sequence; the four PCR products were combined in a single overlap PCR reaction (reaction 5). The full-length recombinant PCR product was purified, self-ligated and electroporated into E. coli.
We evaluated the mutants using a screen similar to that used by Zhang et al.11 in their previous study. E. coli DH5Δlac (DE3) were transformed with the library, and spread on LB agar plates supplemented with kanamycin. After 18 hours of growth at 37 °C, the resulting ~10,000 colonies (~1300 colonies/100 mm plate, eight plates) were adsorbed to nitrocellulose filters and transferred (colony-side up) onto LB plates supplemented with kanamycin, 0.5 mM IPTG, and 80 μg/ml of 5-bromo-4-chloro-3-indoyl-β-d-fucopyranoside (X-fuc). After 1.5 hours of incubation at 37 °C, control colonies expressing the ancestral lacZ and those transformed with pET28a+ (no insert) remained white (Figure 3). Approximately 250 of the ~10,000 colonies expressing mutant lacZ alleles were darker than the ancestral control colonies. We picked the 29 colonies that turned darkest blue and re-streaked them onto LB-kanamycin plates. The plates were incubated overnight at 37 °C, and the resulting colonies were filter-lifted onto LB-kanamycin/IPTG/X-fuc plates. All 29 of the streaks included colonies that turned blue faster than the ancestral controls (data not shown).
Figure 3.
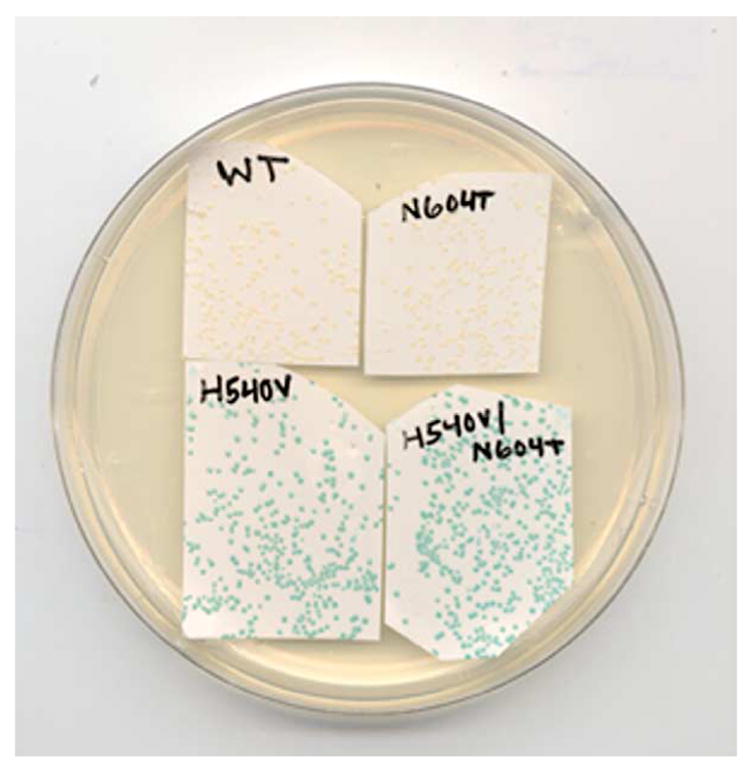
E. coli colonies expressing evolved β-fucosidases. E. coli DH5Δlac(DE3) were transformed with the ancestral, H540V, N604T or H540V/N604T variants of the 6his-lacZ-pET28a+ plasmid. The colonies were propagated on LB-kanamycin agar plates, adsorbed to nitrocellulose filters and transferred colony-site up to LB-kanamycin agar plates supplemented with 0.5 mM IPTG and 80 micrograms/ml X-fuc. This photograph was taken 90 minutes after induction.
The X-fuc filter-lift assay is somewhat narrow in dynamic range, which is to say that it does not enable facile discrimination between good and great β-fucosidases. We therefore compared the selected colonies in a more quantitative assay. We picked single colonies and inoculated 100 μl of LB-kanamycin in a 96-well microplate. The micro-cultures were propagated to saturation on an orbital shaker (250 rpm) for 16 hours at 37 °C. The cells were diluted 100-fold in LB-kanamycin, propagated to mid-log phase for 1.5 hours, and induced with 0.5 mM IPTG for 3 hours. We then reacted 20 μl of each culture with 0.5 mM pNP-gal or pNP-fuc in 200 μl of 50 mM Tris–HCl (pH 7.6) at 25 °C. The formation of the pNP product was monitored continuously in a microplate spectrophotometer at 405 nm over 16 hours (Figure 4). Amongst the 29 variants, 11 exhibited greater β-fucosidase activity than the ancestral cells; we suspect that the other 18 clones were not genetically stable. These 11 variants exhibited greater activity and specificity in reactions with pNP-fuc than the ancestor did with pNP-gal, suggesting that the substrate specificity of BGAL had inverted in a single round of mutagenesis and screening.
Figure 4.
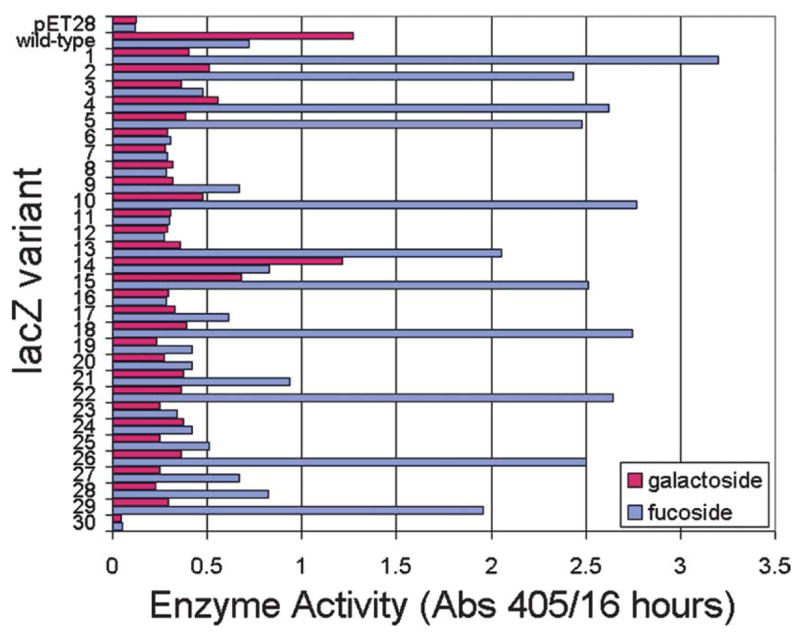
The β-galactosidase and β-fucosidase activities of selected lacZ variants. E. coli cells were transformed with mutated 6his-lacZ-pET28a+ plasmids (or with the ancestral or no insert control plasmids), grown to mid-log stage in LB-kanamycin liquid cultures, and induced for 3 hours with 0.5 mM IPTG. Each culture was split and reacted with either 0.5 mM para-nitrophenyl-β-d-galactopyranoside (native substrate, red) or para-nitrophenyl-β-d-fucopyranoside (novel substrate, blue). The formation of the para-nitrophenol product was monitored continously in a microplate spectrophotometer for 16 h.
We propagated eight strains exhibiting the most β-fucosidase activity, purified the corresponding 6his-lacZ-pET28a+ plasmids, and sequenced the regions about the randomized codons (Table 2). All eight clones contained the H540V and N604T mutations, and the wild-type D201 at the randomized positions. They also shared the silent t570c and N600V mutations, which were encoded in the ancestral template and in the N604X rev primer (by mistake), respectively. We sequenced the entire allele of one lacZ variant (clone 26), and found that it contained a single silent mutation (a309g) in addition to the H540V/N600V/N604T shown in Table 1. In order to assess the contributions of the individual mutations to fitness, we employed site-directed mutagenesis to introduce the H540V, N604T and H540V/N604T mutations into the ancestral 6his-lacZ template. We expressed and purified these mutant proteins, along with the ancestral protein and the H540V/N600V/N604T BGAL variant (clone 26), by immobilized metal affinity chromatography.
Table 2.
Sequences of most active β-fucosidases
| N147G aac → ggc | D164Y g490t | H540V cac → gtt | N600V a1799t | N604T aac → act | |
|---|---|---|---|---|---|
| 1 | X | X | X | ||
| 5 | X | X | X | ||
| 10 | X | X | X | ||
| 13 | X | X | X | ||
| 18 | X | X | X | ||
| 22 | X | X | X | X | |
| 26 | X | X | X | ||
| 29 | X | X | X | X |
Clone 26 was sequenced completely, the others were sequenced for nucleotides 400–1000 and 1600–2100 (wild-type lacZ numbering). The ancestral and selected lacZ variants all contain the silent t570c mutation. The Q600V mutation was accidentally encoded in the lacZ N604T rev primer.
We reacted each of the purified proteins with pNP-gal and pNP-fuc, and determined the steady-state kinetic parameters for each enzyme/substrate reaction (Table 3). The kcat of the ancestral 6his-BGAL in reactions with pNP-gal is ~2.7-fold lower than the previously reported wild-type value, but the KM values are about the same.11,32 The difference in kcat might be based upon structural differences at the N termini.11,33 The ancestral enzyme exhibits a ~15,000-fold preference for pNP-gal (kcat/KM = 723,000 s−1 M−1) over pNP-fuc (kcat/KM = 47 s−1 M−1). The two substrates differ only at the C6 position (–CH2–OH versus –CH3), so this marked substrate preference reflects the importance of H540 and N604 in the wild-type enzyme/substrate complex (Figure 1).
The H540V/N600V/N604T 6his-BGAL protein was less active than its H540V/N604T counterpart (data not shown); this result showed that the N600V mutation, which was encoded accidentally in primer lacZ N604X rev (Table 1), was deleterious. The H540V/N604T 6his-BGAL was 180-fold more reactive with pNP-fuc (kcat/KM = 8365 s−1 M−1), and ~4000-fold less reactive with pNP-gal (kcat/KM = 179 s−1 M−1), than its ancestor. The double mutant exhibited a 46-fold preference for pNP-fuc over pNP-gal, which means that a single round of site-saturation mutagenesis and screening effected a 700,000-fold inversion of specificity (15,000 × 46). In contrast, seven rounds of DNA shuffling and screening produced a multiply mutated BGAL that exhibited a tenfold improvement in pNP-reactivity, and a 1000-fold shift in specificity.11
We characterized the H540V and N604T 6his-BGAL proteins in order to assess the contributions of the individual mutations. Colonies expressing H540V turn dark blue in the X-fuc filter-lift assay, while isogenic colonies expressing the N604T enzyme do not (Figure 3). The N604T mutation is not very beneficial by itself (kcat/KM = 110 s−1 M−1 in reactions with pNP-fuc), although it contributes to specificity in the context of H540V. The H540V protein is apparently faster than even the double mutant in reactions with pNP-fuc (kcat/KM = 10,610 s−1 M−1), and is similarly inactive in reactions with pNP-gal (kcat/KM = 1114 s−1 M−1). We therefore conclude that the H540V mutation accounts for most of the increased β-fucosidase activity exhibited by H540V/N604T.
Discussion
The fixation (ubiquity amongst selected clones) of the H540V/N604T mutations (Table 2) suggests that the D201/V540/T604 combination of amino acids is optimal for β-fucosidase activity, although we cannot exclude the possibility of even better variants in the unscreened portion of the library. It is not obvious, however, why this is the case. The co-crystal structure of the E537Q BGAL/pNP-gal complex showed that the C6 hydroxyl group of pNP-gal forms hydrogen bonds with His540 and Asn604 (Figure 1).30 Since pNP-fuc is missing the C6 hydroxyl group, we expected that the replacement of H540 and/or N604 with larger hydrophobic side-chains would fill the gap and enable novel enzyme/substrate interactions. Contrary to this expectation, both the H540V and N604T mutations resulted in smaller amino acid side-chains and an expansion of the perceived gap. Large hydrophobic residues at positions 540 and/or 604 would also have displaced the active site sodium ion that normally interacts with the C6 hydroxyl group of pNP-gal.30 Since the wild-type D201 residue was retained, we surmise that the sodium ion plays a heretofore under-appreciated role in stabilizing the conformation of the active-site.
The invariability of D201 limits the evolvability of the BGAL active site. The specificity of the H540V/N604T BGAL was greatly altered, but it is much less active against pNP-fuc than is the wild-type enzyme against pNP-gal. We predict that the active site of H540V/N604T is more flexible than that of the wild-type, so as to reduce both substrate discrimination and overall catalytic activity. Our failure to anticipate the functional importance of D201 sharply reduced the effective throughput of our screen, only ~1/32 of the library contained D201, so only ~300 clones (10,000/32) had any chance of surviving our primary screen. Overall, the apparent inflexibility of D201, the relatively modest throughput of our screen and the accidental insertion of the deleterious N600V mutation likely reduced the effectiveness of site-saturation muta-genesis. In spite of these impediments, site-saturation mutagenesis outperformed DNA shuffling in terms of speed, labor intensity and the kinetic parameters of the evolved enzyme variants.
Several obstacles precluded the evolution of the H540V and N604T mutations in the DNA shuffling experiment.11 First, the H540V amino acid change requires at least two nucleotide substitutions (CAC/GTC). A site-directed mutagenesis study showed that the H540F, H540N and H540E variants each exhibited diminished β-fucosidase activity relative to the wild-type enzyme,32 so we suspect that the possible intermediate forms (CTC = leucine, GAC = aspartate) would also have been deleterious. Second, the N604T mutation could be realized by a single transversion (AAC/ACC), but it does not exhibit improved activity in the X-fuc screen (Figure 3). Stepwise adaptation would therefore not have been observed in the context of this screen. Third, mutations that alter substrate specificity generally occur less frequently than those that increase protein expression or solubility. In our experience, constitutive expression systems (such as that used by Zhang et al.) tend to favor mutations that decrease the toxicity of the over-expressed protein. The limited dynamic range of screens based on X-fuc virtually ensures the predominance of over-expression mutations. Thus, subtle differences in the expression system and/or screen can lead to significant differences in outcome. Most of the 13 mutations in the evolved β-fucosidase were silent or in residues distant from the active site,11 and we suppose that many of the latter somehow increased overall activity without altering substrate specificity.
We emphasize that site-saturation mutagenesis will not be the best solution for every directed evolution experiment. This semi-rational technique is a compromise between site-directed and whole gene random mutagenesis; the directed evolution experiment described here exemplifies the conditions that favor this approach. First, the desired activity was amenable to high-throughput screening. The enzyme is readily expressed in E. coli, and chromogenic and histochemical substrates are readily available. Second, the native (X-gal) and novel (X-fuc) substrates differ by a single atom, and likely bind the enzyme active site in the same way. The structure of the BGAL/pNP-gal complex was solved, and the residues that interact directly with the atom in question were identified previously.30 Third, the protein engineers in this case were able to identify functionally important residues, but lacked the insight and confidence to anticipate the H540V/N604T mutations.
Enzyme specificity can also be evolved by random mutagenesis and recombination of entire genes.10,34–37 The major advantage of this approach over site-saturation mutagenesis is greater versatility; whole gene mutagenesis is most useful when structural information is unavailable or difficult to interpret, and can work when native and novel substrates bind the enzyme in different ways.37 The major disadvantage of whole gene mutagenesis is that it requires screens that are more sensitive, precise and broader in dynamic range, and more rounds of evolution. Although site-saturation mutagenesis proved faster, less labor-intensive and effective than whole gene mutagenesis in the single test case described here, we hesitate to recommend the former in all cases. The literature excludes negative results, so it is difficult to assess the success (and failure) rates of any library design strategy. We therefore recommend that investigators develop good high-throughput screens, construct separate libraries by mutagenic PCR and site-saturation mutagenesis, and discover for themselves which strategy works best.
Materials and Methods
Materials
The 6his-lacZ-pET28a(+), also known as induction control E (positive control for the pET28a+ expression vector), was from Novagen (Madison, WI). E. coli strain InvαF′ was from Invitrogen (Carlsbad, CA); DH5Δlac (DE3) has been described.35 DNA purification columns were purchased from Qiagen (Chatsworth, CA). The Butterfly nitrocellulose membranes were from Schleicher and Schuell (Keene, NH). The histochemical β-fucosidase substrate (X-fuc), isopropyl-β-d-thiogalactopyranoside (IPTG), pNP-gal and pNP-fuc were from Sigma Chemicals (St. Louis, MO). BPER was from Pierce (St. Louis, MO). The GeneAmp XL polymerase kit, including the rTth and Vent polymerases, and the BigDye 3.1 DNA sequencing kit were from Perkin-Elmer/Applied Bio-systems (Foster City, CA). All other DNA-modifying enzymes (restriction enzymes, Taq polymerase, phage T4 DNA ligase) were from New England Biolabs (Beverly, MA). Oligonucleotides were synthesized by IDT (Coralville, IA).
Site-saturation mutagenesis
The BGAL gene (lacZ) was mutated randomly at the selected codons (D201, H540, N604), as illustrated in Figure 1. The three short segments (PCR 1–3, encoding BGAL 201–540, 540–604, 604–end) were amplified in PCR reactions that contained 50 ng of the ancestral 6his-lacZ-pET28a+ plasmid, 500 nM primers (Table 1), 200 nM each dNTP, 60 mM Tris–HCl (pH 8.5), 0.5 mM MgCl2, 15 mM (NH4)2SO4 and 2.5 units of Taq polymerase. The reactions were initiated by a “hot start” procedure, and the segments were amplified in 25 cycles of 94 °C for 30 s, 65°C for 30 s, 72 °C for 1 minute.
The long segment (PCR 4, lacZ end–201) was amplified using the mixture of Tth and Vent polymerases provided in the GeneAmp XL polymerase kit. The long PCR reactions included: 50 ng of 6his-lacZ-pET28a+, 500 nM primers lacZ D201X rev and 3′ pETout2 (Table 1), 200 nM each dNTP, ABI XL buffer II, and 0.8 mM, 1.2 mM or 1.5 mM magnesium acetate. The 50 μl reactions were overlaid with light mineral oil and heated to 80 °C in a thermal cycler for a hot start. Then 0.5 μl of Tth/Vent mixture (one unit) was added, and the temperatures were raisedto 94 °C for 1 minute, followed by 25 cycles of 94 °C for 15 s, 68 °C for 3 minutes (1 min/kb). The reaction was further incubated at 72 °C for 10 minutes, and stored at 4 °C.
PCRs (1–4) amplified three short products and one long one. The mineral oil was removed, and each PCR reaction was incubated with 0.5% (w/v) SDS and 50 μg/ml of proteinase K at 65 °C for 15 minutes to eliminate the thermostable polymerases. The PCR product was purified using the Promega Wizard PCR prep kit (Madison, WI) as directed by the manufacturer. The DNA was digested with DpnI to eliminate methylated ancestral template. The PCR products were gel-purified using a Qiaquick spin column as directed by the manufacturer. The concentration of eluted DNA was estimated by agarose gel electrophoresis.
Each of the four double-stranded PCR products overlapped either one (PCR 3 = 601–lacZ end, PCR 4 = lacZ end–201) or two (PCR 1 = 201–540, PCR 2 = 540–604) of the other fragments. They were pooled and amplified in a long overlap PCR reaction using the outside 3′ pET3 and 3′ pETout 2 primers (Table 1, Figure 2). Intra-molecular self-ligation of blunt-ended DNA was used to re-circularize the plasmid.25,33 We reacted 20 fmol of purified recombinant PCR product with one Weiss unit of phage T4 DNA ligase in a 20 μl reaction containing 50 μM each dNTP, 1 mM ATP, 50 mM Tris–HCl (pH 7.6), 10 mM MgCl2, 5 mM DTT, and three units of phage T4 DNA polymerase, at 16 °C for 1 hour. The T4 DNA ligase was heat-killed at 65 °C for 10 min; the DNA was precipitated in butanol,38 and electroporated into freshly prepared E. coli DH5Δlac cells as described.39 The resulting library contained ~21,000 clones.
Protein purification
The 6his-BGAL proteins, which were fused to an N-terminal His6 tag, were expressed in DH5ΔLAC(DE3)/pLysS cells. The N-terminal eight amino acid residues of the native protein were replaced in 6his-BGAL (the ancestral protein used in the study) with a 50 residue peptide containing the His6 tag. The transformants were propagated to mid-log phase in 350 ml of liquid LB culture (25 μg/ml of kanamycin), induced for 3 hours with 0.5 mM IPTG, and lysed with BPER detergent. The proteins were purified to homogeneity (as determined by subsequent SDS-PAGE) by nickel chelate affinity chromatography, and dialyzed overnight in 2 × BGAL storage buffer (100 mM Tris–HCl (pH 7.1), 20 mM MgCl2, 5 mM β-mercaptoethanol). The yields were determined by the Bradford protein assay (data not shown) as described.40 The dialyzed protein (~1 ml) was diluted with one volume of glycerol and stored at −20 °C. All of the enzyme activities were stable for at least two months under these conditions.
Enzyme kinetics
Each of the purified 6his-BGAL proteins (10 nM–1 μM) was added separately to 1 ml of BGAL buffer (30 mM Tes, 140 mM NaCl, 1 mM MgSO4) containing various concentrations (5 μM–6 mM) of substrate. The formation of the pNP product at 25 °C for 1–60 minutes was monitored continuously in a Shimadzu UV-1601 spectrophotometer. The absorption extinction coefficient of para-nitro-phenol (pNP) at 405 nm under these conditions is 10.88 mM−1 cm−1. The kinetic parameters of the wild-type and mutant enzymes were calculated by fitting the steady-state initial velocity values to the Michaelis–Menten equation using a least-squares method and the application Kaleidagraph 3.0.5 (Adelbeck software, Reading, PA). Each of the kcat/KM values was thus derived from at least 18 independent reactions.
Acknowledgments
I.M. made the H540V and N604T mutations and purified the corresponding proteins. We thank Ms. Karen Polizzi for reading the manuscript. M.P. was supported by NSF BIO/MCB (#0109668). I.M. was supported by the NIH/NIAID (1 R21AI054602-01). The FAME Center is supported by NSF/DBI (#0320786).
Abbreviations used
- BGAL
β-galactosidase
- pNP-gal
para-nitrophenyl-β-d-galactopyranoside
- pNP-fuc
para-nitrophenyl-β-d-fucopyranoside
- X-fuc
5-bromo-4-chloro-3-indoyl-β-d-fucopyranoside
- LB
Luria broth
Footnotes
Edited by J. Karn
References
- 1.Cadwell RC, Joyce GF. Randomization of genes by PCR mutagenesis. PCR Methods Appl. 1992;2:28–33. doi: 10.1101/gr.2.1.28. [DOI] [PubMed] [Google Scholar]
- 2.Matsumura I, Ellington AD. Mutagenic polymerase chain reaction of protein-coding genes for in vitro evolution. Methods Mol Biol. 2002;182:259–267. doi: 10.1385/1-59259-194-9:259. [DOI] [PubMed] [Google Scholar]
- 3.Moore JC, Arnold FH. Directed evolution of a para-nitrobenzyl esterase for aqueous-organic solvents. Nature Biotechnol. 1996;14:458–467. doi: 10.1038/nbt0496-458. [DOI] [PubMed] [Google Scholar]
- 4.Crameri A, Raillard SA, Bermudez E, Stemmer WP. DNA shuffling of a family of genes from diverse species accelerates directed evolution. Nature. 1998;391:288–291. doi: 10.1038/34663. [DOI] [PubMed] [Google Scholar]
- 5.Lutz S, Ostermeier M, Benkovic SJ. Rapid generation of incremental truncation libraries for protein engineering using alpha-phosphothioate nucleotides. Nucl Acids Res. 2001;29:E16. doi: 10.1093/nar/29.4.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sieber V, Martinez CA, Arnold FH. Libraries of hybrid proteins from distantly related sequences. Nature Biotechnol. 2001;19:456–460. doi: 10.1038/88129. [DOI] [PubMed] [Google Scholar]
- 7.Harris JL, Craik CS. Engineering enzyme specificity. Curr Opin Chem Biol. 1998;2:127–132. doi: 10.1016/s1367-5931(98)80044-6. [DOI] [PubMed] [Google Scholar]
- 8.Chen R. Enzyme engineering: rational redesign versus directed evolution. Trends Biotechnol. 2001;19:13–14. doi: 10.1016/s0167-7799(00)01522-5. [DOI] [PubMed] [Google Scholar]
- 9.Stemmer WP. DNA shuffling by random fragmentation and reassembly: in vitro recombination for molecular evolution. Proc Natl Acad Sci USA. 1994;91:10747–10751. doi: 10.1073/pnas.91.22.10747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yano T, Oue S, Kagamiyama H. Directed evolution of an aspartate aminotransferase with new substrate specificities. Proc Natl Acad Sci USA. 1998;95:5511–5515. doi: 10.1073/pnas.95.10.5511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang JH, Dawes G, Stemmer WP. Directed evolution of a fucosidase from a galactosidase by DNA shuffling and screening. Proc Natl Acad Sci USA. 1997;94:4504–4509. doi: 10.1073/pnas.94.9.4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.James LC, Tawfik DS. Conformational diversity and protein evolution—a 60-year-old hypothesis revisited. Trends Biochem Sci. 2003;28:361–368. doi: 10.1016/S0968-0004(03)00135-X. [DOI] [PubMed] [Google Scholar]
- 13.Benkovic SJ, Hammes-Schiffer S. A perspective on enzyme catalysis. Science. 2003;301:1196–1202. doi: 10.1126/science.1085515. [DOI] [PubMed] [Google Scholar]
- 14.Suel GM, Lockless SW, Wall MA, Ranganathan R. Evolutionarily conserved networks of residues mediate allosteric communication in proteins. Nature Struct Biol. 2003;10:59–69. doi: 10.1038/nsb881. [DOI] [PubMed] [Google Scholar]
- 15.el Hawrani AS, Moreton KM, Sessions RB, Clarke AR, Holbrook JJ. Engineering surface loops of proteins—a preferred strategy for obtaining new enzyme function. Trends Biotechnol. 1994;12:207–211. doi: 10.1016/0167-7799(94)90084-1. [DOI] [PubMed] [Google Scholar]
- 16.Morley KL, Kazlauskas RJ. Improving enzyme properties: when are closer mutations better? Trends Biotechnol. 2005;23:231–237. doi: 10.1016/j.tibtech.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 17.Hedstrom L. Serine protease mechanism and specificity. Chem Rev. 2002;102:4501–4524. doi: 10.1021/cr000033x. [DOI] [PubMed] [Google Scholar]
- 18.Wilks HM, Hart KW, Feeney R, Dunn CR, Muirhead H, Chia WN, et al. A specific, highly active malate dehydrogenase by redesign of a lactate dehydrogenase framework. Science. 1988;242:1541–1544. doi: 10.1126/science.3201242. [DOI] [PubMed] [Google Scholar]
- 19.Chen R, Greer A, Dean AM. Redesigning secondary structure to invert coenzyme specificity in isopropylmalate dehydrogenase. Proc Natl Acad Sci USA. 1996;93:12171–12176. doi: 10.1073/pnas.93.22.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Onuffer JJ, Kirsch JF. Redesign of the substrate specificity of Escherichia coli aspartate aminotransferase to that of Escherichia coli tyrosine aminotransferase by homology modeling and site-directed mutagenesis. Protein Sci. 1995;4:1750–1757. doi: 10.1002/pro.5560040910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nilsson LO, Gustafsson A, Mannervik B. Redesign of substrate-selectivity determining modules of glutathione transferase A1-1 installs high catalytic efficiency with toxic alkenal products of lipid peroxidation. Proc Natl Acad Sci USA. 2000;97:9408–9412. doi: 10.1073/pnas.150084897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schmidt DMZ, Mundorff EC, Dojka M, Bermudez E, Ness JE, Govindarajan S, et al. Evolutionary potential of (beta/alpha)8-barrels: functional promiscuity produced by single substitutions in the enolase superfamily. Biochemistry. 2003;42:8387–8393. doi: 10.1021/bi034769a. [DOI] [PubMed] [Google Scholar]
- 23.Yokoyama S. Molecular genetic basis of adaptive selection: examples from color vision in vertebrates. Annu Rev Genet. 1997:315–336. doi: 10.1146/annurev.genet.31.1.315. [DOI] [PubMed] [Google Scholar]
- 24.Xia G, Chen L, Sera T, Fa M, Schultz PG, Romesberg FE. Directed evolution of novel polymerase activities: mutation of a DNA polymerase into an efficient RNA polymerase. Proc Natl Acad Sci USA. 2002;99:6597–6602. doi: 10.1073/pnas.102577799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Geddie ML, Matsumura I. Rapid evolution of beta-glucuronidase specificity by saturation mutagenesis of an active site loop. J Biol Chem. 2004;279:26462–26468. doi: 10.1074/jbc.M401447200. [DOI] [PubMed] [Google Scholar]
- 26.Graham LD, Haggett KD, Jennings PA, Le Brocque DS, Whittaker RG, Schober PA. Random mutagenesis of the substrate-binding site of a serine protease can generate enzymes with increased activities and altered primary specificities. Biochemistry. 1993;32:6250–6258. doi: 10.1021/bi00075a019. [DOI] [PubMed] [Google Scholar]
- 27.el Hawrani AS, Sessions RB, Moreton KM, Holbrook JJ. Guided evolution of enzymes with new substrate specificities. J Mol Biol. 1996;264:97–110. doi: 10.1006/jmbi.1996.0626. [DOI] [PubMed] [Google Scholar]
- 28.Santoro SW, Schultz PG. Directed evolution of the site specificity of Cre recombinase. Proc Natl Acad Sci USA. 2002;99:4185–4190. doi: 10.1073/pnas.022039799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chelliserrykattil J, Ellington AD. Evolution of a T7 RNA polymerase variant that transcribes 2′-O-methyl RNA. Nature Biotechnol. 2004;22:1155–1160. doi: 10.1038/nbt1001. [DOI] [PubMed] [Google Scholar]
- 30.Juers DH, Heightman TD, Vasella A, McCarter JD, Mackenzie L, Withers SG, Matthews BW. A structural view of the action of Escherichia coli (lacZ) beta-galactosidase. Biochemistry. 2001;40:14781–14794. doi: 10.1021/bi011727i. [DOI] [PubMed] [Google Scholar]
- 31.Ling MM, Robinson BH. Approaches to DNA mutagenesis: an overview. Anal Biochem. 1997;254:157–178. doi: 10.1006/abio.1997.2428. [DOI] [PubMed] [Google Scholar]
- 32.Roth NJ, Huber RE. The beta-galactosidase (Escherichia coli) reaction is partly facilitated by interactions of His-540 with the C6 hydroxyl of galactose. J Biol Chem. 1996;271:14296–14301. doi: 10.1074/jbc.271.24.14296. [DOI] [PubMed] [Google Scholar]
- 33.Matsumura I, Rowe LA. Whole plasmid mutagenic PCR for directed protein evolution. Biomol Eng. 2005;22:73–79. doi: 10.1016/j.bioeng.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 34.Varadarajan N, Gam J, Olsen MJ, Georgiou G, Iverson BL. Engineering of protease variants exhibiting high catalytic activity and exquisite substrate selectivity. Proc Natl Acad Sci USA. 2005;102:6855–6860. doi: 10.1073/pnas.0500063102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matsumura I, Ellington AD. In vitro evolution of beta-glucuronidase into a beta-galactosidase proceeds through non-specific intermediates. J Mol Biol. 2001;305:331–339. doi: 10.1006/jmbi.2000.4259. [DOI] [PubMed] [Google Scholar]
- 36.Fong S, Machajewski TD, Mak CC, Wong C. Directed evolution of d-2-keto-3-deoxy-6-phosphogluconate aldolase to new variants for the efficient synthesis of d- and l-sugars. Chem Biol. 2000;7:873–883. doi: 10.1016/s1074-5521(00)00035-1. [DOI] [PubMed] [Google Scholar]
- 37.Aharoni A, Gaidukov L, Khersonsky O, Mc QGS, Roodveldt C, Tawfik DS. The “evolvability” of promiscuous protein functions. Nature Genet. 2005;37:73–76. doi: 10.1038/ng1482. [DOI] [PubMed] [Google Scholar]
- 38.Thomas MR. Simple, effective cleanup of DNA ligation reactions prior to electro-transformation of E. coli. Biotechniques. 1994;16:988–990. [PubMed] [Google Scholar]
- 39.Dower WJ, Miller JF, Ragsdale CW. High efficiency transformation of E. coli by high voltage electroporation. Nucl Acids Res. 1988;16:6127–6145. doi: 10.1093/nar/16.13.6127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matsumura I, Wallingford JB, Surana NK, Vize PD, Ellington AD. Directed evolution of the surface chemistry of the reporter enzyme beta-glucuronidase. Nature Biotechnol. 1999;17:696–701. doi: 10.1038/10910. [DOI] [PubMed] [Google Scholar]