

Abstract
A series of putative dipeptide substrates of prostate specific membrane antigen (PSMA) was prepared that explored α- and β/γ-linked acidic residues at the P1 position and various chromophores at the P2 position, while keeping the P1’ residue constant as L-Glu. Four chromophores were examined, including 4-phenylazobenzoyl, 1-pyrenebutyrl, 9-anthracenylcarboxyl-γ-aminobutyrl, and 4-nitrophenylbutyryl. When evaluating these chromophores, it was found that a substrate containing 4-phenylazobenzoyl at the P2 position was consumed most efficiently. Substitution at the P1 position with acidic residues showed that only γ-linked L-Glu and D-Glu were recognized by the enzyme, with the former being more readily proteolyzed. Lastly, binding modes of endogenous substrates and our best synthetic substrate (4-phenylazobenzoyl-Glu-γ-Glu) were proposed by computational docking studies into an X-ray crystal structure of the PSMA extracellular domain.
Keywords: prostate specific membrane antigen, PSMA, glutamate carboxypeptidase II, substrate specificity, molecular docking
1. Introduction
A notable discovery in prostate cancer research has been the identification of an over-expressed membrane-bound cell surface protein on prostate cancer cells, namely, prostate-specific membrane antigen (PSMA). PSMA, also known as folate hydrolase I (FOLH1) and glutamate carboxypeptidase II (GCPII),1–3 is a 750-amino acid type II membrane glycoprotein4 and was discovered during the development of the LNCaP cell line; one which retains most of the known features of prostate cancer.5
Although PSMA is primarily expressed in normal human prostate epithelium, the importance of this enzyme lies with the fact that it is upregulated and strongly expressed in prostate cancer cells, including those of the metastatic disease state.6 PSMA expression has been detected in the endothelium of tumor-associated neovasculature of multiple nonprostatic solid malignancies,7 including metastatic renal carcinoma.8 As such, it is not surprising that PSMA has attracted a great deal of attention as a target for immunotherapy.9–12
In addition to its immunological importance, PSMA is also reported to possess two predominant yet poorly understood enzymatic activities (Figure 1): the hydrolytic cleavage and liberation of glutamate from γ-glutamyl derivatives of folic acid (e.g., 1)13,14 and the proteolysis of the neuropeptide N-acetylaspartylglutamate (NAAG) (2)2. PSMA is highly homologous to NAALADase (N-acetylated alpha-linked L-amino dipeptidase) which is specifically characterized by its ability to hydrolyze the neuropeptide NAAG.15 In contrast to NAALADase which has been extensively studied due to its presumed regulatory role in glutamate neurotransmission, the role of PSMA in prostate cancer remains conjectural.
Figure 1.
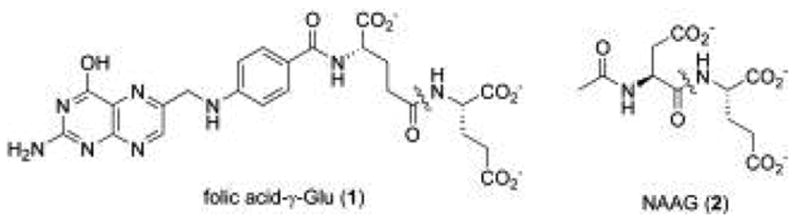
Endogenous substrates for PSMA proteolytic activity, with the key amide bond proteolyzed by the enzyme indicated.
Previous studies on substrate specificity of PSMA or glutamate carboxypeptidase II have indicated that the enzyme tends to prefer, although not exclusively, substrates with acidic residues in the P1 and P1’ position (Figure 2). One such study employed N-acetyl dipeptides to explore the substrate specificity of the NAAG substrate framework for a bacculovirus-expressed extracellular portion of recombinant glutamate carboxypeptidase II. In addition to substrates closely related to NAAG, four novel compounds with non-glutamate P1’residues were identified as modest substrates for GCPII (3–6).16 Mhaka and coworkers examined the substrate specificity of various glutamate and non-glutamate derivatives of methotrexate for a prodrug strategy to liberate methoxtrexate upon proteolysis by PSMA.17 This study identified several novel methotrexate-derived substrates which incorporated NAAG-like α-linked dipeptides (e.g. Asp-α-Glu) (7), as well as folate-like γ-linked dipeptides (Glu-γAsp, Glu-γ-Gln) and poly-γ-Glu peptides (8).
Figure 2.
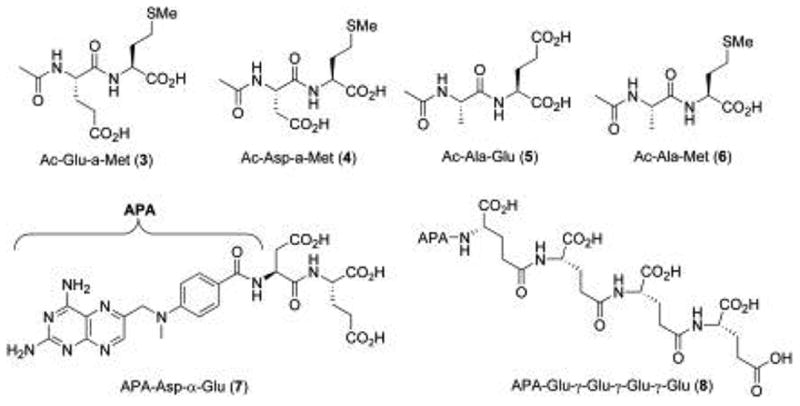
Previously reported substrates of PSMA.
In a previous study, we introduced a HPLC-based assay for PSMA activity to follow the proteolysis of a chromophore-bearing N-acyl-γ-diglutamate substrate to the corresponding cleaved N-acyl-glutamate product.18 The use of HPLC in monitoring PSMA activity is an alternative approach to the more conventional radioisotopic assay which requires ion-exchange chromatography to selectively elute enzymatically liberated, radiolabeled glutamate.
Herein, we explore the substrate specificity for various N-acyl acidic dipeptides using native, full-length PSMA isolated from LNCaP cells, with the end goal of establishing a refined HPLC assay for PSMA activity. In the series of substrates evaluated, γ-linked Glu and α-linked Asp residues were incorporated into the P1 position, making these compounds analogous to the endogenous PSMA substrates folate-γ-Glu (1) and NAAG ( 2). By varying the stereochemistry of the P1 residue as well as the regiochemistry of the peptide bond, we expected to determine the enzyme’s tolerance for variations of the binding determinants of the P1 residue when a large UV-active or fluorescent reporting group was present in the P2 position (Figure 3). Specifically, four chromophores were examined: 4-phenylazobenzoyl (4-Pab), 1-pyrenebutyrl (1-Pyrb), 9-anthracenecarboxyl-γ-aminobutyrl (9-Anth-Abu), and 4-nitrophenylbutyryl (4-Npb). Primarily, these chromophores were selected for there ease of detection by either UV absorption and fluorescence and commercial availability. The chromophores were also chosen to be hydrophobic, in order to maintain resemblance to the benzamide group present in the endogenous substrate folyl-γ-Glu (1), allowing the synthetic substrates to possibly participate in similar interactions near the active site. The 4-Pab chromophore has an additional feature, namely the azo linkage which could possibly participate in H-bonding interactions, possibly experienced by the pteridine heterocycle present in 1. Lastly, computational docking studies were carried out to propose tentative binding models of endogenous and synthetic PSMA substrates. Based upon the data presented herein, designs for novel substrates and inhibitors of PSMA can be refined.
Figure 3.
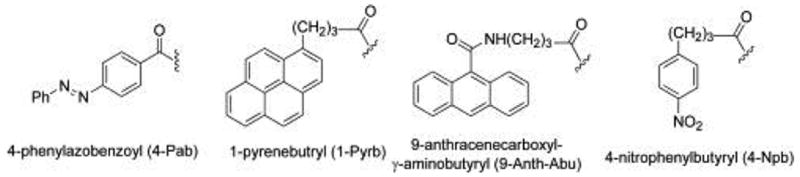
N-Acyl chromophores of PSMA substrate probes.
2. Results and Discussion
The synthesis of PSMA substrates and products was carried out using standard solid-phase peptide synthesis methodology, exemplified by the synthesis of a typical Glu-γ-Glu substrate (Figure 3) using a procedure that we described previously.18 Thus, polymer-supported protected glutamic acid (3) was deprotected and coupled with a second protected Glu residue to generate polymer-bound dipeptide (4). Fmoc-deprotection, followed by solid-supported coupling of the chromophore was affected to generate species (5). Lastly, concomitant acid-mediated deprotection and resin-cleavage was performed to generate the final dipeptide (6). Authentic standards of the expected PSMA-catalyzed proteolysis reaction were synthesized in a similar fashion.
We first chose to evaluate a set of four chromophores attached through an acyl linkage to Glu-γGlu, a dipeptide that comprises the substrate core of poly-γ-glutamylated folic acid (1) (Table 1). Purified yields of both substrates and their putative PSMA-mediated products were approximately 25% in all cases. In each case, reverse phase-HPLC (RP-HPLC) retention times of the dipeptide substrates and proteolytic products were sufficiently distinct to allow quantification (see Experimental section). The series of compounds was then evaluated as substrates for PSMA at 1 mM. As such, they were incubated in the presence of PSMA for 15 minutes before quenching and analysis by RP-HPLC. While each of the four compounds was effectively proteolyzed at the Glu-?-Glu linkage, there was significant variance in the relative turnover velocities of the four different compounds. The 4-Pab substrate (6a) turned over most efficiently, followed by the 9-Anth-Abu (6c), 1-Pyrb (6b), and 4-Npb (6d) conjugated compounds. Because of the greater similarity of the 4-Pab group to the folyl structure of a known PSMA substrate, we tentatively suggested that the 4-Pab substrate, and particularly the azo linkage, may experience favorable interactions in the active site which facilitates binding of this compound. The differential substrate activity of compounds 6b–6d appears less obvious and may be due to differences in hydrophobic interactions with residues in the active site, and may also be due to decreased aqueous solubility of the compounds.
Table 1.
Velocities of PSMA-mediated proteolysis of chromophore-substituted Glu-γ-Glu substrates.
| Substrate | V (nmol/min)* | |
|---|---|---|
| 6a |
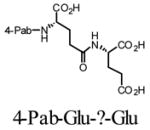
|
236.9 (8.2) |
| 6b |
|
115.2 (0.5) |
| 6c |

|
170.6 (3.1) |
| 6d |

|
90.3 (1.1) |
standard deviation in parenthesis
With the 4-Pab-conjugated Glu-?-Glu substrate (6a) being turned over most efficiently, we then chose to evaluate a series of acidic 4-Pab-labeled dipeptide substrates with variation at the P1 position (Figure 4). In this series of compounds (6e–6j), the P1 position was either Asp or Glu, with L or D stereochemistry, and linked to the P1’ group via the α- or side-chain carboxylate, while P1’ was fixed as L-Glu. As before, the RP-HPLC retention times were distinct with baseline resolution between substrates and the anticipated proteolytic products (see Experimental section). The series of compounds was then evaluated as substrates for PSMA at 1 μM. In this series, only the D-Glu-?-Glu substrate (6f) exhibited substrate activity under these conditions with a relative velocity of 79.8 (±2.6) nmol/min, a notably lower rate than the L-Glu-?-Glu substrates (6a–6d). These results suggest that the PSMA active site accommodates chromophore-substituted L-Glu-?-Glu dipeptide substrates and is not forgiving for similar residues in the P1 position, but stereochemical inversion of the P1 Glu residue is not catastrophic for activity. The absence of substrate activity at 1 μM of 4-Pab-Asp-a-Glu (6h) is curious as this substrate most closely resembles the endogenous substrate NAAG ( 2). We propose that this is due to the chromophore in 6h which alters the binding geometry of this species relative to NAAG. Interestingly, another group found an active Asp-α-Glu substrate linked to a large methotrexate-derived aminopteroyl group (APA) (i.e. APA-Asp-a-Glu).17 However, it is important to recognize that an extended incubation time was used in this study (24 hours) compared to the relatively shorter incubation in our own studies (15 minutes). For the two active 4-Pab conjugated substrates (6a and 6f), Km values were determined and found to be 415 nM (±20 nM) and 643 nM (±23 nm), respectively. These values are not inconsistent with the broad range reported for NAAG (2.6–540 nM).19 Substrates 6a and 6f were more efficiently consumed than other previously reported compounds, including Ac-Glu-α-Met-OH (Km = 53.0 μM), Ac-Asp-α-Met-OH (Km = 24.8 μM), and Ac-Ala-α-Met-OH (Km = 303 μM). 16
Figure 4.
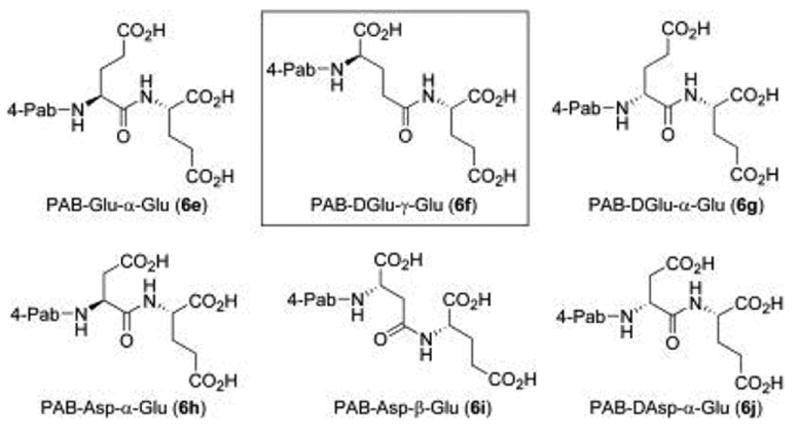
Variation of P1 in a series of potential PSMA inhibitors. At 1 mM, only PAB-DGlu-g-Glu (6f) was proteolyzed by PSMA.
To further understand the relationship of the structural determinants of the endogenous and synthetic substrates to substrate activity, computational docking was carried out on these species. Docking was performed with the FRED2 package (OpenEyes, Inc.) utilizing a recently published structure of recombinant PSMA extracellular domain co-crystallized with glutamate (PDB=2C6G).20 This paper also reported structures with a bound phosphonate inhibitor (PDB=2C6C) and with phosphate ion (PDB=2C6P), which exhibited weak inhibitory potency. Of the three structures, the first was selected as a model for substrate binding as it most closely resembled the structure of the enzyme following a proteolytic event. Folic acid-γ-Glu (1), NAAG (2), as well as our most efficiently consumed substrate (6a), were docked into the active site of PSMA. A restraint in the docking simulation was required in order to filter conformations of substrates bound in the active site that were non-productive for enzymatic activity. This specific restraint was implemented in FRED2 by temporarily converting the substrate dipeptide amide carbonyl (C=O) to a thiocarbonyl (C=S), followed by docking with a pharmacophore rule that the sulfur atom must reside within 2Å of the center of the two active site zinc atoms. After docking, the thiocarbonyl was converted back to a carbonyl, and the protein-ligand complex was re-minimized, without restraint, employing the MMFF94 force field as implemented in SZYBKI (OpenEyes, Inc.).
We first chose to model the binding of the natural substrate folic acid-γ-Glu (1) into the PSMA active site (illustrated in Figure 5a). The key interactions identified were the fitting of the P1’ Glu α-carboxyl into a hydrophilic pocket containing Arg210, Tyr700, and Tyr552, with the γ-carboxyl coordinating with Lys699 and Asn257. The overall orientation of the folic acid P1’ Glu residue is consistent with the orientation of Glu identified in the parent crystal structure.20 The Glu-γ-Glu carboxamide is coordinated to a PSMA zinc atom with a properly positioned water atom (also coordinated to zinc) poised for attack at the amide carboxyl group. The P1 Glu residue α-carboxyl group interacts with polar residues Ser454 and Glu457. The pteroyl benzamide carboxyl makes favorable contacts with Arg534 and Arg536, while the p-aminobenzoyl linking group is oriented for a possible π-stacking interaction with the Tyr700 aromatic system. Lastly, the aminopteridine heterocycle is oriented in a manner to participate in favorable hydrogen bond interactions with the Tyr700 peptide backbone as well as Arg511.
Figure 5.
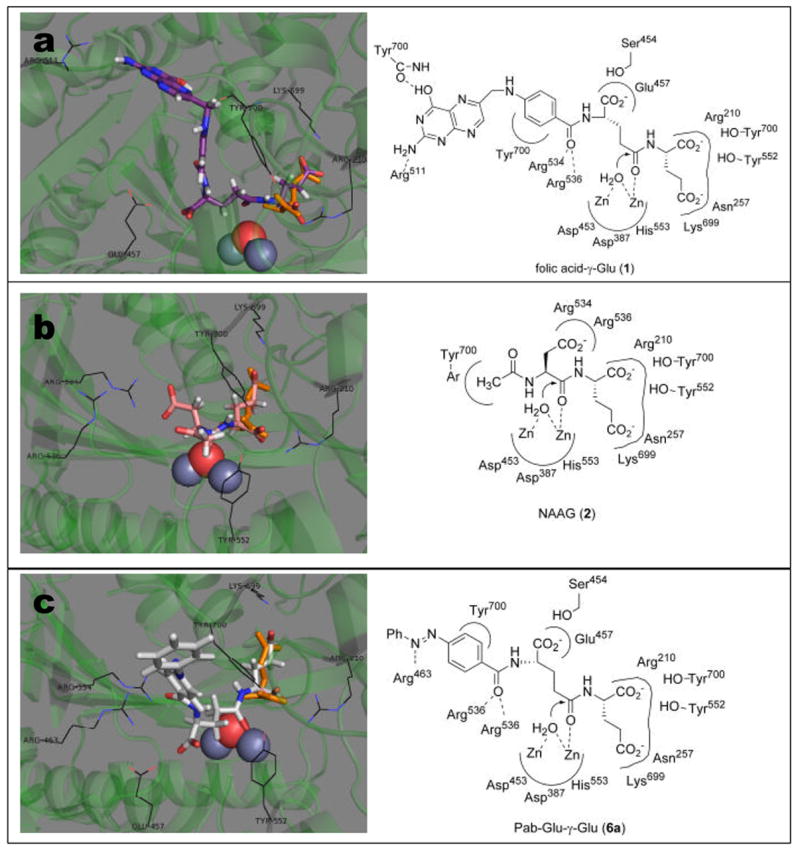
Results from docking natural and synthetic substrates into the active site of PSMA: Panel a: folic acid -γGlu 1 (violet); panel b: NAAG 2 (red); panel c: Pab-Glu-γ-Glu 6a (silver). In each panel, the figure on the left is a graphical representations showing overlay of substrate with glutamate (orange) as is positioned in the co-crystal structure (2C6G); the figure on the right is a cartoon representation showing key interactions of substrates with the PSMA active site.
The natural substrate NAAG ( 2) was also docked into the PSMA active site (Figure 5b). These studies show similar P1’ α- and γ-carboxyl residue interactions that were observed in the folic acid-γGlu docking studies, which are also consistent with the orientation of the co-crystallized Glu residue. The NAAG Asp-α-Glu carboxamide group was oriented in a manner to allow attack by zinc-coordinated water, while the Asp β-carboxyl group makes key favorable interactions with Arg534 and Arg536. It was not surprising that the P1 Glu α-carboxyl in folic acid-γ-Glu (1) may interact with different hydrophilic residues than the NAAG Asp β-carboxyl group, due to the geometrical differences in this dipeptide linkage, as well as the steric requirements of the large pteroyl moiety in folic acid-γ-Glu.
Finally, 4-Pab-Glu-γ-Glu species (6a) was docked into the PSMA active site (Figure 5c). Overall, the binding mode of this molecule shows consistency with our binding model of folic acid-γGlu (1). Notably, the key P1’ interactions are all maintained fairly closely. The geometry of the Glu-γGlu carboxamide carbonyl appears in a rotated orientation to the folic acid Glu-γ-Glu carboxamide, but is at the same time reasonably positioned for a proteolytic hydrolysis event with the zinc-coordinated water molecule. The P1 α-carboxyl group makes polar contacts with Ser454 and Glu457, as were observed for folic acid-γ-Glu (1). The Pab-carboxyl group docks in an orientation consistent with the folic acid-γ-Glu benzamide carboxyl, making hydrogen bond contacts with Arg534 and Arg536, while the Pab-disubstituted phenyl ring makes contact with Tyr700, which may facilitate a pi-stacking interaction. The Pab-azo linkage docks in an orientation allowing a potential hydrogen bond interaction with Arg463.
3. Conclusion
In summary, a series of PSMA substrates was prepared exploring α- and β/γ-linked acidic residues at P1 and chromophore substitution at P2, while keeping the P1’ position held constant as a L-Glu residue. Of the four chromophores examined at the P2 position, 4-Pab-containing substrates were turned over most efficiently by PSMA purified from LNCaP cells. The substitution patterns at the P1 position that were proteolyzed by PSMA were γ-linked L-Glu and D-Glu (e.g. substrates 6a and 6f) while substrates containing other acidic residues at P1 were apparently not recognized by the enzyme under our assay conditions. With regard to the development of new prodrugs requiring activation by PSMA, it is important to note that the kinetic behavior of full-length PSMA purified from LNCaP cells maybe different than PSMA expressed on the surface of intact LNCaP or prostate cancer cells Finally, binding modes of endogenous PSMA substrates folic acid-γ-Glu (1), NAAG (2), and our best synthetic substrate 6a were proposed based on computational docking of these compounds into the active site of PSMA extracellular domain. Not surprisingly, these docking results exhibit considerable consistency in the orientation of the P1’ residue. The similarity in the docking results for folic acid-γ-Glu (1) and the synthetic substrate (6a) are consistent with experimental substrate activity results. The findings from the work are expected to provide additional rationale for the design of new prodrugs and inhibitors of PSMA.
4. Experimental
HPLC Separation and Quantification of PSMA substrates and products
Substrates and their putative hydrolytic products were separated and quantified using an analytical reversed-phase HPLC column (Lichrosphere C18 5 μm, 150 x 4.6 mm; Phenomenex, Torrence, CA) at 1.0 ml/min with mobile phases consisting of ACN/potassium phosphate [25 mM, pH 2.0 (adjusted with H3PO4)] at a following respective isocratic ratios: 6a and 6e–6j: 40:60 (detected at 325 nm); 6b: 48:52 (detected at 252 nm); 6c: 35:65 (detected at 362 nm); 6d: 40:60 (detected at 274). HPLC parameters for 6a–6j are listed in Table 3.
Table 3.
HPLC Parameters of Synthetic PSMA Substrates.
| Retention Time (min) // Capacity Factor (k’) | |||
|---|---|---|---|
| Substrate | Substrate | Product | Selectivity Factor (α) |
| 6a | 4.2 // 2.1 | 6.3 // 3.7 | 1.8 |
| 6b | 3.7 // 1.8 | 5.6 // 3.2 | 1.8 |
| 6c | 4.0 // 2.0 | 5.3 // 2.9 | 1.5 |
| 6d | 3.0 // 1.3 | 3.9 // 1.8 | 1.5 |
| 6e | 4.7 // 2.5 | 6.3 // 2.7 | 1.5 |
| 6f | 4.2 // 2.1 | 6.3 // 3.7 | 1.8 |
| 6g | 4.7 // 2.5 | 6.3 // 3.7 | 1.5 |
| 6h | 4.5 // 2.4 | 5.9 // 3.4 | 1.5 |
| 6i | 4.0 // 2.0 | 6.0 // 3.4 | 1.7 |
| 6j | 4.5 // 2.4 | 5.9 // 3.4 | 1.5 |
Synthesis of PSMA substrates
Reactions were carried out in Biorad Biospin chromatography columns, attached to 3-way solvent-resistant stopcocks. During solid-phase reactions, the vessels were agitated by 360° rotation using a LabQuake rotisserie shaker. 1H and 13C NMR spectra were recorded on either a Bruker 300 or 500 MHz NMR Spectrometer. 1H and 13C NMR chemical shifts are internally referenced to TMS (δ = 0.00 ppm) or the appropriate solvent peak.
Step 1 (Fmoc deprotection)
N-a-Fmoc-L-glutamic acid-a-tert-butyl ester preloaded on Wang resin (typically 0.270mmol; 0.54mmol/g resin 100–200 mesh, Novabiochem San Diego, CA) was deprotected with 5mL piperidine/DMF (20:80, v:v) for 15 min, vacuum filtered, washed, and vacuum filtered using the following solvent wash cycle: 4x3 mL DMF, 4x3 mL CH2Cl, 2x3 mL CH3CN, 2x3 mL CH2Cl2. The presence of free amine was confirmed by a positive Kaiser test.
Step 2 (Amino acid coupling)
A solution of Fmoc-L-glutamic acid-a-tert-butyl ester (2 eq), DIPEA (2 eq), PyBOP (2 eq) and HOBt monohydrate (2 eq) in DMF:CH2Cl2 (1:1) was allowed to pre-activate for two minutes, added to the deprotected resin, and the vessel was agitated for 20 minutes. The resin was vacuum filtered and then subjected to the solvent wash cycle described in step 1. Complete conversion in the coupling reaction was confirmed by a negative Kaiser test.
Step 3 (Fmoc deprotection)
Step 1 was repeated.
Step 4 (Chromophore coupling)
A solution of 4-(phenylazo)benzoic acid (2 eq), DIPEA (2 eq), PyBOP (2 eq) and HOBt monohydrate (2 eq) in DMF:CH2Cl2 (1:1) was allowed to pre-activate for two minutes, added to the deprotected resin, and the vessel was agitated for 20 minutes. The resin was vacuum filtered and then subjected to the solvent wash cycle described in step 1. Complete conversion in the coupling reaction was confirmed by a negative Kaiser test. In cases where coupling appeared incomplete, step 4 was repeated.
Step 5 (Cleavage of product from resin)
A solution of TFA:H2O (4:1, v:v) was added to the resin and the vessel was agitated for 3h. The resin was vacuum filtered then washed with CH2Cl2 (3 x 5 mL). The filtrate was collected and the solvent removed under reduced pressure to provide a reddish brown solid. The products were purified by semi-preperative HPLC (Alltech Semi-Prep Econosil C18- 10l, 250x22 mm) using a mobile phase composed of 65% A and 35% B (Solvent A=0.1% aqueous TFA by volume; Solvent B=CH3CN) to give the final products in typically between 10–30% yield.
N-[4-phenylazobenzoyl]-glutamyl-g-glutamic acid (6a)
Mp 181–182 °C. 1H NMR (300 MHz, D2O): d 1.79–1.93 (m, 2H), 1.96–2.12 (m, 2H), 2.14–2.24(m, 2H), 2.38–2.45 (m, 2H), 4.05–4.09 (m, 1H), 4.27–4.31 (m, 1H), 7.49 (d, 2H, J = 6.6), 7.64–7.68 (m, 5H), 7.80 (d, 2H, J = 8.40). 13C NMR (75 MHz, D2O) : d 28.76, 29.52, 33.37, 35.04, 56.24, 56.46, 123.36, 123.49, 129.28, 130.36, 133.07, 136.46, 152.75, 154.66, 169.61, 175.82, 178.88, 179.65, 182.94. lmax=325nm, e=2.58 x 104 M−1cm−1.
N-[1-pyrenebutyrl]-glutamyl-g-glutamic acid. (6b)
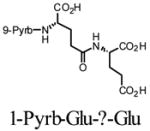
1H NMR (400 MHz,CD3OD): d 1.11–1.17 (t, 2H), 1.23–1.31 (m, 2H), 1.50 (s, 2H), 1.55 (s, 2H), 2.04 – 2.19 (m, 2H), 2.34 – 2.43 (m, 2H), 3.36 – 16 3.40 (m, 2H), 3.87 (m, 1H), 4.39 – 4.40 (m, 1H), 4.74 – 4.86 (m, 5H), 6.90 – 8.36 (m, 9H). 13C NMR (75 MHz, DMSO-d6) : d 6.3, 26.7, 27.6, 30.1, 31.6, 32.1, 34.7, 51.1, 51.6, 123.6, 124.1, 124.2, 124.8, 125.0, 126.1, 126.5, 127.2, 127.4, 127.6, 128.2, 129.3, 130.4, 130.9, 136.6, 171.6, 172.2, 173.4, 173.6, 173.7. HRMS (MALDI-TOF, positive mode) Calc. [M+1] = 547.2080, found [M+1] = 547.2062 for C30H30N2O8.
N-[9-anthracenyl]-aminobutyrl-glutamyl-g-glutamic acid. (6c)
1H NMR (400 MHz, CD3OD) : d 1.88 – 1.97 (m, 2H), 2.07 – 2.16 (m, 2H), 2.14 – 2.23 (m, 2H), 2.35 – 2.41 (m, 4H), 2.46 – 2.49 (m, 2H), 3.64 – 3.68 (m, 1H), 4.38 – 4.44 (m, 2H), 4.86 (s, 6H), 7.48 – 8.57 (m, 9H). 13C NMR (75 MHz, DMSO-d6) : d 25.5, 26.3, 27.0, 30.1, 31.6, 32.8, 51.1, 51.6, 125.3, 125.6, 126.5, 127.1, 127.3, 128.4, 130.7, 133.4, 168.1, 171.6, 172.0, 173.4, 173.5, 173.7. LRMS (MALDI-TOF, positive mode, CD3OD) Calc. [M+1] = 566.2, found [M+1] = 566.9 (plus two other deuteration states: 567.9, and 568.9) for C29H31N3O9.
N-[4-nitrophenylbutyryl]-glutamyl-g-glutamic acid. (6d)
1H NMR (400 MHz, CD3OD) : d 1.88 – 2.01 (m, 4H), 2.10 – 2.24 (m, 4H), 2.26 – 2.27 (m, 2H), 2.27 – 2.40 (m, 2H), 2.79 (t, 2H, J = 12), 4.37 – 4.45 (m, 2H), 7.46 (d, 2H, J = 8), 8.15 (d, 2H, J = 8). 13C NMR (75 MHz, CD3OD) : 25.4, 28.0, 28.2, 28.4, 31.4, 33.3, 36.0, 36.1, 53.2, 53.4, 124.6, 124.7, 130.8, 148.0, 151.5, 175.0, 175.1, 175.9, 176.4. LRMS (ESI-LCMS, positive mode) Calc. [M+1] = 468.16, found [M+1] = 468.17 for C20H25N3O10.
N-[4-phenylazobenzoyl]-glutamyl-a-glutamic acid (6e)
1H NMR (300 MHz, DMSO-d6) : d 1.81 – 2.10 (m, 4H), 2.30 – 2.40 (m, 4H), 4.20 – 4.27 (m, 1H), 4.47 – 4.54 (m, 1H), 7.62 – 7.65 (m, 3H), 7.94 – 7.98 (m, 4H), 8.11 (d, 2H, J = 9), 8.29 (d, 1H, J = 7.7), 8.67 (d, 1H, J = 7.7). 13C NMR (75 MHz, DMSO-d6) : d 26.2, 27.0, 30.1, 30.5, 51.2, 52.8, 122.3, 122.7, 128.4, 128.9, 129.6, 132.0, 136.2, 151.9, 153.3, 165.7, 171.4, 173.1, 173.8, 174.1.
N-[4-phenylazobenzoyl]-D-glutamyl-g-glutamic acid (6f)
1H NMR (300 MHz, DMSO-d6) : d 1.66 – 1.80 (m, 1H), 1.90 – 2.00 (m, 2H), 2.07 – 2.13 (m, 1H), 2.23 – 2.32 (m, 4H), 4.15 – 4.22 (m, 1H), 4. 35 – 4.42 (m, 1H), 7.60 – 7.64 (m, 3H), 7.92 – 7.98 (m, 4H), 8.09 (d, 2H, J = 9), 8.17 (d, 1H, J = 6), 8.87 (d, 1H, J = 6). 13C NMR (75 MHz, DMSO-d6) : d 26.3, 30.1, 31.9, 51.2, 52.6, 122.4, 122.8, 128.8, 129.6, 132.1, 136.2, 151.9,153.4, 165.7, 170.3, 171.8, 173.3, 173.7.
N-[4-phenylazobenzoyl]-D-glutamyl-a-glutamic acid (6g)
1H NMR (300 MHz, DMSO-d6) : d 1.77 – 1.84 (m, 2H), 1.90 – 2.05 (m, 2H), 2.24 – 2.37 (m, 4H), 4.20 – 4.25 (m, 1H), 4.50 – 4.56 (m, 1H), 7.61 – 7.65 (m, 3H), 7.93 – 7.98 (m, 4H), 8.12 (d, 2H, J = 6), 8.26 (d, 1H, J = 6), 8.66 (d, 1H, J = 6). 13C NMR (75 MHz, DMSO-d6) : d 26.5, 27.2, 30.1, 30.5, 51.3, 53.0, 122.3, 122.7, 129.9, 129.6, 132.0, 136.3, 151.9, 153.4, 165.7, 171.3, 173.1, 173.8, 174.0.
N-[4-phenylazobenzoyl]-aspartyl-a-glutamic acid (6h)
1H NMR (300 MHz, DMSO-d6) : d 1.75 – 1.84 (m, 1H), 1.95 – 2.02 (m, 1H), 2.30 (t, 2H, J = 7.6), 2.67 – 2.84 (m, 2H), 4.21 – 4.25 (m, 1H), 4.82 – 4.87 (m, 1H), 7.62 – 7.67 (m, 3H), 7.93 – 7.99 (m, 4H), 8.09 (d, 2H, J = 9), 8.25 (d, 1H, J = 7.8), 8.86 (d, 1H, J = 7). 13C NMR (75 MHz, DMSO-d6) : d 26.0, 30.0, 36.0, 50.2, 51.3, 120.0, 122.3, 122.8, 128.9, 129.6, 132.1, 136.2, 151.9, 153.4, 165.5, 170.9, 171.8, 173.1, 173.8.
N-[4-phenylazobenzoyl]-aspartyl-b-glutamic acid (6i)
1H NMR (500 MHz, CD3OD): d 1.83–1.89 (m, 1H), 2.09–2.16 (m, 1H), 2.27–2.38 (m, 2H), 4.40–4.55 (m, 1H), 4.91–4.95 (m, 1H), 7.45–7.51 (m, 2H), 7.81–7.97 (m, 7H). 13C NMR (125 MHz, CD3OD ): d 28.0, 31.2, 38.1, 51.4, 53.0, 123.9, 124.2, 129.8, 130.5, 133.0, 137.3, 154.1, 155.9, 169.2, 172.7, 174.2, 174.9, 176.4.
N-[4-phenylazobenzoyl]-D-aspartyl-a-glutamic acid (6j)
1H NMR (500 MHz, DMSO-d6): d 1.97–2.02 (m, 1H), 2.14–2.24 (m, 1H), 2.34–2.42 (m, 2H), 2.78–2.86 (dd, 1H, J1 = 7.4 Hz, J2 = 16.6 Hz ), 2.95–3.03 (dd, 1H, J1 = 6.3 Hz, J2 = 16.6 Hz , 4.40–4.49 (m, 1H), 4.97–5.03 (m, 1H), 7.50–7.57 (m, 2H), 7.90–8.03 (m, 7H). 13C NMR (75 MHz, CD3OD): d 28.0, 31.2, 36.8, 52.0, 53.4, 123.8, 124.21, 130.0, 130.5, 133.0, 137.4, 154.1, 155.9, 169.6, 173.1, 174.2, 174.7, 176.6.
PSMA substrate activity assay
The purification of full-length PSMA from cultured LNCaP cells has been described previously.1 Enzyme activity assays for various substrates was based upon a method previously reported with slight modifications.18 Working solutions of the substrates were made in TRIS buffer (50 mM, pH 7.5). A typical incubation mixture (final volume 250 μL) was prepared by the addition of 25 μL of a solution of purified PSMA to 200 μL TRIS buffer (50 mM, pH 7.4). The enzymatic reaction was initiated by the addition of 25 μL substrate (10 μM) giving a final substrate concentration of 1 μM. The reaction was allowed to proceed for 15 min with constant shaking at 37 °C and was terminated by the addition of 25 μL methanolic TFA (2% trifluoroacetic acid by volume in methanol) followed by vortexing and centrifugation (10 min at 7000g). An 85 μL aliquot of the resulting supernatant was subsequently quantified by HPLC as described above. Under the assay conditions described, it was noted that the initial substrate concentration was not substantially depleted during the time course of the incubation (e.g., less than 10 % conversion to product was observed.
Computational modeling
Prior to docking, ligands were minimized in the MMFF94 force field using SZYBKI (OpenEyes) and then converted into conformational libraries using OMEGA (OpenEyes).21 Docking was then performed with FRED2 (OpenEyes) employing a structure of recombinant PSMA extracellular domain co-crystallized with glutamate (PDB=2C6G).20 Partial charges were assigned with MOLCHARGE (OpenEyes) using the MMFF94 model for the protein, and 19 the AM1BCC model for ligands. The active site was defined as the residues falling within a 16 angstrom sphere around the PSMA catalytic zinc atoms. Substrate conformations non-productive for PSMA proteolytic activity were filtered by utilizing a restraint in the docking model. Namely, the dipeptide amide carbonyl, known to be proteolyzed by PSMA, was converted to a thioamide (C=S) group and then a SMARTS constraint was specified requiring the substrate sulfur atom to be within 2 angstroms of the center of the two PSMA zinc atoms. For each substrate, the top scoring consensus pose was identified using two FRED2 scoring functions which consider metal interactions between the protein and ligand (ChemGauss2 and PLP). For this pose, the C=S group was converted to C=O and the protein-ligand complex was re-minimized in the MMFF94 force field as implemented in SZYBKI, without restraint. The final protein-ligand complexes were visualized using PYMOL (DeLano Scientific).
Supplementary Material
Figure 3.
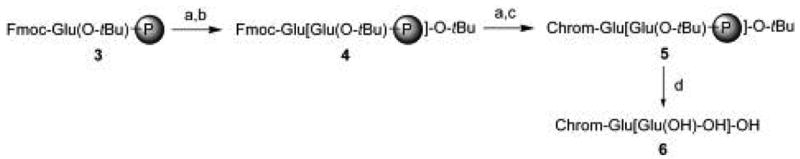
Synthesis of glutamate-based dipeptide substrates of PSMA. Reagents and conditions: (a) piperidine, DMF, 15 min, RT. (b) Fmoc-Glu(OtBu)-OH, PyBOP, HOBt, DIEA, DMF:CH2Cl2 (1:1) 20 min, RT. (c) chromophore-containing carboxylic acids, PyBOP, HOBt, DIEA, DMF:CH2Cl2, 20 min, RT. (d) TFA:H2O (4:1 v:v), 3h, RT.
Acknowledgments
This work was supported in part by grants from the National Institutes of Health, MBRS SCORE Program-NIGMS (Grant No. S06-GM052588) and the National Cancer Institute, U56-Program (Grant No. CA 96217). The authors also extend their gratitude to W. Tam and the NMR facility at SFSU for their expert assistance.
Footnotes
Supporting Information
1H and 13C NMR spectra are provided for compounds 6a–6j.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Drugs in R & D. 2004;5(6) [Google Scholar]
- 2.Carter RE, Feldman AR, Coyle JT. Proc Natl Acad Sci U S A. 1996;93:749–53. doi: 10.1073/pnas.93.2.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tiffany CW, Lapidus RG, Merion A, Calvin DC, Slusher BS. Prostate. 1999;39:28–35. doi: 10.1002/(sici)1097-0045(19990401)39:1<28::aid-pros5>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 4.Holmes EH, Greene TG, Tino WT, Boynton AL, Aldape HC, Misrock SL, Murphy GP. Prostate Suppl. 1996;7:25–9. [PubMed] [Google Scholar]
- 5.Horoszewicz JS, Leong SS, Kawinski E, Karr JP, Rosenthal H, Chu TM, Mirand EA, Murphy GP. Cancer Res. 1983;43:1809–18. [PubMed] [Google Scholar]
- 6.Bacich DJ, Pinto JT, Tong WP, Heston WD. Mamm genome. 2001;12(2):117–23. doi: 10.1007/s003350010240. [DOI] [PubMed] [Google Scholar]
- 7.Chang SS, O'Keefe DS, Bacich DJ, Reuter VE, Heston WD, Gaudin PB. Clin Cancer Res. 1999;5(10):2674–81. [PubMed] [Google Scholar]
- 8.Chang SS, Reuter VE, Heston WD, Gaudin PB. Urology. 2001;57:801–5. doi: 10.1016/s0090-4295(00)01094-3. [DOI] [PubMed] [Google Scholar]
- 9.Tasch J, Gong M, Sadelain M, Heston WD. Crit Rev Immunol. 2001;21:249–61. [PubMed] [Google Scholar]
- 10.Salit RB, Kast WM, Velders MP. Frontiers in bioscience : a journal and virtual library. 2002;7:e204–13. doi: 10.2741/salit. [DOI] [PubMed] [Google Scholar]
- 11.Lu J, Celis E. Cancer Res. 2002;62(20):5807–12. [PubMed] [Google Scholar]
- 12.Fracasso G, Bellisola G, Cingarlini S, Castelletti D, Prayer-Galetti T, Pagano F, Tridente G, Colombatti M. The Prostate. 2002;53(1):9–23. doi: 10.1002/pros.10117. [DOI] [PubMed] [Google Scholar]
- 13.Heston WD. Urology. 1997;49:104–12. doi: 10.1016/s0090-4295(97)00177-5. [DOI] [PubMed] [Google Scholar]
- 14.Pinto JT, Suffoletto BP, Berzin TM, Qiao CH, Lin S, Tong WP, May F, Mukherjee B, Heston WD. Clin Cancer Res. 1996;2:1445–51. [PubMed] [Google Scholar]
- 15.Robinson MB, Blakely RD, Couto R, Coyle JT. J Biol Chem. 1987;262:14498–506. [PubMed] [Google Scholar]
- 16.Barinka C, Rinnova M, Sacha P, Rojas C, Majer P, Slusher BS, Konvalinka J. J Neurochem. 2002;80:477–87. doi: 10.1046/j.0022-3042.2001.00715.x. [DOI] [PubMed] [Google Scholar]
- 17.Mhaka A, Gady AM, Rosen DM, Lo KM, Gillies SD, Denmeade SR. Cancer Biol Ther. 2004;3:551–8. doi: 10.4161/cbt.3.6.846. [DOI] [PubMed] [Google Scholar]
- 18.Maung J, Mallari JP, Girtsman TA, Wu LY, Rowley JA, Santiago NM, Brunelle AN, Berkman CE. Bioorg Med Chem. 2004;12:4969–79. doi: 10.1016/j.bmc.2004.06.031. [DOI] [PubMed] [Google Scholar]
- 19.Several representative references and Km values for NAAG with various sources of GCPII follow. Km=2.6 nM: Tang H, Brown M, Ye Y, Huang G, Zhang Y, Wang Y, Zhai H, Chen X, Shen TS, Tenniswood M. Biochem Biophys Res Commun. 2003;307:8. doi: 10.1016/s0006-291x(03)01119-7.Km=73 nm: Luthi-Carter R, Barczak AK, Speno H, Coyle JT. Brain Res. 1998;795:341–348. doi: 10.1016/s0006-8993(98)00244-3.Km=87, 100, and 146 nM: Luthi-Carter R, Barczak AK, Speno H, Coyle JT. J Pharmacol Exp Ther. 1998;286:1020–1025.Km=130 nM: Rojas C, Frazier ST, Flanary J, Slusher BS. Anal Biochem. 2002;310:50. doi: 10.1016/s0003-2697(02)00286-5.Km=200 nM: Tiffany CW, Lapidus RG, Merion A, Calvin DC, Slusher BS. Prostate. 1999;39:28. doi: 10.1002/(sici)1097-0045(19990401)39:1<28::aid-pros5>3.0.co;2-a.Km=427 nM: Barinka C, Rinnova M, Sacha P, Rojas C, Majer P, Slusher BS, Konvalinka JJ. Neurochem. 2002;80:477. doi: 10.1046/j.0022-3042.2001.00715.x.Km=540 nM: Robinson MB, Blakely RD, Couto R, Coyle JT. J Biol Chem. 1987;262:14498.
- 20.Mesters JR, Barinka C, Li W, Tsukamoto T, Majer P, Slusher BS, Konvalinka J, Hilgenfeld R. Embo J. 2006;25:1375–84. doi: 10.1038/sj.emboj.7600969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bostrom J, Greenwood JR, Gottfries J. J Mol Graph Model. 2003;21:449–62. doi: 10.1016/s1093-3263(02)00204-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.