

Abstract
Aims
Whereas cortical EEG effects of benzodiazepines are well characterized, information about benzodiazepine effects in other areas of the central nervous system is sparse. This study investigated the action of midazolam and its active metabolite α-hydroxy-midazolam on different parts of the auditory pathway in six healthy volunteers in a randomized, double-blind, three-way cross-over study.
Methods
Acoustically evoked short (SLP) and middle (MLP) latency potentials, transitory evoked otoacoustic emissions (TEOAE), and EEG power spectra were analysed after short i.v. injections of placebo, or 0.15 mg kg−1 midazolam, or α-hydroxy-midazolam, respectively.
Results
All subjects fell asleep during the 4 min infusion of active drug. SLP showed a significant transient increase of Jewett wave V 10 min after injection for midazolam and α-hydroxy-midazolam while the latency of wave I was unchanged. Both benzodiazepines induced a marked and long-lasting MLP amplitude decrease for 240 min with slow recovery over the following 360 min. No changes of TEOAE were observed. In agreement with earlier reports, increases in EEG beta activity and decreases in alpha activity were observed after administration of either drug.
Conclusions
Systemically administered benzodiazepines modulate the auditory pathway above the level of the cochlea. While SLP changes were closely associated with sedation and high plasma benzodiazepine concentrations, MLP effects persisted for hours after sedation even at low benzodiazepine plasma levels. Evoked potentials may therefore be more sensitive than EEG as a tool to monitor benzodiazepine effects.
Keywords: acoustically evoked potentials, α-hydroxy-midazolam, electroencephalogram, healthy volunteers, midazolam, pharmacokinetics, transiently evoked otoacoustic emissions
Introduction
Almost all pharmacological effects of benzodiazepines result from their actions on the central nervous system. The electroencephalogram (EEG) and evoked potentials are validated methods to monitor pharmacological effects of centrally acting drugs. In addition to the well-known EEG effects of benzodiazepines with decreases in alpha and increases in beta activity [1], amplitude reductions of cortical somatosensory-evoked potentials and latency shifts have also been described [2]. However, the effects of benzodiazepines on different qualities of auditory evoked potentials (AEP) have not been thoroughly investigated in humans and little is known about the effect of benzodiazepines on different areas of the auditory pathway [3, 4].
The aim of this placebo-controlled, double-blind study was to assess the effects of midazolam and its active metabolite α-hydroxy-midazolam on the functional integrity of the auditory pathway, which can be monitored from the cerebral cortex to the cochlear periphery by objective methods [5]. These include short latency brainstem evoked potentials (SLP), middle latency potentials (MLP), and transiently evoked otoacoustic emissions (TEOAE). TEOAE reflect the activity of the contractile outer hair cells of the cochlea [6], SLP the basal generation pattern of the cochlea, eighth nerve, and brainstem, and MLP upper brainstem, thalamus, subcortical, and early cortical sections of the auditory pathway [7]. In general MLP (10–50 ms) are of neurogenic or myogenic origin. In this study, mostly myogenic responses were evaluated because only the myogenic crossed acoustic response (CAR) was recorded, since this can easily be monitored in a clinical setting. CAR differs from neurogenic MLP by early onset, a five to tenfold increased potential amplitude, and the characteristic that it is abolished by coadministration of muscle relaxants (e.g. curare) [8]. Similar to the stapedial reflex, the afferent limb of the CAR includes the inner ear, the vestibular organ, the eighth nerve, and the upper olivary complex in the brainstem. The salivatory nucleus, the seventh nerve, and the posterior auricular muscle form the efferent limb of the reflex [9, 10].
Methods
Subjects
Six healthy normal hearing male volunteers (3 smokers) with a mean age of 29 years (range 26–32 years), weighing 70 kg (range 63–83 kg) participated in the study after giving written informed consent. They were not taking any medications and had no history of significant medical disease. All had normal clinical and electrocardiographic examinations and normal laboratory values (blood chemistry, complete blood cell count, urinalysis). The protocol was reviewed and approved by the Ethics Committee of the University Hospital of Basel, Switzerland.
Protocol
This was a randomized, single dose, three-way crossover study. The three treatment conditions were midazolam (0.15 mg kg−1), α-hydroxy-midazolam (0.15 mg kg−1), and placebo (normal saline) given intravenously at least 1 week apart. With the exception of the first study in volunteer 1 (pilot study using midazolam) all studies were done in a double-blind manner. The selected doses were sufficiently high to produce peak plasma concentrations in a clearly sedating but still well tolerated range. These estimations were based on earlier studies in healthy subjects using the same benzodiazepines [1, 11].
After an overnight fast, subjects were admitted to a quiet study room with dimmed lights in the Medical Intensive Care Unit. During the experiment, the volunteers were in a relaxed supine position. A cannula for blood sampling was inserted into a forearm vein and kept patent by continuous infusion of normal saline (1000 ml over approximately 4 h). The drugs were infused over 4 min through a separate intravenous cannula in the opposite arm. Blood samples were taken before drug administration and after 10, 30, 50, 70, 90, 120, 150, 180, 210, 240, 300, 360, 420, 480, 560, 600, 660, and 720 min. The plasma was separated within 1 h and kept frozen at −20° C until analysed. A urine sample was obtained before drug administration and thereafter urine was collected from 0 to 2, 2–4, 4–6, 6–12, and 12–24 h. An aliquot of each urine sample was stored deep-frozen until analysed. The volunteers fasted from 8 h before until 4 h after drug injection and remained in bed throughout the study. After 4 h, a standard meal was served. Beverages containing methylxanthines or alcohol were not allowed during the trial.
At approximately 50 and 10 min before drug administration, two EEG and AEP baseline values were recorded. Respiratory rate and capillary oxygen saturation (measured by finger pulse oximetry) were monitored continuously throughout the study. The EEG power spectrum, respiratory rate, and capillary oxygen saturation were always recorded immediately before taking each blood sample, AEP measurements were obtained immediately thereafter.
Drug concentrations and pharmacokinetic analysis
Plasma and urine concentrations of midazolam and α-hydroxy-midazolam were measured by a specific high-performance liquid chromatography method as previously described [12]. Briefly, 0.5 ml plasma and 0.1 ml (0.5 ml for urine) borate buffer 0.1 m were added to a glass tube containing 100 ng of Ro 05–6669 (7-chloro-5-(4-methoxyphenyl)-1-methyl-3H-1,4-benzodiazepine- 2(1H)-one) as internal standard. After extraction with diethylether, the aqueous phase was discarded and the organic phase was evaporated to dryness. The residue was dissolved in the mobile phase (methanol: isopropylalcohol:HClO4 7.5 10−3m) (90:40:30 v/v) and 150 ml was injected into the h.p.l.c. column. The effluent was monitored with a u.v. detector (245 nm). At a flow rate of 1.7 ml min−1 the retention times of internal standard (Ro 05–6669), 1,4-dihydroxy-midazolam, α-hydroxy-midazolam, 4-hydroxy-midazolam, and midazolam were 4.85, 6.7, 7.9, 9.8, and 10.1 min, respectively. Peak height ratios were used for quantification. The extraction recoveries of α-hydroxy-midazolam and midazolam were 95±5%. Intra-and interassay coefficients of variation were less than 6% for plasma samples and less than 10% for urine samples. The lower limit of detection was 2.5 ng ml−1 in 0.5 ml plasma or urine for all compounds.
Iterative nonlinear least-squares regression was performed with SIPHAR®, an integrated computer program for pharmacokinetic analysis (SIMED, Créteil, France). Drug kinetics were described by one- or two-compartment open models. The slope of the terminal log-linear phase was determined by linear regression of the log-transformed plasma concentration-time data. The area under the plasma concentration-time curve was calculated using the trapezoidal rule with extrapolation to infinity (AUC(0–∞)). Volumes of distribution (V), total body and renal clearances were estimated by standard procedures [13].
EEG analysis
Sixteen standard disc electrodes were attached at positions according to the International Ten-Twenty System and connected by a common reference lead to a 1.5 MΩ resistance. The EEG was recorded on a 16-channel apparatus (Encephaloscript ES 16000®, Picker International, München, Germany), using a time constant of 0.3 s and a high-frequency filter of 30 Hz. Impedances were maintained below 1000 Ω. The data were stored on a floppy disc. Sixty-second periods free of artefacts and sleep spindles were chosen for power spectra analysis (Brain Surveyor®, Picker International, München, Germany). The frontal leads F3 and F4 were used for analysis of beta-activity, the occipital leads O1 and O2 for delta-, theta-, and alpha-activity. The mean of the two baseline values was set to zero percent and the data were expressed as percentage changes over baseline activity. The frequency ranges were defined as follows: delta (0.5–6 Hz), theta (6–8.5 Hz), alpha (8.5–13 Hz), and beta (13.5–15.5 Hz). For comparison, beta activity was also evaluated in the range from 13 to 30 Hz.
SLP
SLP were elicited as previously described [14] with rarefaction clicks of 100 μs duration with an intensity of 115 dB peak equivalent sound pressure level. Masking white noise of an intensity of 50 dB below the stimulus intensity was presented to the contralateral ear. The recordings from scalp EEG electrodes, placed at the vertex (Cz) and the mastoids (A1 and A2), were amplified differentially with respect to a forehead reference electrode and band-pass filtered between 150 Hz and 3 kHz. AEP interpretation was based on differences in amplitudes (μV) and latencies of waves I and V (ms) of individual waveforms. The stimulation and recording equipment was a Nicolet Compact Four System (Nicolet Biomedical, Madison, Wisconsin, USA).
MLP
CAR was elicited by rarefaction clicks as previously described [15]. The stimulus presentation rate was 9 Hz for the 50 ms middle latency reaction time analysis window. The presentation level was 115 dB peak equivalent sound pressure level. CAR interpretation was based on individual amplitude differences (μV). The same electrode placement and recording equipment was used as for the measurement of SLP.
TEOAE
An ILO88 software/hardware package (Otodynamics Ltd, Hatfield, UK) run on a portable computer was used to measure TEOAE [16]. A small probe with an integrated microphone and loudspeaker was fixed in the ear canal using an ear plug. A stimulus arrangement cancelling the linearly growing portion of the responses (‘nonlinear stimulus’ of ILO88) was used. Stimuli were clicks of 85 dB sound pressure level. The final result was the average of 256 responses which were Fourier analysed and stored on disks for later analysis. Responses were evaluated by subtracting levels on the basis of the overall level (dB) and frequency specific windows (0–2, 2–4, 4–6 kHz) of the click response.
Pure tone audiogram
Pure tone testing at 0.125, 0.25, 0.5, 1, 2, 4, 6, and 8 kHz was performed before and after the trial in a soundproof cabin with a Madsen audiometer (Madsen Electronic, Taastrup, Denmark).
Statistical analysis
Data are expressed as means±s.e. mean unless indicated otherwise. Net drug effects were evaluated after subtraction of the corresponding placebo values. Differences between the pharmacokinetic parameters of the two drugs or between observed drug effects and baseline values were evaluated by Student's t-test or Wilcoxon Signed Rank test as appropriate. anova for repeated measures with Bonferroni/Dunn correction was used to calculate effects of midazolam and α-hydroxy-midazolam on MLP and TEOAE. A P value <0.05 was considered statistically significant.
Results
All six subjects fell asleep during the 4 min injections of benzodiazepines whereas no subject fell asleep during the placebo administration. The only side-effects observed were transient hiccups occurring immediately after drug administration in three subjects (twice after midazolam, once after the metabolite) which disappeared after few minutes. Occurrence of hiccups after intravenous administration of midazolam or α-hydroxy-midazolam has been reported previously [11].
Pharmacokinetics
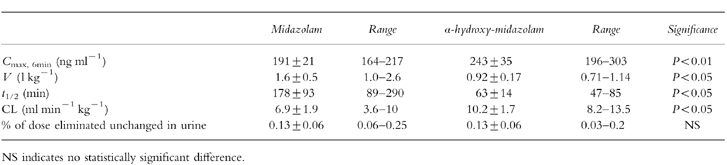
After administration of midazolam, peak plasma concentrations of midazolam were in the therapeutic range [17, 18] in all subjects (Figure 1). α-Hydroxy-midazolam was detectable in the first blood sample, which was drawn 10 min after the start of the 4 min infusion of midazolam. After administration of the metabolite, no midazolam was detected in any blood sample. Individual plasma concentration-time profiles of midazolam and α-hydroxy-midazolam were described by mono- or biexponential functions. The terminal elimination half-life of α-hydroxy-midazolam was significantly shorter than that of midazolam, its clearance was significantly higher, and its volume of distribution significantly smaller (Table 1). After 12 h, unconjugated benzodiazepine levels were below the detection limit (<2.5 ng ml−1) in all urine samples.
Figure 1.
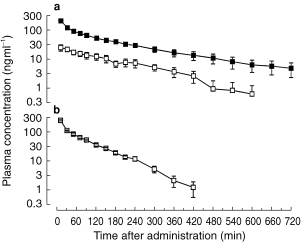
Semi-logarithmic plot of the mean concentration-time curves of midazolam (▪) and α-hydroxy-midazolam (□) after intravenous administration of midazolam (a) or α-hydroxy-midazolam (b) (0.15 mg kg−1 over 4 min) in six healthy volunteers.
Table 1.
Mean±s.e. mean maximum plasma concentration, volume of distribution, elimination half-life, clearance and urinary recovery in six healthy volunteers after intravenous administration of 0.15 mg kg−1 of midazolam or α-hydroxy-midazolam.
Respiratory effects
In accordance with earlier studies [19, 20], there were transient decreases in oxygen saturation immediately after midazolam administration (before midazolam: 96.1%±0.3, after midazolam: 92.2%±2; P < 0.05). There was no significant change in oxygen saturation after placebo administration (before placebo: 96.0%±0.4, after placebo: 95.7±0.8; P = 0.9). However, compared with placebo the changes after midazolam and after α-hydroxy-midazolam did not reach statistical significance (P = 0.08) and also changes in respiratory rate were not different.
EEG
In all subjects, increases in EEG beta and delta activity and a decrease in alpha activity were found after administration of the benzodiazepines. No consistent changes in theta activity were observed. Analysis of the whole beta range and of a smaller frequency band revealed that the changes in beta activity in the range from 13.5 to 15.5 Hz were much more pronounced than in the range from 13 to 30 Hz. Relationships between plasma benzodiazepine concentration and EEG beta activity are shown in Figure 2. The two curves for midazolam and α-hydroxy-midazolam were statistically not different.
Figure 2.
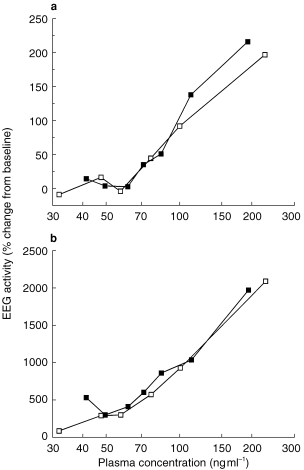
(a) Relationship between plasma concentration and percentage change in EEG beta activity (13–30 Hz) after intravenous administration of midazolam (▪) and α-hydroxy-midazolam (□), respectively. Data are expressed as means of six healthy volunteers. (b) Relationship between plasma concentration and percentage change in EEG activity in the frequency range from 13.5 to 15.5 Hz after intravenous administration of midazolam (▪) and α-hydroxy-midazolam (□), respectively. Data are expressed as means of six healthy volunteers. Changes in the lower beta range (13.5–15.5 Hz) were much more pronounced compared with the total change in the range from 13 to 30 Hz.
SLP
Latency and amplitude of wave I did not show any significant change under either benzodiazepine. At the time of benzodiazepine peak concentrations (10 min) wave V latency was increased from 5.81±0.07 ms to 5.97±0.07 after midazolam and from 5.84±0.09 to 5.87±0.11 after α-hydroxy-midazolam (P < 0.05 compared with placebo). No significant amplitude changes of wave V were observed.
MLP
Whereas CAR amplitudes in the placebo group remained essentially unchanged, both midazolam and α-hydroxy-midazolam induced a marked decrease in CAR amplitudes for 240 min with slow recovery over the following 360 min to normal values (Figure 3). The benzodiazepine effect-time curves were significantly different from the placebo curves (midazolam: P < 0.003, α-hydroxy-midazolam: P < 0.008). Individual CAR waveforms of one volunteer are displayed in Figure 4 for the three different therapy modalities: placebo, midazolam and α-hydroxy-midazolam.
Figure 3.
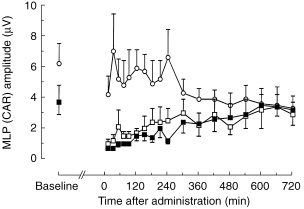
Averaged CAR middle latency changes over time after intravenous injection of 0.15 mg kg−1 midazolam (▪), α-hydroxy-midazolam (□), or placebo (○). Data are expressed as means of six healthy volunteers. The benzodiazepine time-effect curves were significantly different from the placebo curves (midazolam: P < 0.003, α-hydroxy-midazolam: P < 0.008; anova for repeated measures).
Figure 4.
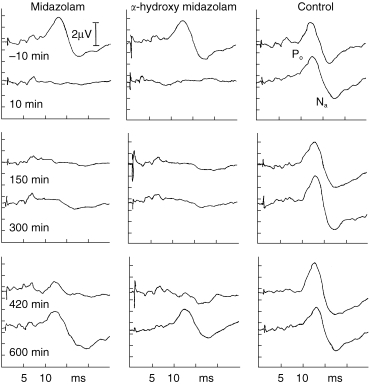
Individual MLP-waveforms (CAR) of one volunteer at different time points after systemic administration of placebo, α-hydroxy-midazolam (0.15 mg kg−1), or midazolam (0.15 mg kg−1), respectively.
TEOAE
The overall response level of TEOAE showed no statistically significant change after midazolam and α-hydroxy-midazolam treatment compared with the placebo curve. Analysis of the cochlear response according to three frequency ranges revealed no drug effect.
Pure tone audiogram
Pre-and post-test pure tone audiograms were similar and no temporary or permanent threshold shift was observed in any volunteer during or after the experiment.
Discussion
The primary aim of this study in healthy subjects was to evaluate the pharmacologic effect of midazolam and of its short-lived, more hydrophilic metabolite α-hydroxy-midazolam on different structures of the auditory pathway and to relate these to plasma benzodiazepine concentrations. As a control, we simultaneously assessed the effect on the EEG, which is well established.
Several previous studies describing the pharmacokinetics of α-hydroxy-midazolam reported ‘elimination half-lives’ of α-hydroxy-midazolam similar to those of the parent compound, i.e. values substantially higher than found in this study [21–23]. In all these earlier studies metabolite half-lives were calculated from the concentration-time course of the metabolite after administration of the parent compound. Under these conditions, however, the terminal plasma concentration-time course of the metabolite does not reflect the elimination rate of α-hydroxy-midazolam but is mainly determined by the rate of metabolite formation from midazolam. It is therefore impossible to appropriately estimate elimination rate and half-life of the metabolite without taking the metabolite formation rate into consideration, which seems to be rate-limiting in the case of midazolam.
EEG measurements after benzodiazepine administration revealed similar results as earlier reported [24]. Because benzodiazepine-induced EEG changes mainly occur in the lower beta range with maximum effects approximately at 15 Hz, we have evaluated the whole beta range and changes in its lowest segment (13.5–15.5 Hz) [1]. This analysis confirmed that changes in the lower beta range are much more pronounced than above 20 Hz and account for most of the drug-induced variation in total beta activity. Analysis of a much smaller frequency range revealed similar results for both compounds suggesting that monitoring of this small segment is sufficient to construct concentration-effect curves for benzodiazepines.
While the EEG reflects the spontaneous electrical activity, AEP are the average time-locked responses to external stimulation [25]. They are, thus, suitable to assess the functional integrity of afferent neuronal pathways from peripheral sensory organs to subcortical and cortical areas or muscles [26]. Whereas cochlear TEOAE responses remained unchanged, the functional brainstem areas of the auditory pathway were modified by low, non sedative benzodiazepine plasma concentrations. The increase in wave V latency early after benzodiazepine injection coincided with benzodiazepine peak concentrations and with a trend to oxygen desaturation, which suggests the involvement of deep vital brainstem structures including the colliculus inferior region as the putative generator of wave V. In previous reports, which all lack determination of benzodiazepine concentrations, possible benzodiazepine effects on SLP were denied [2, 27]. However, these previous studies are likely to have missed the transient effect of wave V prolongation because no measurements were obtained at the time of maximum concentrations. The fact that the short latency wave I, which reflects the basal generation pattern of the cochlea, was unchanged under both benzodiazepines is in accordance with preserved (cochlear) TEOAE responses. These findings suggest that the outer hair cells of the cochlea maintain their activity even under higher, sedative plasma benzodiazepine concentrations. Olivocochlear innervation has been postulated to regulate active mechanical processes in the mammalian cochlea over transmembranous GABA/benzodiazepine receptors. These inhibitory receptors prevail in the apical turn, which represents the lower hearing frequencies and produces the strongest TEOAE responses [28]. Our data suggest that the benzodiazepine doses did not access these putative intracochlear receptors because even in the most susceptible low frequency window no effect on TEOAE was observed. A tight blood–cochlea barrier may explain these diverging findings.
While myogenic MLP are supposedly generated in the upper brainstem, the origin of neurogenic MLP lies in the thalamus and the primary auditory cortex of the temporal lobe [29]. No marked benzodiazepine induced changes on neurogenic MLP were found by Schwender et al. [30]. Our study is the first to assess the effect of benzodiazepines on myogenic MLP (CAR). While in normal sleep CAR is not altered [31], both curarization and local anaesthesia of the facial nerve may blunt it [8, 32]. Unlike curare and local anaesthetics, benzodiazepines have neither a direct effect on the motor endplate nor on the peripheral nerve. Because benzodiazepine induced muscle relaxation is caused by inhibition of polysynaptic reflexes [33], a polysynaptic reflex mechanism may be postulated for the generation of CAR. Based on the short latency time of 10–12 ms, CAR has been considered a monosynaptic reflex [10]. Both benzodiazepines caused transient brainstem effects as assessed by changes of SLP and MLP. While the short-lasting SLP effect always concurred with sedation, the long-lasting effect on MLP persisted for hours after sedation. Hence, processing of auditory information by the cochleovestibular organ is clearly altered hours after the end of sedation at benzodiazepine plasma concentrations (<30 ng ml−1) which are not expected to cause clinically apparent effects [17]. Interestingly, no such effect is seen at the cortical level as evaluated by EEG.
At least two factors may explain the long persistence of MLP changes. The putative brainstem structures involved could be more sensitive to benzodiazepines than cortical structures responding already to lower plasma concentrations. The design of this study with rapid initial drug administration was not suitable to evaluate whether MLP changes already occur at lower concentrations or whether they may only be observed after administration of a sedative dose. To answer this question benzodiazepines should be given as an infusion over at least 30 min to allow repetitive measurements at low plasma benzodiazepine concentrations. Secondly, clearance of benzodiazepines from the site of action could be slower in the brainstem than in the cortex. However, if drug distribution would indeed be a limiting factor, one would also expect a considerable lag time between peak plasma concentration and maximum effect, which was not the case. In the future, once the procedure has reached the high resolution required for the evaluation of small brainstem structures, this question might be addressed using positron emission tomography (PET) [34]. Finally, although conceivable in principle, the short duration of action of midazolam, established in numerous time-response analyses including PET studies [34], argues against an agonist-induced modification of the benzodiazepine receptor as a cause for prolonged MLP effects.
In summary, these studies have shown that the function of several areas of the auditory pathway is modulated by single doses of systemically administered benzodiazepines. These effects were still observed when sedation and EEG effects had returned to baseline values. Stimulated effect parameters such as the easily measurable CAR, which has previously been shown to detect neurologically mute brainstem lesions [35, 36], may therefore be a more sensitive tool than EEG to monitor benzodiazepine effects. Whether it is also suited to quantify benzodiazepine effects after long-term administration (e.g. in intensive care or psychiatric patients) requires further investigation.
Acknowledgments
We are grateful for the excellent technical assistance of Helene Zehnder and the staff of the Medical Intensive Care Unit. Dr Haefeli was supported by the Schweizerische Stiftung für Medizinisch-Biologische Stipendien and by the Professor Dr Max Cloëtta Stiftung.
References
- 1.Breimer LTM, Hennis PJ, Burm AGL, et al. Quantification of the EEG effect of midazolam by aperiodic analysis in volunteers. Pharmacokinetic/pharmacodynamic modelling. Clin Pharmacokinet. 1990;18:245–253. doi: 10.2165/00003088-199018030-00006. [DOI] [PubMed] [Google Scholar]
- 2.Loughnan BL, Sebel PS, Thomas D, Rutherfoord CF, Rogers H. Evoked potentials following diazepam or fentanyl. Anaesthesia. 1987;42:195–198. doi: 10.1111/j.1365-2044.1987.tb02999.x. [DOI] [PubMed] [Google Scholar]
- 3.Thürauf N, Ditterich W, Kobal G. Different sensitivity of pain-related chemosensory potentials evoked by stimulation with CO2 tooth pulp event-related potentials and acoustic event-related potentials to the tranquilizer diazepam. Br J Clin Pharmacol. 1994;38:545–555. doi: 10.1111/j.1365-2125.1994.tb04396.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Milligan KR, Lumsden J, Howard RC, Howe JP, Dundee JW. Use of auditory evoked responses as a measure of recovery from benzodiazepine sedation. J Royal Soc Med. 1989;82:595–597. doi: 10.1177/014107688908201009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lehnhardt E. Praxis der Audiometrie. 7 Auflage, Stuttgart-New York: Thieme; 1997. pp. 251–313. [Google Scholar]
- 6.Probst R, Lonsbury-Martin BL, Martin GK. A review of otoacoustic emissions. J Acoust Soc Am. 1991;89:2027–2067. doi: 10.1121/1.400897. [DOI] [PubMed] [Google Scholar]
- 7.Moller AR, Janetta PJ. Neural generators of the auditory brainstem response. In: Jacobson JT, editor. The Auditory Brainstem Response. London: Taylor & Francis Ltd; 1985. pp. 13–31. [Google Scholar]
- 8.Cody DTR, Jacobson Jl, Walker JL, Bickford RG. Averaged evoked myogenic and cortical potentials to sound in man. Ann Otol Rhinol Laryngol. 1964;73:763–777. doi: 10.1177/000348946407300315. [DOI] [PubMed] [Google Scholar]
- 9.Gibson WPR. Essentials of clinical electric response audiometry. Edinburg, London New York: Churchill Livingstone; 1978. pp. 132–156. [Google Scholar]
- 10.Stöhr M, Dichgans J, Diener HC, Buetner UW. Evozierte Potentiale. Berlin, Heidelberg New York: Springer Verlag; 1982. pp. 371–376. [Google Scholar]
- 11.Ziegler WH, Shalch E, Leishman B, Eckert M. Comparison of the effects of intravenously administered midazolam, triazolam and their hydroxy metabolites. Br J Clin Pharmacol. 1983;16:63S–69S. doi: 10.1111/j.1365-2125.1983.tb02272.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ha HR, Rentsch KM, Kneer J, Vonderschmitt DJ. Determination of midazolam and its α-hydroxy metabolite in human plasma and urine by high-performance liquid chromatography. Ther Drug Monit. 1993;15:338–343. doi: 10.1097/00007691-199308000-00013. [DOI] [PubMed] [Google Scholar]
- 13.Gibaldi M, Perrier D. Pharmacokinetics. 2. Vol. 15. New York: Dekker; 1982. pp. 199–219.pp. 319–353.pp. 445–449. [Google Scholar]
- 14.Hotz MA, Allum JHJ, Kaufmann G, Follath F, Pfaltz CR. Shifts in auditory brainstem reponse latencies following plasma level controlled aminoglycoside therapy. Eur Arch Otorhinolaryngol. 1990;247:202–205. doi: 10.1007/BF00178984. [DOI] [PubMed] [Google Scholar]
- 15.Mendel MI. Influence of stimulus level and sleep stage on the early components of the averaged electroencephalic response to clicks during all night sleep. J Speech Res. 1974;17:5–17. doi: 10.1044/jshr.1701.05. [DOI] [PubMed] [Google Scholar]
- 16.Kemp DT. Stimulated acoustic emissionsfrom within the human auditory system. J Acoust Soc Am. 1978;64:1386–1391. doi: 10.1121/1.382104. [DOI] [PubMed] [Google Scholar]
- 17.Persson MP, Nilsson A, Hartvig P. Relation of sedation and amnesia to plasma concentrations of midazolam in surgical patients. Clin Pharmacol Ther. 1988;43:324–331. doi: 10.1038/clpt.1988.39. [DOI] [PubMed] [Google Scholar]
- 18.Michalk S, Moncorge C, Fichelle A, et al. Midazolam infusion for basal sedation in intensive care: absence of accumulation. Intens Care Med. 1988;15:37–41. doi: 10.1007/BF00255634. [DOI] [PubMed] [Google Scholar]
- 19.Bell GD, Antrobus JHL, Lee J, Coady T, Morden A. Bolus or slow titrated injection of midazolam prior to upper gastrointestinal endoscopy? Relative effect on oxygen saturation and prophylactic value of supplemental oxygen. Aliment Pharmacol Ther. 1990;4:393–401. doi: 10.1111/j.1365-2036.1990.tb00485.x. [DOI] [PubMed] [Google Scholar]
- 20.Bell GD, Reeve PA, Moshiri M, et al. Intravenous midazolam: a study of the degree of oxygen desaturation occurring during upper gastrointestinal endoscopy. Br J Clin Pharmacol. 1987;23:703–708. doi: 10.1111/j.1365-2125.1987.tb03104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bornemann LD, Min BH, Crews T, et al. Dose dependent pharmacokinetics of midazolam. Eur J Clin Pharmacol. 1985;29:91–95. doi: 10.1007/BF00547375. [DOI] [PubMed] [Google Scholar]
- 22.Heizmann P, Eckert M, Ziegler WH. Pharmacokinetics and bioavailability of midazolam in man. Br J Clin Pharmacol. 1983;16:43S–49S. doi: 10.1111/j.1365-2125.1983.tb02270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vree TB, Baars AM, Booij LHD, Driessen JJ. Simultaneous determination and pharmacokinetics of midazolam and its hydroxymetabolites in plasma and urine of man and dog by means of high-performance liquid chromatography. Arzneimittelforschung/Drug Res. 1981;31:2215–2219. [PubMed] [Google Scholar]
- 24.Mandema J, Tuk B, van Stenveninck AL, Breimer DD, Cohen AF, Danhof M. Pharmacokinetic-pharmacodynamic modeling of the central nervous system effects of midazolam and its main metabolite α-hydroxymidazolam in healthy volunteers. Clin Pharmacol Ther. 1992;51:715–728. doi: 10.1038/clpt.1992.84. [DOI] [PubMed] [Google Scholar]
- 25.Jones JG, Konieczko E. Hearing and memory in anaesthetised patients. Br Med J. 1986;292:1291–1293. doi: 10.1136/bmj.292.6531.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kochs E. Schulte am Esch J. Neurophysiologisches Monitoring und Benzodiazepinwirkung. Anästh Intensivther Notfallmed. 1988;23:145–152. [PubMed] [Google Scholar]
- 27.Schulte am Esch J, Koch SE. Midazolam and flumazenil in neuroanaesthesia. Acta Anaesthesiol Scand. 1990;34:96–102. doi: 10.1111/j.1399-6576.1990.tb03194.x. [DOI] [PubMed] [Google Scholar]
- 28.Plinkert PK, Möhler H, Zenner HP. A subpopulation of outer hair cells possessing GABA receptors with tonotopic organization. Arch Otorhinolaryngol. 1989;246:417–422. doi: 10.1007/BF00464301. [DOI] [PubMed] [Google Scholar]
- 29.Picton TW, Hillyard SA, Krausz HI, Galambos R. Human evoked potentials. I. Evaluation of components. Electroenceph Clin Neurophysiol. 1974;36:179–190. doi: 10.1016/0013-4694(74)90155-2. [DOI] [PubMed] [Google Scholar]
- 30.Schwender D, Klasing S, Madler C, Poppel E, Peter K. Effects of benzodiazepines on midlatency auditory evoked potentials. Can J Anaesth. 1993;40:1148–1154. doi: 10.1007/BF03009604. [DOI] [PubMed] [Google Scholar]
- 31.Mendel MI, Goldstein R. Early component of the averaged electro-encephalic response to constant clicks during all-night sleep. J Speech Res. 1971;14:829–840. doi: 10.1044/jshr.1404.829. [DOI] [PubMed] [Google Scholar]
- 32.Graham JM, Hurron JNF, Beagley HA. Postauricular myogenic response during traces tympanic electrocochleotrophy. Br J Audiol. 1975;9:116–127. [Google Scholar]
- 33.Hobbs WR, Rall TW, Verdoorn TA. Hypnotics and sedatives; ethanol. In: Hardman JG, Goodman Gilman A, Limbird LE, editors. The Pharmacological Basis of Therapeutics. 9. New York: MacGraw-Hill; 1996. pp. 365–366. [Google Scholar]
- 34.Malizia AL, Gunn RN, Wilson SJ, et al. Benzodiazepine site pharmacokinetic/pharmacodynamic quantification in man: direct measurement of drug occupancy and effects on the human brain in vivo. Neuropharmacol. 1996;35:1483–1491. doi: 10.1016/s0028-3908(96)00072-x. [DOI] [PubMed] [Google Scholar]
- 35.Douek EE, Ashcroft PB, Humphries KN. The clinical value of the postauricular myogenic crossed acoustic responses in neuro-otology. In: Stephens SDG, editor. Disorders of Auditory Function, II. London: Academic Press; 1976. pp. 139–143. [Google Scholar]
- 36.Clifford-Jones RE, Clarke GP, Mayles P. Crossed acoustic response combined with visual and somatosensory evoked responses in the diagnosis of multiple sclerosis. J Neurol Neurosurg Psychiatry. 1979;42:749–752. doi: 10.1136/jnnp.42.8.749. [DOI] [PMC free article] [PubMed] [Google Scholar]