

Abstract
Aims
The aim of this open-label, placebo-controlled, randomized, four-period crossover study was to determine the effects of cimetidine and ranitidine on the pharmacokinetics and pharmacodynamics of a single dose of dofetilide.
Methods
Twenty healthy male subjects received 100 or 400 mg twice daily of cimetidine, 150 mg twice daily of ranitidine, or placebo for 4 days. On the second day, a single oral 500 μg dose of dofetilide was administered immediately after the morning doses of cimetidine, ranitidine, or placebo. Treatment periods were separated by 1–2 weeks. Pharmacokinetic parameters were determined from plasma and urinary dofetilide concentrations; prolongation of the QTc interval was determined from three-lead electrocardiograms.
Results
Ranitidine did not significantly affect the pharmacokinetics or pharmacodynamics of dofetilide; however, a dose-dependent increase in exposure to dofetilide was observed with cimetidine. When dofetilide was administered with 100 and 400 mg of cimetidine, the area under the plasma concentration-time curve of dofetilide increased by 11% and 48% and the maximum plasma dofetilide concentration increased by 11% and 29%, respectively. The respective cimetidine doses reduced renal clearance of dofetilide by 13% and 33% and nonrenal clearance by 5% and 21%. Dofetilide-induced prolongation of the QTc interval was enhanced by cimetidine; the mean maximum change in QTc interval from baseline was increased by 22% and 33% with 100 and 400 mg of cimetidine, respectively. However, the relationship between the prolongation of the QTc interval and plasma dofetilide concentrations was unaffected by cimetidine or ranitidine; a 1 ng ml−1 increase in plasma dofetilide concentration produced a 17–19 ms prolongation of the QTc interval. Dofetilide was well tolerated, with no treatment-related adverse events or laboratory abnormalities.
Conclusions
These results suggest that cimetidine increased dofetilide exposure by inhibiting renal tubular dofetilide secretion, whereas ranitidine did not. This effect is not an H2-receptor antagonist class effect but is specific to cimetidine. If therapy with an H2-receptor antagonist is required, it is recommended that cimetidine at all doses be avoided; since ranitidine has no effect on dofetilide pharmacokinetics or prolongation of the QTc interval, it can be seen as a suitable alternative.
Keywords: antiarrhythmic drug, cimetidine, dofetilide, pharmacodynamics, pharmacokinetics, ranitidine
Introduction
Dofetilide is a potent and effective class III antiarrhythmic agent that selectively inhibits the rapid component of the cardiac delayed-rectifier potassium current (IKr) without affecting the fast inward sodium current [1, 2]. As a result, dofetilide increases the effective atrial and ventricular refractory period by prolonging action potential duration; however, it does not affect conduction [3–5].
Dofetilide has demonstrated efficacy in the treatment of supraventricular reentrant tachydysrhythmias in well-controlled clinical trials. Intravenous dofetilide converted both sustained atrial flutter and atrial fibrillation (AF) into normal sinus rhythm and was especially effective against atrial flutter [6–8]. Oral dofetilide is effective in the maintenance of sinus rhythm after cardioversion from AF [9].
Pharmacokinetic and pharmacodynamic modelling indicate that the ability of dofetilide to prolong the QTc interval is related linearly to plasma dofetilide concentration [10]. Following oral administration, dofetilide is well absorbed, with systemic bioavailability in excess of 90% [10, 11]. Maximum plasma concentrations (Cmax) are achieved within 2.5–3.5 h, and the elimination half-life (t1/2) from plasma is 7–9 h [10, 11]. Dofetilide undergoes both renal and hepatic clearance, with renal excretion representing 50% to 83% of total elimination (Pfizer Inc, data on file) [11, 12]. Renal clearance of dofetilide appears to take place through both active renal tubular secretion and passive glomerular filtration [12].
Cimetidine and ranitidine are H2-receptor antagonists that are used widely in the treatment of gastrointestinal disorders caused by oversecretion of gastric acid. These agents may affect drug absorption by increasing gastric pH, or they may impair hepatic or renal drug clearance by one of several mechanisms, including altered cytochrome P450 hepatic drug metabolism, reduced hepatic blood flow, and lowered renal tubular secretion [13, 14]. In terms of renal drug clearance, cimetidine and ranitidine undergo proximal tubular secretion via the cationic transport system and may compete with other drugs that are also cleared by this process [15, 16]. As a potent inhibitor of the renal secretion of several organic cations and as a nonspecific inhibitor of the cytochrome P450 oxidase system, cimetidine has the potential to interact with dofetilide elimination by these two mechanisms. Ranitidine competes with cimetidine for active tubular secretion and may interact with dofetilide in a similar manner. However, compared with cimetidine, ranitidine has a greatly reduced effect on the cytochrome P450 system [17]. Cimetidine has also been shown to significantly alter both the pharmacokinetics and pharmacodynamics of other antiarrhythmic agents, but its interaction with dofetilide remains unknown [13, 18–20].
The present study was designed to ascertain whether cimetidine and ranitidine affect the pharmacokinetics and pharmacodynamics of a single dose of dofetilide in healthy male subjects.
Methods
Study design
The study was conducted at the Pfizer Clinical Research Unit, Canterbury, and was approved by the Research Ethics Committee, Kent and Canterbury Hospital. After providing written informed consent, subjects entered the clinic on the evening before the first dose of study medication and received a full physical examination. Subjects were to receive twice-daily treatment with 100 mg cimetidine, 400 mg cimetidine, 150 mg ranitidine, or placebo for 4 days (days 1–4) in a four-way crossover design and in random order. The two doses of cimetidine were selected to evaluate an over-the-counter dose (100 mg) and a prescription dose (400 mg); the ranitidine dose corresponded to a prescription dose. Subjects fasted overnight and until 4 h after dosing on days 1–3 but were allowed to drink water. On day 2, a single 500 μg capsule of dofetilide was administered immediately after the morning dose of cimetidine, ranitidine, or placebo. Subjects were required to remain in the clinic until 48 h after this dose of dofetilide. Serial blood samples and 12-lead electrocardiograms (ECG) were obtained during these 48 h. Subjects were then permitted to leave the clinic. The cimetidine and ranitidine doses given on days 3 and 4 were intended to maximize their potential interaction with dofetilide. All drug doses were administered at the clinic except for the last dose of cimetidine, ranitidine, and placebo, which was taken at home. Each treatment period began at successive 2-week intervals, leaving a 10-day washout period between treatments.
All observed or volunteered adverse events that occurred within 7 days of dosing were coded by the investigator's assessment of their relationship to treatment. Hematologic studies, clinical chemistry, and urinalysis were performed during each treatment period before dosing and before subjects were discharged from the clinic and again 2–3 weeks after the last dofetilide dose. Safety was also assessed by continuous ambulatory electrocardiogram (ECG) measurement, starting 30 min before dofetilide dosing and continuing for 8 h after, and by 12-lead ECGs and vital sign measurements obtained before dosing on days 1 and 2 and at 2, 4, 8, 24, and 48 h after. A follow-up examination, including a physical examination, laboratory tests, and a 12-lead ECG, was done 2–3 weeks after completion of the study. The study was monitored in accordance with Standard Operating Procedures and Good Clinical Practice.
Subjects
Twenty healthy male Caucasian subjects between 19 and 42 years of age (mean, 29 years), weighing 59–89 kg (mean, 79 kg) participated in this open-label, placebo-controlled, randomized, four-way crossover study. All subjects satisfied the following inclusion and exclusion criteria: within 10% of their ideal weight for their height and frame, supine heart rate between 45 and 100 beats min−1, supine blood pressure between 90/60 and 160/95 mmHg, and calculated creatinine clearance (CLCr)>60 ml min−1 (method of Cockcroft & Gault) [21]. Subjects with any clinically significant disease or laboratory abnormality, a family history of sudden death before the age of 40 years, or a history of cardiac dysrhythmias or fainting were excluded. Subjects who smoked more than five cigarettes per day, had any clinically significant abnormality in resting ECG or 24 h ambulatory ECG, or had a QTc interval >420 ms were also ineligible. Subjects were not allowed to take any prescription or over-the-counter drug, with the exception of paracetamol, for 2 weeks before the study. All 20 subjects completed the study.
Measurements
Pharmacokinetics
On day 2 of each treatment period, blood samples were collected in heparinized tubes before dofetilide administration and at 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 16, 24, 36, 48, and 72 h after dosing. Samples were centrifuged within 60 min of collection, and the plasma was stored frozen (−20° C) in polypropylene tubes until assayed. Urine samples were collected for the 12 h period before dofetilide administration and for each 12 h period after dosing up to 48 h. The total volume of each 12 h urine sample was measured, and a 20 ml aliquot was removed and stored frozen (−20° C) until assayed. Plasma dofetilide concentrations were measured by a validated automated double-antibody radioimmunoassay with a lower limit of quantification of 0.05 ng ml−1 [22]. Urinary dofetilide concentrations were measured by a validated reversed-phase high-performance liquid chromatography procedure that was calibrated over the range of 5–300 ng ml−1 [23]. Cimetidine and ranitidine were shown not to interfere with the dofetilide measurements.
Pharmacodynamics
Duplicate three-lead ECGs were performed on day 1 of each treatment period before cimetidine, ranitidine, and placebo administration, and at 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, and 16 h after dosing. Duplicate three-lead ECGs were performed at these same times on day 2 before and after dofetilide administration and at 24, 36, 48, and 72 h after dosing. The QTc interval was calculated from the ECG recordings using Bazett's formula, QTc=QT (1/RR)1/2, where RR is the interval in seconds between the two preceding R waves.
Data analysis
Pharmacokinetics
The Cmax and the time of its occurrence (tmax) were identified from measured dofetilide concentrations. The area under the plasma concentration-time curve over 48 h (AUC(0,48h)) was calculated using the linear trapezoidal rule. The apparent terminal phase rate constant (λz) was calculated using log-linear regression analysis for all subjects. In some cases, the λz could not be determined because the plasma concentration profile did not adequately define the terminal phase. The AUC between the final measurable plasma concentration (Ct) and infinity was calculated from Ct/λz and was added to AUC(0,48h) to calculate AUC.
The total clearance (CL/F) of dofetilide was calculated from the ratio of dose to AUC(0,48h). AUC(0,48h) was used instead of AUC, as several AUC values were missing because of the inability to evaluate λz in some subjects. Since the difference between AUC(0,48h) and AUC is minimal for dofetilide, this difference was not deemed to affect significantly the conclusions of the study. Renal clearance (CLr) of dofetilide was calculated from the ratio of cumulative urinary dofetilide excreted during 48 h to the AUC(0,48h), whereas nonrenal clearance (CLnr/F) was calculated from the difference between CL/F and CLr.
The Cmax and AUC(0,48h) (both log-transformed), CLr, CLnr/F, and λz were analysed by an analysis of variance with terms for sequence, subject (within sequence), period, treatment, and carryover using SAS (SAS Institute Inc.). The results were summarized by mean differences and ratios between means, with 90% confidence intervals used to indicate statistical significance for the ratios between means.
Pharmacodynamics
The mean maximum change in QTc interval from baseline to 12 h after dosing with dofetilide and the area under the change in QTc-time curve (AUEC(0,12h)) from baseline QTc interval to 12 h after dosing were summarized by means (±s.d.) and subjected to an analysis of variance with terms for sequence, subject (within sequence), period, treatment, and carryover using SAS (SAS Institute Inc.).
For each subject, QTc interval at each time point was plotted against the corresponding plasma dofetilide concentration; the slope of this QTc-plasma concentration curve was determined by linear regression. In some cases, the plot exhibited evidence of an anticlockwise hysteresis, as shown in Figure 1. In such cases, the plasma data were modelled using a two-compartment model with first-order absorption, and then an effect compartment model was used to relate the pharmacokinetic and pharmacodynamic data. The slope of the QTc-effect compartment concentration was then determined by linear regression.
Figure 1.
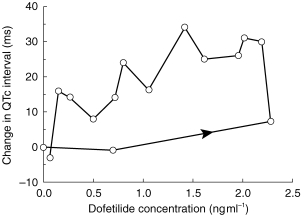
Change in QTc interval vs plasma dofetilide concentration obtained during treatment with cimetidine (100 mg twice daily) in a representative patient showing a hysteresis.
Results
Pharmacokinetics
Figure 2 shows mean plasma concentrations of dofetilide when 500 μg dofetilide was administered with 100 mg cimetidine, 400 mg cimetidine, 150 mg ranitidine, or placebo. Higher plasma concentrations of dofetilide were achieved and maintained when the drug was administered with 400 mg cimetidine than with placebo or the other drugs.
Figure 2.
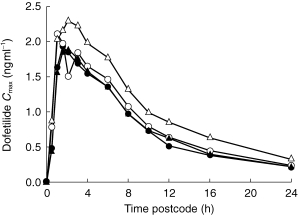
Mean plasma concentrations of dofetilide after single 500 μg dose administered with twice-daily placebo (•), 150 mg of ranitidine (▴), 100 mg cimetidine (○), and 400 mg cimetidine (▵).
Pharmacokinetic measurements were obtained for each subject; the mean value for each parameter is presented in Table 1. No statistically or clinically significant changes in any of the dofetilide pharmacokinetic parameters (Cmax, tmax, CL/F, CLr, and CLnr/F) occurred after treatment with ranitidine compared with placebo. However, significant changes in several parameters occurred with cimetidine.
Table 1.
Mean (±s.d.) pharmacokinetic parameters of dofetilide after administration of a single 500 μg oral dose with cimetidine, ranitidine, and placebo.
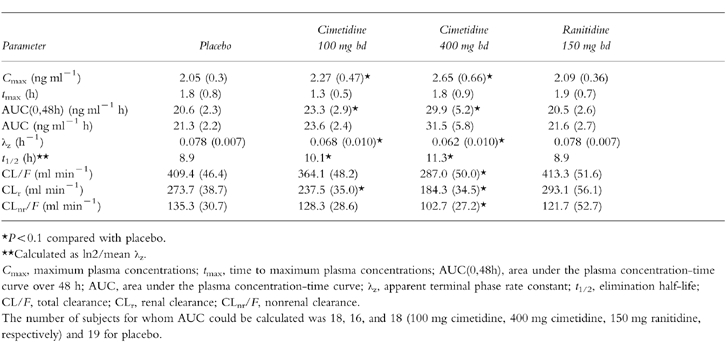
The ratio of mean Cmax after 100 mg cimetidine to mean Cmax after placebo was 109.6% (90% confidence intervals, 101.8%, 118.0%), and the ratio of the mean AUC(0,48h) values was 112.7% (108.7%, 116.8%). The ratio of the mean Cmax after 400 mg of cimetidine to mean Cmax after placebo was 126.8% (117.8%, 136.6%), and the ratio of mean AUC(0,48h) values was 143.9% (138.8%, 149.2%). These changes were statistically significant. A plot of mean AUC(0,48h), an indicator of total exposure to dofetilide, as a function of the respective doses of cimetidine (including a zero dose during placebo) shows a linear relationship between cimetidine dose and AUC(0,48h) (Figure 3).
Figure 3.
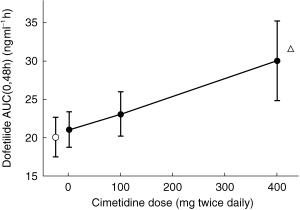
Effects (mean±s.d.) of cimetidine and ranitidine on dofetilide exposure (AUC(0,48h)) after a single 500 μg dose. The relationship between AUC(0,48h) and cimetidine dose was linear (•), with the 0 mg data point representing the AUCt during the placebo period. ○ = AUC(0,48h) during ranitidine. ▵ = AUC(0,48h) predicted for a 750 μg dose of dofetilide alone.
The CLr of dofetilide administered with placebo, measured directly from urinary concentrations, was 273.7 ml min−1. The CLr of dofetilide was reduced after both doses of cimetidine compared with placebo (Table 1, Figure 4), with a ratio of means of 87.7% (82.7%, 92.8%) for 100 mg cimetidine and 65.6% (60.7%, 70.6%) for 400 mg cimetidine. Ranitidine compared with placebo did not reduce CLr as indicated by a ratio of means of 106.7% (101.7%, 111.7%).
Figure 4.
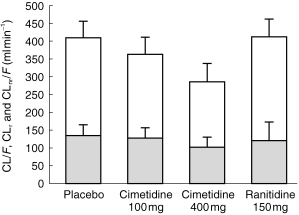
Effects (mean±s.d.) of placebo, cimetidine, and ranitidine on dofetilide CLr, CLnr/F, and CL/F. Non-renal clearance, CLnr/F, is represented by the shaded area. Renal clearance, CLr, is represented by the unshaded area. Total clearance, CL/F, is represented by the total height of the bar for each treatment, the sum of CLnr/F and CLr.
The mean CL/F of dofetilide was 409.4 ml min−1 during the placebo period and was not significantly affected by either 100 mg cimetidine or 150 mg ranitidine (Table 1, Figure 4). However, 400 mg cimetidine compared with placebo lowered CL/F.
The mean CLnr/F of dofetilide was 135.3 ml min−1 during the placebo period and was not significantly affected by 100 mg cimetidine but was significantly reduced with 400 mg cimetidine (Table 1, Figure 4). The ratio of mean CLnr/F after 400 mg of cimetidine compared with placebo was 75.6% (65.8%, 85.4%).
The mean λz of dofetilide was 0.078 h−1 during the placebo period, corresponding to a mean t1/2 of 8.9 h. Ranitidine did not affect the λz of dofetilide, whereas cimetidine significantly reduced λz, with a consequent increase in t1/2. The ratios of mean λz with 100 mg and 400 mg cimetidine compared with placebo were 86.9% (82.0%, 91.7%) and 81.2% (76.0%, 86.3%), respectively.
Pharmacodynamics
On day 1, the mean baseline QTc interval for each treatment group ranged from 366 to 374 ms. The mean maximum changes from baseline QTc interval following administration of the first dose of cimetidine, ranitidine, or placebo (all in the absence of dofetilide) ranged from 10.3 to 15.2 ms (Table 2). Further, the mean AUEC(0,12h) on day 1 did not change significantly following administration of cimetidine, ranitidine, and placebo and ranged from −33.0±175.8 to −80.9±214.0. Thus, there was no evidence that cimetidine or ranitidine affects QTc interval.
Table 2.
Mean (±s.d.) maximum changes from baseline QTc interval after a single oral dose of 500 μg of dofetilide.
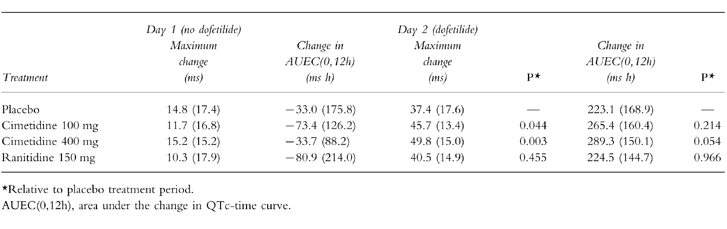
In comparison, dofetilide prolonged the QTc interval during each treatment period (Table 2). On day 2 before dofetilide administration, the mean baseline QTc interval for each treatment group ranged from 361 to 364 ms. Following dofetilide administration, the mean maximum changes from baseline QTc interval were 37.4 ms (placebo), 45.7 ms (cimetidine 100 mg), 49.8 ms (cimetidine 400 mg), and 40.5 ms (ranitidine). Ranitidine did not significantly affect the action of dofetilide on QTc interval; however, both doses of cimetidine significantly increased the mean maximum dofetilide-induced prolongation of the QTc interval. Whereas the mean AUEC(0,12h) was increased by cimetidine, only the 400 mg dose approached statistical significance (P = 0.054).
Pharmacokinetic–pharmacodynamic relationship
Prolongation of the QTc interval was related to the plasma dofetilide concentration. Neither cimetidine nor ranitidine altered the sensitivity of QTc prolongation to plasma dofetilide concentration, as judged by comparison of the slopes of this relationship. Figure 5 shows a representative example of a subject with a linear pharmacokinetic-pharmacodynamic relationship.
Figure 5.
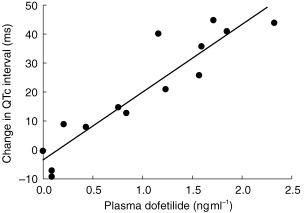
Pharmacokinetic-pharmacodynamic relationship of dofetilide: Change in QTc interval vs plasma dofetilide concentration obtained during treatment with placebo in a representative subject. Slope = 23.3; r2 = 0.86 (determined from linear regression).
The slope of change in QTc interval from baseline vs plasma dofetilide was determined from 16 subjects whose data were suitable for analysis. There were no statistical differences between the mean (±s.d.) slopes for placebo (18.5±3.8 ms ng−1 ml−1), 100 mg of cimetidine (18.5± 4.42 ms ng−1 ml−1), 400 mg of cimetidine (19.3±4.9 ms ng−1 ml−1), and ranitidine (17.1±4.5 ms ng−1 ml−1).
Adverse events
In this study, the incidence of adverse events was similar when cimetidine or ranitidine were administered alone or in combination with dofetilide. There were no discontinuations from treatment, and none of the adverse events was considered by the investigator to be treatment related. There were no clinically significant electrophysiologic events, and the QTc interval did not exceed 450 ms in any individual. There were no treatment-related laboratory abnormalities.
Discussion
The results of this study demonstrate that cimetidine increases exposure to single doses of dofetilide by reducing the renal tubular secretion of dofetilide. Further, the effect of cimetidine does not represent a class effect of H2-receptor antagonists, because ranitidine did not significantly affect either the pharmacokinetics or pharmacodynamics of dofetilide.
Pharmacokinetics
The 400 mg dose of cimetidine produced a significant reduction in CL/F of dofetilide because of a significant decrease in the CLr of dofetilide and a modest reduction in CLnr, resulting in increased exposure to dofetilide. The mean Cmax also increased. No clinically or statistically significant differences in CLr or CLnr/F were observed with 150 mg ranitidine compared with placebo.
In the present study, CLnr/F was calculated as the difference between two measurements and was therefore subject to the added variability of each measurement. During administration of placebo, CLnr/F accounted for 30% of CL/F, which is consistent with previous reports [11, 12]. Cimetidine may affect hepatic clearance by inhibiting cytochrome P450-mediated metabolism, most notably the pathways that involve N-dealkylation, aromatic hydroxylation, and sulphoxidation [14]. Although N-dealkylation and N-oxidation represent only a small component of dofetilide metabolism, with only low levels of inactive metabolites detected in plasma [12], the reduction of CLnr/F by 400 mg of cimetidine may reflect an inhibitory effect on the P450-mediated dofetilide metabolism. This is consistent with the report that cimetidine increased exposure to flecainide (another antiarrhythmic drug) by 28%, without affecting CLr but by decreasing the biotransformation of flecainide [24].
Pharmacodynamics
The effect of cimetidine on dofetilide pharmacokinetics was mirrored in the prolongation of the QTc interval. Ranitidine did not affect the pharmacokinetics, and similarly it did not affect the maximum change in QTc interval from baseline or the AUEC(0,12h).The 30% increase in Cmax produced by 400 mg cimetidine was associated with a 33% increase in the mean maximum change in QTc interval from baseline. The 45% increase in AUC(0,48h) produced by 400 mg cimetidine was associated with an increase in AUEC(0,12h) that approached statistical significance (P = 0.054).
Effects of cimetidine and ranitidine on pharmacokinetics and pharmacodynamics of other antiarrhythmic drugs
Cimetidine similarly alters the pharmacokinetics and pharmacodynamics of other antiarrhythmic agents. Cimetidine, 300 mg four times daily, has been shown to reduce the CL/F of quinidine by 37%, resulting in a 20% increase in Cmax and a 55% increase in the mean t1/2 [18]. While QTc interval was prolonged, the relationship between blood concentration and QTc interval remained unchanged [18]. Baciewicz & Baciewicz reported that the AUC of 1 g procainamide was increased by 35% with 1 g cimetidine, and the CLr was reduced by 42% [13]. This was attributed to the inhibition of the renal tubular secretion of procainamide by cimetidine [13, 20]. Cimetidine doses of 400 mg tid increased Cmax of the class IC antiarrhythmic drug propafenone by 24%, although there was wide intersubject variability. A statistically significant prolongation of the QRS duration resulted [19].
In the present study, ranitidine did not reduce the CLr of dofetilide. These results are consistent with its effects on the CLr of procainamide, as reported by Rodvold et al. [20] but in contrast to the findings of Somogyi & Bochner [25] with ranitidine and procainamide. They reported that 150 mg of ranitidine significantly reduced the CL/F and CLr of procainamide (measured for 12 h instead of 48 h, as in the present study) but without a significant increase in Cmax, tmax, or t1/2.
Pharmacokinetic-pharmacodynamic relationship
Cimetidine and ranitidine did not affect the pharmacokinetic-pharmacodynamic relationship between plasma dofetilide concentration and prolongation of the QTc interval, since the mean slope of the relationship was similar to that of placebo. The slopes seen in this study were between 18.5 and 19.3 ms ng−1 ml−1, which are similar to those obtained in normal subjects, subjects with renal impairment (Pfizer Inc, data on file), and subjects with ischaemic heart disease (Pfizer Inc, data on file). An increase of 1 ng ml−1 of dofetilide in plasma prolonged the QTc interval by 17–19 ms.
Conclusions
These results suggest that cimetidine increases dofetilide exposure by inhibiting renal tubular dofetilide secretion but ranitidine does not. This effect is not an H2-receptor inhibitor class effect but is specific to cimetidine. When cimetidine is needed in clinical practice, ranitidine may be substituted with no effect on dofetilide pharmacokinetics or pharmacodynamics.
References
- 1.Gwilt M, Blackburn KJ, Burges RA, Higgins AJ, Milne AA, Solca AM. Electropharmacology of dofetilide, a new class III agent, in anaesthetised dogs. Eur J Pharmacol. 1992;215:137–144. doi: 10.1016/0014-2999(92)90021-u. [DOI] [PubMed] [Google Scholar]
- 2.Carmeliet E. Voltage- and time-dependent block of the delayed K+ current in cardiac myocytes by dofetilide. J Pharmacol Exp Ther. 1992;262:809–817. [PubMed] [Google Scholar]
- 3.Bashir Y, Thomsen P-EB, Kingma JH, et al. Electrophysiologic profile and efficacy of intravenous dofetilide (UK-68,798), a new class III antiarrhythmic drug, in patients with sustained monomorphic ventricular tachycardia. Am J Cardiol. 1995;76:1040–1044. doi: 10.1016/s0002-9149(99)80293-8. [DOI] [PubMed] [Google Scholar]
- 4.Sedgwick ML, Rasmussen HS, Cobbe SM. Effects of the class III antiarrhythmic drug dofetilide on ventricular monophasic action potential duration and QT interval dispersion in stable angina pectoris. Am J Cardiol. 1992;70:1432–1437. doi: 10.1016/0002-9149(92)90295-a. [DOI] [PubMed] [Google Scholar]
- 5.Sedgwick ML, Dalrymple I, Rae AP, Cobbe SM. Effects of the new class III antiarrhythmic drug dofetilide on the atrial and ventricular intracardiac monophasic action potential in patients with angina pectoris. Eur Heart J. 1995;16:1641–1646. doi: 10.1093/oxfordjournals.eurheartj.a060790. [DOI] [PubMed] [Google Scholar]
- 6.Crijns HJGM, Van Gelder IC, Kingma JH, Dunselman PHJM, Gosselink ATM, Lie KI. Atrial flutter can be terminated by a class III antiarrhythmic drug but not by a class IC drug. Eur Heart J. 1994;15:1403–1408. doi: 10.1093/oxfordjournals.eurheartj.a060402. [DOI] [PubMed] [Google Scholar]
- 7.Falk RH, Pollak A, Singh SN, Friedrich T for the Intravenous Dofetilide Investigators. Intravenous dofetilide, a class III antiarrhythmic agent, for the termination of sustained atrial fibrillation or flutter. J Am Coll Cardiol. 1997;29:385–390. doi: 10.1016/s0735-1097(96)00506-2. [DOI] [PubMed] [Google Scholar]
- 8.Suttorp MJ, van Polak PE, ’T Hof A, Rasmussen HS, Dunselman PH, Kingma JH. Efficacy and safety of a new selective class III antiarrhythmic agent dofetilide in paroxysmal atrial fibrillation or atrial flutter. Am J Cardiol. 1992;69:417–419. doi: 10.1016/0002-9149(92)90247-v. [DOI] [PubMed] [Google Scholar]
- 9.Singh SN, Berk MR, Yellen LG, et al. Efficacy and safety of oral dofetilide in maintaining normal sinus in patients with atrial fibrillation/flutter: a multicenter study [abstract 2145] Circulation. 1997;96(8) Suppl:I-383. Abstract. [Google Scholar]
- 10.Le Coz F, Funck-Brentano C, Morell T, Ghadanfar MM, Jaillon P. Pharmacokinetic and pharmacodynamic modeling of the effects of oral and intravenous administrations of dofetilide on ventricular repolarization. Clin Pharmacol Ther. 1995;57:533–542. doi: 10.1016/0009-9236(95)90038-1. [DOI] [PubMed] [Google Scholar]
- 11.Tham TCK, MacLennan BA, Burke MT, Harron DWG. Pharmacodynamics and pharmacokinetics of the class III antiarrhythmic agent dofetilide (UK-68,798) in humans. J Cardiovasc Pharmacol. 1993;21:507–512. doi: 10.1097/00005344-199303000-00024. [DOI] [PubMed] [Google Scholar]
- 12.Smith DA, Rasmussen HS, Stopher DA, Walker DK. Pharmacokinetics and metabolism of dofetilide in mouse, rat, dog and man. Xenobiotica. 1992;22:709–719. doi: 10.3109/00498259209053133. [DOI] [PubMed] [Google Scholar]
- 13.Baciewicz AM, Baciewicz FA., Jr Effect of cimetidine and ranitidine on cardiovascular drugs. Am Heart J. 1989;118:144–154. doi: 10.1016/0002-8703(89)90085-9. [DOI] [PubMed] [Google Scholar]
- 14.Somogyi A, Muirhead M. Pharmacokinetic interactions of cimetidine 1987. Clin Pharmacokinet. 1987;12:321–366. doi: 10.2165/00003088-198712050-00002. [DOI] [PubMed] [Google Scholar]
- 15.van Crugten J, Bochner F, Keal J, Somogyi A. Selectivity of the cimetidine-induced alterations in the renal handling of organic substrates in humans. Studies with anionic, cationic and zwitterionic drugs. J Pharmacol Exp Ther. 1986;236:481–487. [PubMed] [Google Scholar]
- 16.McKinney TD, Speeg KV., Jr Cimetidine and procainamide secretion by proximal tubules in vitro. Am J Physiol. 1982;11:F672–F680. doi: 10.1152/ajprenal.1982.242.6.F672. [DOI] [PubMed] [Google Scholar]
- 17.Rendic S, Kajfez F, Ruf H-H. Characterization of cimetidine, ranitidine, and related structures interaction with cytochrome P-450. Drug Metab Dispos. 1983;11:137–142. [PubMed] [Google Scholar]
- 18.Hardy BG, Zador IT, Golden L, Lalka D, Schentag JJ. Effect of cimetidine on the pharmacokinetics and pharmacodynamics of quinidine. Am J Cardiol. 1983;52:172–175. doi: 10.1016/0002-9149(83)90091-7. [DOI] [PubMed] [Google Scholar]
- 19.Pritchett ELC, Smith WM, Kirsten EB. Pharmacokinetic and pharmacodynamic interactions of propafenone and cimetidine. J Clin Pharmacol. 1988;28:619–624. doi: 10.1002/j.1552-4604.1988.tb03185.x. [DOI] [PubMed] [Google Scholar]
- 20.Rodvold KA, Paloucek FP, Jung D, Gallastegui J. Interaction of steady-state procainamide with H2-receptor antagonists cimetidine and ranitidine. Ther Drug Monit. 1987;9:378–383. doi: 10.1097/00007691-198712000-00003. [DOI] [PubMed] [Google Scholar]
- 21.Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16:31–41. doi: 10.1159/000180580. [DOI] [PubMed] [Google Scholar]
- 22.Walker DK, Aherne GW, Arrowsmith JE, et al. Measurement of the class III antidysrhythmic drug, UK-68,798, in plasma by radioimmunoassay. J Pharm Biomed Anal. 1991;9:141–149. doi: 10.1016/0731-7085(91)80137-x. [DOI] [PubMed] [Google Scholar]
- 23.Walker DK, Smith DA, Stopher DA. Liquid–liquid extraction and high-performance liquid chromatography for the determination of a novel antidysrhythmic agent (UK-68,798) in human urine. J Chromatogr B Biomed Appl. 1991;568:475–480. doi: 10.1016/0378-4347(91)80186-g. [DOI] [PubMed] [Google Scholar]
- 24.Tjandra-Maga TB, Van Hecken A, Van Melle P, Verbesselt R, De Schepper PJ. Altered pharmacokinetics of oral flecainide by cimetidine. Br J Clin Pharmacol. 1986;22:108–110. [PMC free article] [PubMed] [Google Scholar]
- 25.Somogyi A, Bochner F. Dose and concentration dependent effect of ranitidine on procainamide disposition and renal clearance in man. J Clin Pharmacol. 1984;18:175–181. doi: 10.1111/j.1365-2125.1984.tb02450.x. [DOI] [PMC free article] [PubMed] [Google Scholar]