

Abstract
Aims
To predict the drug interactions of amiodarone and other drugs, the inhibitory effects and inactivation potential for human cytochrome P450 (CYP) enzymes by amiodarone and its N-dealkylated metabolite, desethylamiodarone were examined.
Methods
The inhibition or inactivation potency of amiodarone and desethylamiodarone for human CYP activities were investigated using microsomes from B-lymphoblastoid cell lines expressing CYP1A1, CYP1A2, CYP2A6, CYP2B6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4. The in vivo drug interactions of amiodarone and desethylamiodarone were predicted in vitro using the 1+Iu/Ki values.
Results
Amiodarone weakly inhibited CYP2C9, CYP2D6, and CYP3A4-mediated activities with Ki values of 45.1–271.6 μm. Desethylamiodarone competitively inhibited the catalytic activities of CYP2D6 (Ki = 4.5 μm) and noncompetitively inhibited CYP2A6 (Ki = 13.5 μm), CYP2B6 (Ki = 5.4 μm), and CYP3A4 (Ki = 12.1 μm). The catalytic activities of CYP1A1 (Ki = 1.5 μm, α = 5.7), CYP1A2 (Ki = 18.8 μm, α = 2.6), CYP2C9 (Ki = 2.3 μm, α = 5.9), and CYP2C19 (Ki = 15.7 μm, α = 4.5) were inhibited by desethylamiodarone with mixed type. The 1+Iu/Ki values of desethylamiodarone were higher than those of amiodarone. Amiodarone inactivated CYP3A4, while desethylamiodarone inactivated CYP1A1, CYP1A2, CYP2B6, and CYP2D6.
Conclusions
The interactions between amiodarone and other drugs might occur via the inhibition of CYP activities by its N-dealkylated metabolite, desethylamiodarone, rather than by amiodarone itself. In addition, the inactivation of CYPs by desethylamiodarone as well as by amiodarone would also contribute to the drug interactions.
Keywords: amiodarone, cytochrome P450, desethylamiodarone, inactivation, inhibition
Introduction
Amiodarone, a member of a new class of antiarrhythmic drugs with predominantly Class III (Vaughan Williams' classification) effects, is used for life-threatening supraventricular and ventricular dysrhythmias, such as ventricular fibrillation or haemodynamically unstable ventricular tachycardia [1]. Amiodarone has been shown to have a huge distribution and a correspondingly long serum elimination half-life of 40–50 days [2–4]. Owing to the slow onset caused by its cumulative property in tissues, it often takes weeks to months to attain the desired antiarrhythmic action without intravenous or oral loading doses [2, 5]. It has been reported that amiodarone is mainly metabolized to an active metabolite, desethylamiodarone by cytochrome P450 (CYP) 3A in humans (Figure 1) [6, 7]. The blood concentration of desethylamiodarone has been known to be comparable with that of amiodarone [8, 9] with an elevating trough level during the first 6 months [10]. Although there are large interindividual variations in mean values, the terminal elimination half-life of desethylamiodarone is generally longer than that of amiodarone after long-term oral treatment [4].
Figure 1.
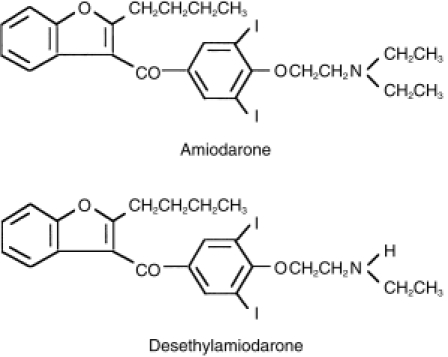
Chemical structures of amiodarone and desethylamiodarone.
CYP consists of a superfamily of heme-containing monooxygenases and is responsible for the oxidation of many drugs, environmental chemicals, and endogenous substances [11]. Three families (CYP1, CYP2, and CYP3) are currently thought to be responsible for most drug metabolism. While pharmacokinetic drug interactions can occur during the absorption, distribution, metabolism, and elimination phases after initial administration, interference with drug metabolism by CYP appears to be the predominant mechanism. Amiodarone has been reported to interact with a number of other therapeutic agents such as phenytoin [12], warfarin [13, 14], dextromethorphan [15], flecainide [16], and cyclosporin A [17, 18] in some clinical situations. These drugs are substrates of CYP2C9, CYP2D6, or CYP3A4 [19]. However, there have been few in vitro studies in which the inhibitory effects of amiodarone and desethylamiodarone on CYP activities were determined [7, 20, 21].
It is well known that chemicals which possess several common moieties such as a tertiary amine function [22, 23], furan ring [24, 25], and acetylene function [26, 27] are metabolized by CYPs and bind to the same enzyme covalently to form a CYP-metabolite complex and thereby inactivate the enzyme. Since amiodarone contains a tertiary amine and furan ring in its structure, its role as a mechanism-based inhibitor has been postulated. Therefore, in the current study, the inhibitory effects of amiodarone and desethylamiodarone on each human CYP activity was investigated. We also investigated the possibility of an inactivation of each human CYP isoform by amiodarone and desethylamiodarone. The purpose of our study was to predict the in vivo drug interactions of amiodarone from in vitro data and to understand the roles of desethylamiodarone in the mechanism of the inhibitory effects of amiodarone.
Methods
Chemicals
Amiodarone hydrochloride[2-butyl-3-benzofuranyl 4-[2- (diethylamino)ethoxy]-3,5-diiodophenyl ketone hydrochloride] and desethylamiodarone[2-butyl-3-benzofuranyl 4-[2-(monoethylamino)ethoxy]-3,5-diiodophenyl ketone] were kindly provided by Taisho Pharmaceutical (Tokyo, Japan). 7-Ethoxyresorufin, 7-benzyloxyresorufin, resorufin and chlorzoxazone were purchased from Sigma (St Louis, MO). Phenacetin, acetaminophen, coumarin and 7-hydroxycoumarin were purchased from Wako Pure Chemical Industries (Osaka, Japan). S-(−)-Warfarin, 7-hydroxywarfarin, S-(+)-mephenytoin (+/−)-4′-hydroxymephenytoin (+/−)-bufuralol hydrochloride, 1′-hydroxybufuralol maleate, and 6-hydroxychlorzoxazone were from Ultrafine-chemicals (Manchester, UK). Testosterone, 6β-hydroxytestosterone and 11β-hydroxytestosterone were from Steraloids (Wilton, NH). NADP+, glucose-6-phosphate and glucose-6-phosphate dehydrogenase were purchased from Oriental Yeast (Tokyo, Japan). Other chemicals were of the highest grade commercially available.
Enzyme preparations
Microsomes from human B-lymphoblastoid cells expressing CYP1A1, CYP1A2, CYP2A6, CYP2B6, CYP2C9 (Arg), CYP2C19, CYP2D6 (Val), CYP2E1, and CYP3A4 were obtained from Gentest (Woburn, MA). Except for CYP1A2, CYP2B6 and CYP2C19, these were coexpressed with NADPH-cytochrome P450 reductase. The CYP contents of these microsomes were provided in the data sheets by the manufacturer.
Enzyme assays
7-Ethoxyresorufin O-dealkylase activity (EROD) in microsomes from B-lymphoblastoid cells expressing CYP1A1 were determined as described previously [28]. The substrate concentration was 2 μm for the determination of the IC50 values, and ranged from 100 to 1000 nm for the determination of Ki values. Phenacetin O-deethylase activity (POD) in microsomes from B-lymphoblastoid cells expressing CYP1A2 was determined as described previously [29]. The substrate concentration was 10 μm for the determination of the IC50 values, and ranged from 5 to 40 μm for the determination of the Ki values. Coumarin 7-hydroxylase activity (COH) in microsomes from B-lymphoblastoid cells expressing CYP2A6 was determined as described previously [30] with slight modifications. The incubation mixture contained 50 mm potassium phosphate buffer (pH 7.4), the NADPH-generating system, 0.05 mg ml−1 microsomal protein, with coumarin as a substrate. The substrate concentration was 50 μm for the determination of the IC50 values, and ranged from 1 to 10 μm for the determination of the Ki values. The reaction was initiated by the addition of the NADPH-generating system, following a 2 min preincubation at 37° C. After incubation for 10 min, the reaction was terminated by adding 10 μl of ice-cold 60% perchloric acid. After removal of protein by centrifugation at 10 000 rev min−1 for 5 min, a 20 μl portion of the supernatant was injected into a h.p.l.c. system. The mobile phase for COH was 45% CH3CN, 20 mm NaClO4 (pH 2.5). The flow rate was 0.7 ml min−1 and the column temperature was 35° C. The formed product was detected fluorometrically (excitation: 338 nm, emission: 458 nm), and the quantification of 7-hydroxycoumarin was performed by comparing the h.p.l.c. peak heights with those of an authentic standard. 7-Benzyloxyresorufin O-dealkylase activity (BROD) in microsomes from B-lymphoblastoid cells expressing CYP2B6 was determined as described previously [28]. The substrate concentration was 2 μm for the determination of the IC50 values, and ranged from 0.5 to 4 μm for the determination of the Ki values. S-Warfarin 7-hydroxylase activity (S-WFOH) in microsomes from B-lymphoblastoid cells expressing CYP2C9 was determined as described previously [31]. The substrate concentration was 10 μm for the determination of the IC50 values, and ranged from 1 to 10 μm for the determination of the Ki values. S-Mephenytoin 4′-hydroxylase activity (S-MPOH) in microsomes from B-lymphoblastoid cells expressing CYP2C19 was determined as described previously [32]. The substrate concentration was 100 μm for the determination of the IC50 values, and ranged from 50 to 500 μm for the determination of the Ki values. Bufuralol 1′-hydroxylase activity (BFOH) in microsomes from B-lymphoblastoid cells expressing CYP2D6 was determined as described previously [28]. The substrate concentration was 1 μm for the determination of the IC50 values, and ranged from 0.5 to 10 μm for the determination of the Ki values. Chlorzoxazone 6-hydroxylase activity (CZXOH) in microsomes from B-lymphoblastoid cells expressing CYP2E1 was determined as described previously [33]. The substrate concentration was 50 μm for the determination of the IC50 values. Testosterone 6β-hydroxylase activity (TESOH) in microsomes from B-lymphoblastoid cells expressing CYP3A4 was determined as described previously [34]. The substrate concentration was 100 μm for the determination of the IC50 values, and ranged from 25 to 200 μm for the determination of the Ki values. For the determination of the Ki values toward various CYP enzymes, the ranges of amiodarone and desethylamiodarone concentrations were 0–200 μm and 0–30 μm, respectively.
With the exception of 7-ethoxyresorufin and 7-benzyloxyresorufin, which were dissolved in dimethyl sulfoxide, the substrates and inhibitors were dissolved in methanol so that the final concentration of solvent in the incubation mixture was <1%.
H.p.l.c. analysis
H.p.l.c. analyses were performed using an L-7100 pump (Hitachi), L-7400 UV detector (Hitachi), F-1080 fluorescence detector, L-7200 autosampler (Hitachi), L-7500 integrator (Hitachi), and 865-CO column oven (Jasco, Tokyo, Japan) equipped with a Capcell Pak C18 UG120 (4.6×250 mm; 4 μm) column (Shiseido, Tokyo, Japan).
Mechanism-based inactivation of human CYPs
Microsomes from B-lymphoblastoid cells expressing human CYP were preincubated at 37° C for the inactivation of CYP activities with various concentrations of amiodarone or desethylamiodarone in the presence of the NADPH-generating system (NADPH for CYP1A1 and CYP2B6). The preincubation time was 10, 20, and 30 min for EROD, BROD, POD and S-WFOH and 5, 10, 15, and 20 min for COH, S-MPOH, BFOH, and 2.5, 5, 7.5, and 10 min for TESOH, respectively. After the preincubation, typical substrates were added and the corresponding marker activities were measured according to the method described in the previous section. The incubation time was modified to 10 min for BROD, 15 min for POD, S-WFOH, and S-MPOH, 3 min for BFOH, respectively.
Data analysis
The IC50 values were determined by a nonlinear regression analysis. For the determination of the type of inhibition, the Lineweaver-Burk plot and Dixon plot were adopted [35], and then the kinetic parameters were determined by a nonlinear regression analysis using a computer program (K.cat, BioMetallics, Princeton, NJ).
For the determination of Kinact values (maximum rate constant for inactivation) and KI values (dissociation constant for the enzyme-inactivator), linear regression analysis was used to determine the Kobs values (initial rate constants of inactivation) [36]. The Kobs values were obtained as slopes of an initial linear phase plotting logarithm of the remaining activity against the preincubation time. The Kinact and KI values were determined by a nonlinear regression analysis using a computer program of KaleidaGraph (Synergy Software, Reading, PA). All data were analysed using the mean of duplicate determinations.
Prediction of drug interactions at clinical doses from in vitro data
If an enzyme reaction proceeds with a single enzyme and it is inhibited competitively or noncompetitively by other drugs, when the substrate concentration is much lower the Km, the change of the intrinsic clearance (CLint) is expressed by the following equation [37]:
where Iu is the unbound concentrations of inhibitor and Ki is the inhibition constant. In the present study, the changes of the CLint caused by amiodarone or desethylamiodarone were predicted using the unbound concentrations in plasma or unbound concentrations in the liver estimated by plasma binding ratio of amiodarone (96%) [38]. Since the tissue binding of amiodarone and desethylamiodarone and the plasma binding of desethylamiodarone have not been reported, we assumed that these are the same as the plasma binding of amiodarone.
Results
Inhibition of human CYP activities by amiodarone and desethylamiodarone
Using microsomes from human B-lymphoblastoid cells expressing each CYP isoform, the inhibitory effects of amiodarone and desethylamiodarone on human CYP activities were examined. As shown in Figure 2, amiodarone showed weak inhibition against all CYP activities examined in the present study. The IC50 values were 54 μm for CYP2D6, 57 μm for CYP2A6, 78 μm for CYP2C9, and >100 μm for the other activities. In contrast, the inhibitory effects of desethylamiodarone on each CYP activity were stronger than amiodarone. The IC50 values were 4.5 μm for CYP2D6, 4.9 μm for CYP2B6, 5.5 μm for CYP2C9, 5.6 μm for CYP1A1, 9.4 μm for CYP2A6, 14 μm for CYP1A2, 17 μm for CYP3A4, and 19 μm for CYP2C19. The inhibition of CYP2E1 by desethylamiodarone was relatively weak (IC50>100 μm).
Figure 2.

Inhibitory effects of amiodarone (○) and desethylamiodarone (•) on human CYP activities. (a) EROD by recombinant CYP1A1 was determined at a 7-ethoxyresorufin concentration of 2 μm. The control activity was 10.8 pmol min−1 pmol−1 CYP. (b) POD by recombinant CYP1A2 was determined at a phenacetin concentration of 10 μm. The control activity was 0.5 pmol min−1 pmol−1 CYP. (c) COH by recombinant CYP2A6 was determined at a coumarin concentration of 50 μm. The control activity was 6.7 pmol min−1 pmol−1 CYP. (d) BROD by recombinant CYP2B6 was determined at a 7-benzyloxyresorufin concentration of 2 μm. The control activity was 0.4 pmol min−1 pmol−1 CYP. (e) S-WFOH by recombinant CYP2C9 was determined at a S-warfarin concentration of 10 μm. The control activity was 0.05 pmol min−1 pmol−1 CYP. (f) S-MPOH by recombinant CYP2C19 was determined at a S-mephenytoin concentration of 100 μm. The control activity was 1.3 pmol min−1 pmol−1 CYP. (g) BFOH by recombinant CYP2D6 was determined at a bufuralol concentration of 1 μm. The control activity was 1.3 pmol min−1 pmol−1 CYP. (h) CZXOH by recombinant CYP2E1 was determined at a chlorzoxazone concentration of 50 μm. The control activity was 2.0 pmol min−1 pmol−1 CYP. (I) TESOH by recombinant CYP3A4 was determined at a testosterone concentration of 100 μm. The control activity was 13.5 pmol min−1 pmol−1 CYP. Each data point represents the mean of duplicate determinations. The IC50 values of amiodarone (AMD) and desethylamiodarone (DEA) are shown as μm.
As theIC50 value varies with the substrate concentration, we next determined the Ki values, which is a better parameter to define the interaction of an inhibitor with a particular enzyme in an inhibition study. Since it has been reported that amiodarone interacts with drugs which are substrates for CYP2C9, CYP2D6, and CYP3A4 such as phenytoin, S-warfarin, flecainide and cyclosporin A [12–18], the Ki values of amiodarone for S-WFOH, BFOH, and TESOH were determined. S-WFOH catalysed by CYP2C9 was noncompetitively inhibited by amiodarone with the Ki values of 94.6 μm (Table 1). BFOH catalysed by CYP2D6 was inhibited with the mixed type of competitive and noncompetitive components (Table 1) and the Ki value was 45.1 μm (α = 1.8). TESOH catalysed by CYP3A4 was noncompetitively inhibited by amiodarone (Ki = 271.6 μm).
Table 1.
Inhibitory types and kinetic constants of amiodarone and desethylamiodarone for human CYP activities.*.
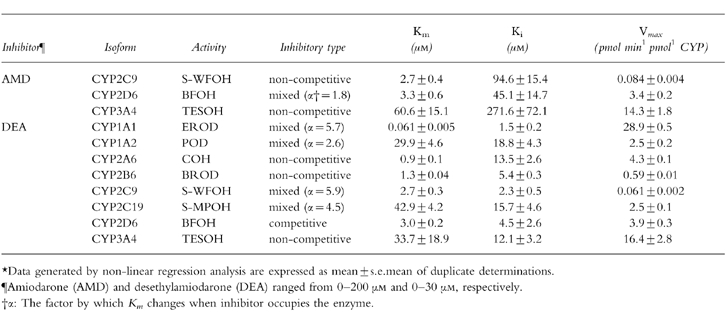 |
The Ki values of desethylamiodarone for each CYP isoform-specific activity except CYP2E1 were determined (Table 1). The inhibitory pattern of desethylamiodarone for EROD catalysed by CYP1A1 exhibited the mixed type of competitive and noncompetitive components with a Ki value of 1.5 μm (α = 5.7). POD catalysed by CYP1A2 was also inhibited by desethylamiodarone with mixed type inhibition and a with Ki value of 18.8 μm (α = 2.6). COH catalysed by CYP2A6 and BROD catalysed by CYP2B6 showed noncompetitive inhibition by desethylamiodarone, with Ki values of 13.5 μm and 5.4 μm, respectively. The inhibition of S-WFOH catalysed by CYP2C9 and S-MPOH catalysed by CYP2C19 was of mixed type with Ki values of 2.3 μm (α = 5.9) and 15.7 μm (α = 4.5), respectively. BFOH catalysed by CYP2D6 was competitively inhibited by desethylamiodarone (Ki value = 4.5 μm). TESOH catalysed by CYP3A4 was noncompetitively inhibited by desethylamiodarone with Ki value of 12.1 μm.
Mechanism-based inactivation of human CYP enzymes by amiodarone and desethylamiodarone
Amiodarone inhibited TESOH catalysed by CYP3A4 with NADPH-, in a time-and concentration-dependent manner (Table 2). The Kinact value was 0.06 min−1, and the KI value was 13.4 μm. In contrast, amiodarone did not inactivate BFOH catalysed by CYP2D6 (Table 2). EROD catalysed by CYP1A1 was inactivated by desethylamiodarone with a Kinact value of 0.03 min−1, and a KI value of 1.0 μm (Table 2). Desethylamiodarone inactivated CYP1A2 activity with a Kinact value of 0.03 min−1, and a KI value of 11.6 μm. BROD catalysed by CYP2B6 was inactivated by desethylamiodarone with a Kinact value of 0.02 min−1, and a KI value of 0.6 μm. Especially, desethylamiodarone exhibited potent inactivation of BFOH by CYP2D6 (Figure 3). The Kinact value was 0.12 min−1, and the KI value was 1.3 μm. CYP2A6, CYP2C9, CYP2C19, and CYP3A4 activities were not inactivated by desethylamiodarone.
Table 2.
Inactivation constants for amiodarone and desethylamiodarone of human CYP activities.*.
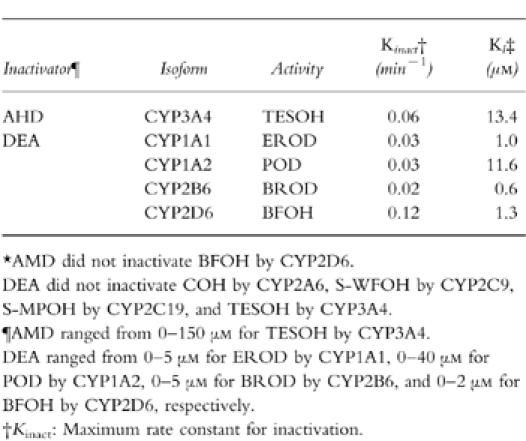 |
Figure 3.
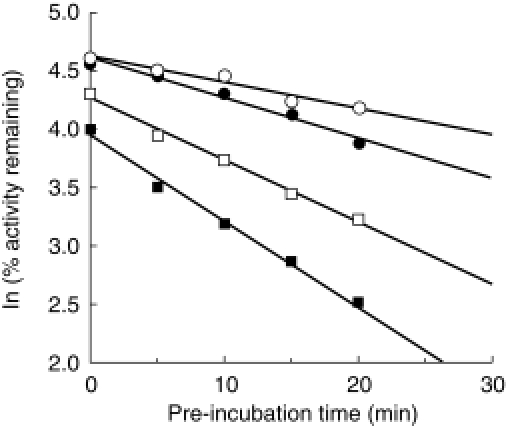
Mechanism-based inactivation of CYP2D6 by desethylamiodarone. The recombinant CYP2D6 was preincubated with 0 μm (○), 0.5 μm (•), 1 μm (□), and 2 μm (▪) desethylamiodarone for 0, 5, 10, 15, and 20 min at 37 °C in the presence of an NADPH-generating system. After preincubation, bufuralol was added to the reaction mixture and BFOH was determined.
Predicted changes of the clearance of the coadministered drugs by amiodarone and desethylamiodarone from in vitro data
To predict the possibility of drug interactions via a metabolic pathway between amiodarone and other drugs, the values of 1+Iu/Ki were calculated using Iu in plasma or Iu in liver estimated by plasma binding ratio and the Ki values shown in Table 1. The concentrations of amiodarone and desethylamiodarone in plasma were reported to range between 0.9–3.6 μm (0.6–2.3 mg l−1) and 0.6–5.7 μm (0.4–3.5 mg l−1), respectively, in postmortem samples [39]. The plasma binding ratio of amiodarone was reported to be 96% [38]. Therefore, the Iu values in plasma of amiodarone and desethylamiodarone were calculated to be 0.04–0.14 μm and 0.03–0.23 μm, respectively. As the Ki values of amiodarone for S-WFOH catalysed by CYP2C9, BFOH catalysed by CYP2D6, and TESOH catalysed by CYP3A4 were determined to be 94.6 μm, 45.1 μm, and 271.6 μm, respectively (Table 1), the 1+Iu/Ki values using Iu in plasma for CYP2C9, CYP2D6, and CYP3A4 were calculated as 1.00 (Table 3). Similarly, the 1+Iu/Ki values were calculated using Iu in plasma and the Ki values of desethylamiodarone as follows: CYP1A1 (1.02–1.15), CYP1A2 (1.00–1.01), CYP2A6 (1.00–1.02), CYP2B6 (1.00–1.04), CYP2C9 (1.01–1.10), CYP2C19 (1.00– 1.01), CYP2D6 (1.01–1.05), and CYP3A4 (1.00–1.02).
Table 3.
Predicted changes of the clearance of coadministered drugs by amiodarone and desethylamiodarone from in vitro data.
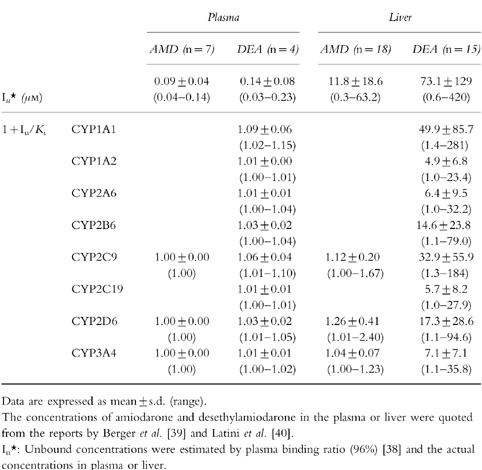 |
The concentrations of amiodarone and desethylamiodarone in liver were reported to range between 7.1–1379.2 μm (4.6–890 mg kg−1) and 15.4–10530.2 μm (9.5–6500 mg kg−1), respectively, in postmortem samples [39]. In another study [40], the concentrations of amiodarone and desethylamiodarone in liver were reported to be 1580.6 μm (1020 mg kg−1) and 8181.2 μm (5050 mg kg−1), respectively, with biopsy samples from patients treated chronically with amiodarone. According to the treatment duration and the maintenance dose, the hepatic levels of amiodarone and desethylamiodarone showed large interindividual difference. The unbound drug concentrations in liver were estimated by plasma binding ratio. Therefore, the Iu values in liver of amiodarone and desethylamiodarone were estimated to range between 0.3–63.2 μm and 0.6–421.2 μm, respectively. The 1+Iu/Ki values using Iu in liver and the Ki values of amiodarone for CYP2C9, CYP2D6, and CYP3A4 were calculated as 1.00–1.67, 1.01–2.40, and 1.00–1.23, respectively (Table 3). Similarly, the 1+Iu/Ki values were calculated using Iu in plasma and the Ki values of desethylamiodarone as follows: CYP1A1 (1.4–281), CYP1A2 (1.0–23.4), CYP2A6 (1.0–32.2), CYP2B6 (1.1–79.0), CYP2C9 (1.3–184), CYP2C19 (1.0–27.9), CYP2D6 (1.1–94.6), and CYP3A4 (1.1–35.8).
Discussion
Amiodarone, a drug used clinically for the treatment of life-threatening supraventricular and ventricular dysrhythmias, is generally administered in combination with other drugs. Coadministration of a number of drugs often causes drug interactions leading to severe side-effects [41]. One of the most important mechanisms of drug interactions is the inhibition of hepatic metabolism catalysed by CYP(s). Drug metabolism catalysed by CYP(s) can be inhibited by any of the following three mechanisms. The first is competitive inhibition caused by coadministration of drugs metabolized by the same CYP isoform. The second is the inactivation of CYP by the metabolite of a drug which covalently binds to the enzyme to form a complex with CYP, leading to irreversible inhibition [36]. In this case, as a drug has to be metabolically activated by an enzyme and covalently binds to the same enzyme, inactivation affects only the CYP isoform that is involved in the drug metabolism. The third is inhibition by the binding of imidazole or a hydrazine group to the heme portion of CYP and this causes mainly nonspecific inhibition of many CYP isoforms [42]. In the present study, the first two inhibitory mechanisms of amiodarone and desethylamiodarone were investigated.
Owing to the high lipophilicity, amiodarone is extensively distributed into tissues during long-term therapy [39]. In humans, amiodarone is principally metabolized to desethylamiodarone [7, 39]. Liver is known as a tissue where amiodarone and desethylamiodarone are highly accumulated [39]. The concentration of desethylamiodarone in the liver is higher than that of amiodarone in humans [39]. In addition, it has been reported that the concentrations of amiodarone and desethylamiodarone in the liver are two or three orders of magnitude higher than those in plasma or blood [39]. Previous reports showed that amiodarone interacts with a number of drugs metabolized by CYP2C9, CYP2D6, and CYP3A4 [12–18]. Therefore, we investigated the inhibitory effects of amiodarone and desethylamiodarone on human CYP activities including these isoforms, since the inhibition of CYP would be responsible for the mechanism of the drug interactions.
The major metabolic pathway of amiodarone, N-deethylation to desethylamiodarone, has been reported to be catalysed by CYP3A with a Kmvalue of 0.33±0.11 mm in human liver microsomes [7]. In the previous report [7], it was shown that amiodarone competitively inhibited nifedipine metabolism catalysed by CYP3A4 with a Ki value of ~0.57 mm in human liver microsomes. However, we obtained the result that amiodarone noncompetitively inhibited TESOH with a Ki value of 271.6 μm in recombinant CYP3A4 from human B-lymphoblastoid cells (Table 1). Although the reason for the inconsistency in the inhibition manner of amiodarone is unknown, it might be due to a difference in the substrate, source of enzyme and/or CYP3A4 properties. In another study [20], amiodarone was reported to inhibit phenytoin hydroxylation (CYP2C9) in human liver microsomes (IC50 = 25 μm), but not bufuralol hydroxylation (CYP2D6) and felodipine oxidation (CYP3A4) in human liver microsomes (IC50>100 μm). Furthermore, Jaruratanasirikul & Hortiwakul [21] reported that dextromethorphan O-demethylation (CYP2D6) was inhibited by amiodarone and desethylamiodarone with Ki values of 52.7 μm and 34.4 μm, respectively. The results in our inhibition study with amiodarone toward CYP2C9, CYP2D6, and CYP3A4 were similar to those of these previous studies, in terms of the weak inhibitory effects. It has been suggested that desethylamiodarone exhibited a stronger inhibition of CYP2D6 activity than did amiodarone [21]. In the present study, we showed that desethylamiodarone inhibit other CYP activities such as CYP1A1, CYP1A2, CYP2A6, CYP2B6, and CYP2C19 with more potent effects than amiodarone.
The possibility of amiodarone and desethylamiodarone as mechanism-based inactivators of human CYP was determined, since these contain structures of a tertiary amine and a furan ring, which are reported to bind with CYP covalently after metabolism by the CYP [22–27]. Previously, Larrey et al. [43] reported that amiodarone administered to rats, mice, and hamsters formed an inactive CYP Fe(II)-metabolite complex in vivo, leading to irreversible inactivation of the enzyme. However, it was not determined whether the N-deethylated metabolites of amiodarone are further activated by CYP enzymes to form active metabolites that inhibit CYP-dependent drug oxidations in humans. Therefore, we also examined the inactivation of each CYP isoform by amiodarone and desethylamiodarone. The inactivation kinetics of human CYP isoforms by typical mechanism-based inactivators have been reported previously. For example, L-754,394, an HIV-1 protease inhibitor, has been reported to inactivate TESOH catalysed by CYP3A4 with a Kinact value of 1.62 min−1 and KI value of 7.5 μm [44]. It has been reported that furafylline inactivated R-warfarin 6-hydroxylation catalysed by CYP1A2 with the Kinact value of 0.87 min−1 and KI value of 23 μm [45], and that gestodene inactivated nifedipine oxidation catalysed by CYP3A4 with a Kinact value of 0.39 min−1 and KI value of 46 μm [26]. In addition, erythromycin has also been reported to inactivate midazolam metabolism via CYP3A4 with a Kinact value of 0.064 min−1 and KI value of 19 μmfor α-hydroxylation pathway and Kinact value of 0.058 min−1 and KI value of 22 μmfor 4-hydroxylation pathway [Sugiyama et al. unpublished data]. The Kinact values of amiodarone and desethylamiodarone for several CYP activities obtained in this study were comparable with those of previous reports. Therefore, it was suggested that amiodarone and desethylamiodarone would be potent mechanism-based inactivators of human CYPs. This type of interaction requires more attention than the common type of inhibition, because the inhibitory effects remain after the elimination of the inhibitor from blood and tissue. The terminal elimination half lives of amiodarone and desethylamiodarone after long-term oral treatment are approximately 40 days or longer [3, 4]. Therefore, the inactivation in addition to the common inhibition might cause severe drug interactions.
When we discuss drug interactions via the inhibition of CYP activities using the value of 1+Iu/Ki, Iu should be the unbound concentration of the inhibitor around the CYP enzyme in liver. However, it is impossible to measure directly this in vivo. Many drugs are transported into the liver by passive diffusion, allowing one to assume that the unbound concentration in liver equals that in plasma. This means that the estimation using the unbound concentration in plasma may be proper for some drugs, although this assumption is not valid exactly. Firstly, the unbound amiodarone and desethylamiodarone concentrations in plasma were adopted to calculate the 1+Iu/Ki, values in the present study. On the other hand, in the case of drugs which are actively transported into the liver, the unbound concentration in liver may be higher than that in plasma. Since amiodarone and desethylamiodarone are highly accumulative in liver, the possibility that these are actively transported into the liver would be implied. Therefore, the unbound concentrations in liver estimated using the plasma binding ratio (96%) were adopted to calculate the 1+Iu/Ki, values. As expected, the 1+Iu/Ki, values using Iu in liver were higher than those using Iu in plasma.
In an in vivo study, it has been reported that cyclosporin clearance was decreased by more than 2-fold after the amiodarone therapy in a heart transplant patient [17]. In addition, it has been also reported that the clearances of phenytoin [12] and theophylline [46] were decreased by approximately 1.3-fold and 1.2–1.9-fold, respectively, with coadministration of amiodarone. These changes of in vivo clearance are higher than the 1+Iu/Ki, values using Iu in plasma. Accordingly, it was suspected that the unbound concentrations of amiodarone and desethylamiodarone are higher than those in plasma. Furthermore, the possibility of interactions of amiodarone with other drugs metabolized by CYP2A6, CYP2B6, and CYP2C19 was also suggested from the inhibition data by desethylamiodarone.
In conclusion, it was shown that desethylamiodarone, a major metabolite of amiodarone, exhibited more potent inhibitory effects on human CYP activities than did amiodarone. Accordingly, it is suggested that the drug interactions of amiodarone might be caused by inhibition of the metabolic pathway via CYP by desethylamiodarone.
Acknowledgments
This work was supported in part by a Health Sciences Research Grant from the Ministry of Health and Walfare in Japan. We thank Taisho Pharmaceutical for providing amiodarone and desethylamiodarone and acknowledge Mr Brent Bell for reviewing the manuscript.
References
- 1.Gill J, Heel RC, Fitton A. Amiodarone. An overview of its pharmacological properties, and review of its therapeutic use in cardiac arrhythmias. Drugs. 1992;43:69–110. doi: 10.2165/00003495-199243010-00007. [DOI] [PubMed] [Google Scholar]
- 2.Kannan R, Nademanee K, Hendrickson JA, Rostami HJ, Singh BN. Amiodarone kinetics after oral doses. Clin Pharmacol. 1982;31:438–444. doi: 10.1038/clpt.1982.57. [DOI] [PubMed] [Google Scholar]
- 3.Stäubli M, Bircher J, Galeazzi RL, Remund H, Studer H. Serum concentrations of amiodarone during long term therapy. Relation to dose, efficacy and toxicity. Eur J Clin Pharmacol. 1983;24:485–494. doi: 10.1007/BF00609891. [DOI] [PubMed] [Google Scholar]
- 4.Holt DW, Tucker GT, Jacson PR, Storey GCA. Amiodarone pharmacokinetics. Am Heart J. 1983;106:840–847. doi: 10.1016/0002-8703(83)90006-6. [DOI] [PubMed] [Google Scholar]
- 5.Andreasen F, Agerbæk H, Bjerregaard P, Gøtzsche H. Pharmacokinetics of amiodarone after intravenous and oral administration. Eur J Clin Pharmacol. 1981;19:293–299. doi: 10.1007/BF00562807. [DOI] [PubMed] [Google Scholar]
- 6.Trivier J-M, Libersa C, Belloc C, Lhermitte M. Amiodarone N-deethylation in human liver microsomes: involvement of cytochrome P4503A enzymes (first report) Life Sci. 1993;52:91–96. doi: 10.1016/0024-3205(93)90523-6. [DOI] [PubMed] [Google Scholar]
- 7.Fabre G, Julian B, Saint-Aubert B, Joyeux H, Berger Y. Evidence for CYP3A-mediated N-deethylation of amiodarone in human liver microsomal fractions. Drug Metab Dispos. 1993;21:978–985. [PubMed] [Google Scholar]
- 8.Heger JJ, Prystowsky EN, Zipes DP. Relationship between amiodarone dosage, drug concentrations, and adverse side effect. Am Heart J. 1983;106:931–935. doi: 10.1016/0002-8703(83)90018-2. [DOI] [PubMed] [Google Scholar]
- 9.Marchiset D, Bruno R, Djiane P, et al. Amiodarone and desethylamiodarone elimination kinetics following withdrawal of long-term amiodarone maintenance therapy. Biopharm Drug Dispos. 1985;6:209–215. doi: 10.1002/bdd.2510060211. [DOI] [PubMed] [Google Scholar]
- 10.Stäubi M, Troendle A, Schmid B, et al. Pharmacokinetics of amiodarone, desethylamiodarone and other iodine-containing amiodarone metabolites. Eur J Clin Pharmacol. 1985;29:417–423. doi: 10.1007/BF00613455. [DOI] [PubMed] [Google Scholar]
- 11.Nelson DR, Koymans L, Kamataki T, et al. P450 superfamily: update on new sequence, gene mapping, accession numbers and nomenclature. Pharmacogenetics. 1996;6:1–42. doi: 10.1097/00008571-199602000-00002. [DOI] [PubMed] [Google Scholar]
- 12.Nolan PE, Marcus FI, Hoyer GL, Bliss M, Gear K. Pharmacokinetic interaction between intravenous phenytoin and amiodarone in healthy volunteers. Clin Pharmacol Ther. 1989;46:43–50. doi: 10.1038/clpt.1989.104. [DOI] [PubMed] [Google Scholar]
- 13.O'Reilly RA, Tranger WF, Rettie AE, Goulart DA. Interaction of amiodarone with racemic warfarin and its separated enantiomorphs in human. Clin Pharmacol Ther. 1987;42:290–294. doi: 10.1038/clpt.1987.149. [DOI] [PubMed] [Google Scholar]
- 14.Heimark LD, Wienkers L, Kunze K, et al. The mechanism of the interaction between amiodarone and warfarin in humans. Clin Pharmacol Ther. 1992;51:398–407. doi: 10.1038/clpt.1992.39. [DOI] [PubMed] [Google Scholar]
- 15.Funck-Brentano C, Jacqz-Aigrain E, Leenhardt A, et al. Influence of amiodarone on genetically determined drug metabolism in humans. Clin Pharmacol Ther. 1991;50:259–266. doi: 10.1038/clpt.1991.135. [DOI] [PubMed] [Google Scholar]
- 16.Funck-Brentano C, Becquemont L, Kroemer KH, et al. Variable disposition kinetics and electrocardiographic effects of flecainide during repeated dosing in humans: Contribution of genetic factors, dose-dependent clearance, and interaction with amiodarone. Clin Pharmacol Ther. 1994;55:256–269. doi: 10.1038/clpt.1994.26. [DOI] [PubMed] [Google Scholar]
- 17.Nicolau DP, Uber WE, Crumbley AJ III, Strange C. Amiodarone–cyclosporine interaction in a heart transplant patient. J Heart Lung Transplant. 1992;11:564–568. [PubMed] [Google Scholar]
- 18.Chitwood KK, Abdul-Haqq AJ, Heim-Duthoy KL. Cyclosporine–amiodarone interaction. Ann Pharmacother. 1993;27:569–571. doi: 10.1177/106002809302700506. [DOI] [PubMed] [Google Scholar]
- 19.Smith G, Stubbins J, Harries LW, Wolf CR. Molecular genetics of the human cytochrome P450 monooxygenase superfamily. Xenobiotica. 1998;28:1129–1165. doi: 10.1080/004982598238868. [DOI] [PubMed] [Google Scholar]
- 20.Walker DK, Alabaster CT, Congrave GS, et al. Significance of metabolism in the disposition and action of the antidysrhythmic drug, dofetilide. Drug Metab Dispos. 1996;24:447–454. [PubMed] [Google Scholar]
- 21.Jaruratanasirikul S, Hortiwakul R. The inhibitory effect of amiodarone and desethylamiodarone on dextromethorphan O-demethylation in human and rat liver microsomes. J Pharm Pharmacol. 1994;46:953–955. doi: 10.1111/j.2042-7158.1994.tb05721.x. [DOI] [PubMed] [Google Scholar]
- 22.Larry D, Funk-Brentano C, Breil P, et al. Effect of erythromycin on hepatic drug-metabolizing enzymes in humans. Biochem Pharmacol. 1983;32:1063–1068. doi: 10.1016/0006-2952(83)90626-3. [DOI] [PubMed] [Google Scholar]
- 23.Ohmori S, Ishii I, Kuriya S, et al. Effects of clarithromycin and its metabolites on the mixed function oxidase system in hepatic microsomes of rat. Drug Metab Dispos. 1993;21:358–363. [PubMed] [Google Scholar]
- 24.Khojasteh-Bakht SC, Koenigs LL, Peter RM, Trager WF, Nelson SD. (R) -(+)-Menthofuran is a potent, mechanism-based inactivator of human liver cytochrome P450 2A6. Drug Metab Dispos. 1998;26:701–704. [PubMed] [Google Scholar]
- 25.He K, Iyer RK, Hayes NR, et al. Inactivation of cytochrome P450 3A4 by bergamottin, a component of grapefruit juice. Chem Res Toxicol. 1998;11:252–259. doi: 10.1021/tx970192k. [DOI] [PubMed] [Google Scholar]
- 26.Guengerich FP. Mechanism-based inactivation of human liver microsomal cytochrome P-450 IIIA4 by gestodene. Chem Res Toxicol. 1993;3:363–371. doi: 10.1021/tx00016a015. [DOI] [PubMed] [Google Scholar]
- 27.He K, Woof TF, Hollenberg F. Mechanism-based inactivation of cytochrome P-450–3A4 by mifepristone (RU486) Drug Metab Dispos. 1999;288:791–797. [PubMed] [Google Scholar]
- 28.Nakajima M, Kobayashi K, Shimada N, et al. Involvement of CYP1A2 in mexiletine metabolism. Br J Clin Pharmacol. 1998;46:55–62. doi: 10.1046/j.1365-2125.1998.00048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kobayashi K, Nakajima M, Chiba K, et al. Inhibitory effects of antiarrhythmic drugs on phenacetin O-deethylation catalysed by human CYP1A2. Br J Clin Pharmacol. 1998;45:361–368. doi: 10.1046/j.1365-2125.1998.t01-1-00692.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yamazaki H, Tanaka M, Shimada T. Highly sensitive high-performance liquid chromatographic assay for coumarin 7-hydroxylation and 7-ethxycoumarin O-deethylation by human liver cytochrome P450 enzyme. J Chromatogr B Biomed Appl. 1999;721:13–19. doi: 10.1016/s0378-4347(98)00472-1. [DOI] [PubMed] [Google Scholar]
- 31.Lang D, Döcker R. Highly sensitive and specific high-performance liquid chromatograghic analysis of 7-hydroxywarfarin, a marker for human cytochrome P-4502C9 activity. J Chromatogr B Biomed Appl. 1995;672:305–309. doi: 10.1016/0378-4347(95)00222-5. [DOI] [PubMed] [Google Scholar]
- 32.Nakajima M, Inoue T, Shimada N, et al. Cytochrome P4502C9 catalyzes indomethacin O-demethylation in human liver microsomes. Drug Metab Dispos. 1998;26:261–266. [PubMed] [Google Scholar]
- 33.Court MH, Von-Moltke LL, Shader RI, Greenblatt DJ. Biotransformation of chlorzoxazone by hepatic microsomes from humans and ten other mammalian species. Biopharm Drug Dispos. 1997;18:213–226. doi: 10.1002/(sici)1099-081x(199704)18:3<213::aid-bdd15>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 34.Arlotto MP, Trant JM, Estabrook RW. Measurement of steroid hydroxylation reactions by high-performance liquid chromatography as indicator of P450 identity and function. Meth Enzymol. 1991;206:454–462. doi: 10.1016/0076-6879(91)06114-i. [DOI] [PubMed] [Google Scholar]
- 35.Segel Ih. Enzyme Kinetics. New York: A Wiley-Interscience Publication; 1993. Rapid equilibrium partial and mixed-type inhibition; pp. 161–226. [Google Scholar]
- 36.Silverman RB. Chemistry and Enzymology. Vol. 1. Boca Raton: CRC Press; 1988. Mechanism-based enzyme inactivation; pp. 3–30. [Google Scholar]
- 37.Ito K, Iwatsubo T, Kanamitsu S, Ueda K, Suzuki H, Sugiyama Y. Prediction of pharmacokinetic alteration caused by drug–drug Interactions: Metabolic interaction in the liver. Pharmacol Rev. 1998;50:387–411. [PubMed] [Google Scholar]
- 38.Lalloz MRA, Byfield PGH, Greenwood RM, Himsworth RL. Binding of amiodarone by serum proteins and the effects of drugs, hormones and other interacting ligands. J Pharm Pharmacol. 1984;36:366–372. doi: 10.1111/j.2042-7158.1984.tb04400.x. [DOI] [PubMed] [Google Scholar]
- 39.Berger Y, Harris L. Pharmacokinetics. In: Harris L, Roncucci R, editors. Amiodarone: Pharmacology, Pharmacokinetics, Toxicology, and Clinical Effects. Paris: Medecine et Science Internationales; 1986. pp. 45–98. [Google Scholar]
- 40.Latini R, Tognoni G, Kates ER. Clinical pharmacokinetics of amiodarone. Clin Pharmacokinet. 1984;9:136–156. doi: 10.2165/00003088-198409020-00002. [DOI] [PubMed] [Google Scholar]
- 41.Jafari-Fesharaki M, Scheinman MM. Adverse effect of amiodarone. Pace. 1998;21:108–120. doi: 10.1111/j.1540-8159.1998.tb01068.x. [DOI] [PubMed] [Google Scholar]
- 42.Somogyi A, Muirhead M. Pharmacokinetic interaction of cimetidine. Clin Pharmacokinet. 1987;12:321–366. doi: 10.2165/00003088-198712050-00002. [DOI] [PubMed] [Google Scholar]
- 43.Larrey D, Tinel M, Letteron P, et al. Formation of an inactive cytochrome P-450Fe (II)-metabolite complex after administration of amiodarone in rats, mice and hamsters. Biochem Pharmacol. 1986;35:2213–2220. doi: 10.1016/0006-2952(86)90594-0. [DOI] [PubMed] [Google Scholar]
- 44.Chiba M, Nishime JA, Lin JH. Potent and selective inactivation of human liver microsomal cytochrome P-450 isoforms by L-754,394, an investigational human immune deficiency virus protease inhibitor. J Pharmacol Exp Ther. 1995;275:1527–1535. [PubMed] [Google Scholar]
- 45.Kunze KL, Trager WF. Isoform-selective mechanism-based inhibition of human cytochrome P450 1A2 by furafylline. Chem Res Toxicol. 1993;6:649–656. doi: 10.1021/tx00035a009. [DOI] [PubMed] [Google Scholar]
- 46.Hirsch A, Saccar C, McGeady SJ. Interaction between theophylline and amiodarone. Ann Allergy. 1993;70:68. [Google Scholar]