

Abstract
Aims
To characterize the nonspecific binding to human liver microsomes of drugs with varying physicochemical characteristics, and to develop a model for the effect of nonspecific binding on the in vitro kinetics of drug metabolism enzymes.
Methods
The extent of nonspecific binding to human liver microsomes of the acidic drugs caffeine, naproxen, tolbutamide and phenytoin, and of the basic drugs amiodarone, amitriptyline and nortriptyline was investigated. These drugs were chosen for study on the basis of their lipophilicity, charge, and extent of ionization at pH 7.4. The fraction of drug unbound in the microsomal mixture, fu(mic), was determined by equilibrium dialysis against 0.1 m phosphate buffer, pH 7.4. The data were fitted to a standard saturable binding model defined by the binding affinity KD, and the maximum binding capacity Bmax. The derived binding parameters, KD and Bmax, were used to simulate the effects of saturable nonspecific binding on in vitro enzyme kinetics.
Results
The acidic drugs caffeine, tolbutamide and naproxen did not bind appreciably to the microsomal membrane. Phenytoin, a lipophilic weak acid which is mainly unionized at pH 7.4, was bound to a small extent (fu(mic) = 0.88) and the binding did not depend on drug concentration over the range used. The three weak bases amiodarone, amitriptyline and nortriptyline all bound extensively to the microsomal membrane. The binding was saturable for nortriptyline and amitriptyline. Bmax and KD values for nortriptyline at 1 mg ml−1 microsomal protein were 382 ± 54 µm and 147 ± 44 µm, respectively, and for amitriptyline were 375 ± 23 µm and 178 ± 33 µm, respectively. Bmax, but not KD, varied approximately proportionately with the microsome concentration. When KD is much less than the Km for a reaction, the apparent Km based on total drug can be corrected by multiplying by fu(mic). When the substrate concentration used in a kinetic study is similar to or greater than the KD (Km ≥ KD), simulations predict complex effects on the reaction kinetics. When expressed in terms of total drug concentrations, sigmoidal reaction velocity vs substrate concentration plots and curved Eadie Hofstee plots are predicted.
Conclusions
Nonspecific drug binding in microsomal incubation mixtures can be qualitatively predicted from the physicochemical characteristics of the drug substrate. The binding of lipophilic weak bases is saturable and can be described by a standard binding model. If the substrate concentrations used for in vitro kinetic studies are in the saturable binding range, complex effects are predicted on the reaction kinetics when expressed in terms of total (added) drug concentration. Sigmoidal reaction curves result which are similar to the Hill plots seen with cooperative substrate binding.
Keywords: human liver microsomes, in vitro clearance prediction, in vitro drug metabolism kinetics, nonspecific binding
Introduction
In vitro techniques are increasingly being used during new drug development to predict human in vivo drug metabolism and disposition. The qualitative assessment of the environmental and genetic factors potentially affecting the metabolism of a new drug, including the potential for metabolic drug interactions, is well established [1–3]. Quantitative predictions of in vivo drug clearance and the magnitude of potential drug interactions are also important to enable the development of drugs with optimal pharmacokinetic properties [4–6].
Kinetic studies in vitro, most commonly with human liver microsomes, allow determination of the maximal velocity (Vmax) and Michaelis constant (Km) for a metabolic pathway. Under linear conditions, the ratio of Vmax to Km represents the metabolic intrinsic clearance, which is a measure of the efficiency of drug metabolism by the liver, and is the key parameter for extrapolation of in vitro kinetic data to in vivo metabolic clearance.
While this approach has been reasonably successful, large discrepancies sometimes occur between hepatic clearance predicted from in vitro data and that measured in vivo, with the in vivo value usually being higher than predicted [7]. A number of factors could contribute to this including metabolism by extrahepatic tissues, incorrect assumptions about the equilibrium of drug between blood and hepatocyte, incorrect fraction of drug unbound used in the in vivo clearance model, nonrepresentative liver samples used in vitro, and incorrect determination of Vmax and/or Km.
Determination of Km and Ki is critical in the quantitative extrapolation of in vivo drug clearance and inhibitory drug interactions, respectively. In vivo, the unbound drug in plasma is in equilibrium with the unbound drug in the hepatocyte cytosol, which accesses the active sites of drug metabolizing enzymes. The in vitro correlate of this is the unbound drug in the microsomal incubation. Most drugs are lipid soluble organic compounds that bind nonspecifically to the lipid-protein milieu of the microsomal membrane. Despite this, almost all workers have used the total (added), rather than the unbound, drug concentration in kinetic experiments to determine Km and Ki. This results in overestimation of these parameters and underestimation of in vivo drug clearance and extent of inhibition. Some groups have recently used a correction factor, the fraction of drug unbound in the microsomal incubation (fu(mic)), to correct for this [7–9], but have assumed that fu(mic) is independent of substrate concentration.
The present study characterizes the nonspecific binding to human liver microsomes of a range of weak acids and bases, with varying physicochemical characteristics (log D7.4, and extent of ionization). A model for the nonspecific binding of drugs to microsomes has been developed, and the impact of binding on in vitro enzyme kinetics has been modelled.
Methods
Human liver microsomes
Microsomes were prepared from one human liver sample obtained from a renal transplant donor with the consent of the next-of-kin and the Flinders Medical Centre Committee on Clinical Investigation [10]. Microsomal pellets were suspended in 0.1 m phosphate buffer containing 20% (v/v) glycerol and stored at −70° C. Protein content was measured by the method of Lowry et al. [11] using bovine serum albumin as the standard. Microsomes were diluted to the desired protein concentration in 0.1 m phosphate buffer. This liver is representative of others in our liver bank.
Equilibrium dialysis
Drug binding to microsomes was determined by equilibrium dialysis. A Dianorm apparatus was used with Teflon dialysis cells of 1.2 ml capacity per side and Spectrapor#4 dialysis membrane (molecular weight cut off 12 000–14 000 Da) purchased from Spectrum Medical Industries Inc. (Los Angeles, CA, USA). The sample volume on each side of the cell was 1 ml. The dialysis membrane was prepared by soaking overnight in 0.1 m phosphate buffer, pH 7.4 at 4° C. Each individual drug was initially placed in the microsomal compartment and dialysed against 0.1 m phosphate buffer, pH 7.4. The complete assembly of dialysis cells was immersed in a water bath maintained at 37° C, and rotated at 12 rev min−1 for 3 h. Each dialysis experiment had buffer/buffer (i.e. buffer on both sides of the dialysis cell) and microsome/microsome controls at a high (500 µm) and low (50 µm) drug concentration to ensure that equilibrium was reached. The ratios of concentrations of drug on the two sides of the cell were within the range 0.8–1.2 indicating that equilibrium was reached in the 3 h dialysis time.
The concentrations of each drug used in dialysis experiments were selected to span the reported apparent Km value for that compound. Each drug was initially dialysed at varying concentrations against 1 mg ml−1 microsomal protein.
With the exception of amiodarone, each of the drugs was prepared as an aqueous solution and diluted 1 : 100 upon addition to the dialysis cells. Naproxen was solubilized in 0.1 m phosphate buffer. Phenytoin, and tolbutamide were solubilized by dropwise addition of an aqueous solution of 1.0 m NaOH. Amiodarone was dissolved in DMSO so that the final concentration in the dialysis mixture did not exceed 1% v/v.
The octanol/buffer partition coefficients at pH 7.4 (log D7.4) were determined for each drug using the shake flask method [12], with equal volumes of octanol and 0.1 m phosphate buffer pH 7.4.
Analytical methods
Drug concentrations were determined by h.p.l.c. analysis, comparing peak heights to a standard curve. Standard curves were performed out of buffer and microsomes and were linear with r2 values ranging from 0.983 to 0.999. With the exception of caffeine, 0.2 ml aliquots of samples recovered from each side of the dialysis apparatus were mixed with 0.4 ml of acetonitrile. Tubes were vortex mixed for 1 min, and centrifuged (1600 g for 5 min) to pellet protein. Amiodarone, amitriptyline and nortriptyline were injected directly (0.1 ml) onto the h.p.l.c. column without changing peak shape or retention time. However, dilution of samples with distilled water (1 :5) was necessary for the analysis of S-naproxen, phenytoin, and tolbutamide given the lower proportion of acetonitrile in the mobile phase. In the case of amiodarone, the buffer sides of the cells were unloaded into a microsome suspension and standard curves were constructed out of microsomes. This gave good (> 80%) recovery of amiodarone.
The very low proportion of acetonitrile in the caffeine mobile phase precluded the use of the acetonitrile precipitation method described above, and an extraction procedure was used. Aliquots (0.2 ml) of solution from either side of the dialysis cell were added to a 15 ml glass culture tube containing 8-chlorotheophylline (internal standard; 0.1 ml of a 1 mm solution) and HCl (0.4 m, 0.1 ml). The mixture was extracted with dichloromethane by vortex mixing for 1 min. Culture tubes were centrifuged (1600 g for 5 min) and the aqueous layer was aspirated, and discarded. The organic phase was transferred to a conical glass tube and evaporated to dryness under N2. The residue was reconstituted in the assay mobile phase (0.3 ml) and an aliquot (0.1 ml) injected onto the h.p.l.c. column. Chromatography conditions for the various analytes are shown in Table 1.
Table 1.
H.p.l.c. conditions for drug assays.
| Drug | Mobile phase | H.p.l.c. column | Mobile phase flow rate (ml min−1) | Detector wavelength (nm) | Retention time of analyte (min) |
|---|---|---|---|---|---|
| Caffeine (internal standard) | 95% Na acetate buffer (pH 4.0, 1.7 mm)/5% acetonitrile | C-18 | 2 | 276 | 8.5 (10.2) |
| Amiodarone | 58% distilled water with 0.01 m NH4CIO4/42% acetronitrile (adjusted to pH 3.0 with perchloric acid) | C-8 | 2 | 242 | 8.75 |
| Amitriptyline | 60% distilled water with 3 mm octanesulphonic acid, and 0.5 mm NNN’N′-TMED/40% acetonitrile (adjusted to pH 2.5 with H3PO4) | C-8 | 2 | 230 | 7.0 |
| Nortriptyline | As for amitriptyline | 7.75 | |||
| Naproxen | 85% 20 mm phosphate buffer (pH 7.0)/15% acetonitrile | C-18 | 1.8 | 254 | 5.5 |
| Tolbutamide | 70% 10 mm Na acetate/ 30% acetonitrile (adjusted to pH 4.3 with glacial acetic acid) | C-18 | 2 | 254 | 6.75 |
| Phenytoin | As for tolbutamide | C-18 | 2 | 254 | 5.2 |
C-18: Waters, Nova Pak, particle size 4 micron, 3.9 mm (id) × 150 mm C-8: Beckman Ultrasphere (Octyl), particle size 5 micron, 4.6 mm (id) × 25 cm.
The within day precision of the analytical methods was assessed by triplicate determinations from buffer and microsomes at three drug concentrations and a microsomal protein concentration of 1 mg ml−1. Coefficients of variation were < 10% in all cases.
The unbound fraction (fu(mic)) of drug in microsomal compartment was expressed as the free drug concentration (concentration in the buffer compartment of the dialysis cell) divided by the total drug concentration (concentration in the microsome compartment). The within day precision for the measurement of the microsomal binding of each drug was assessed by triplicate measurements of fu(mic) at two different drug concentrations (20 µm and 100 µm). The coefficient of variation was less than 10% for each of the drugs at both the high and low concentrations.
Theoretical considerations
The nonspecific binding of a drug to the microsomal membrane is given by
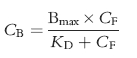 |
(1) |
where CB is the concentration of drug bound, CF is the free drug concentration, Bmax is the maximal binding capacity and KD is the dissociation constant. Data were inspected graphically as a Scatchard plot, to obtain initial estimates of the binding parameters Bmax and KD. These values were then used as the initial estimates in the non linear extended least squares regression modellingprogram, MK model [13], to calculate ‘best-fit’ Bmax and KD values.
If it is assumed that only free drug is available for metabolism in an in vitro incubation, the concentration term in the standard equation describing reaction velocity must be replaced by CF as shown below.
| (2) |
For simulations of the effects of binding on in vitro kinetics, the free drug concentration was calculated from equation 1 (with CB = CT−CF) by solving the quadraticexpression
| (3) |
Results
The physicochemical characteristics and binding of the drugs studied are shown in Table 2. The hydrophilic weak acids naproxen, tolbutamide and caffeine showed no microsomal membrane binding, whereas phenytoin, a lipophilic weak acid mostly unionized at pH 7.4, was bound slightly to microsomal membrane with an fu(mic) independent of drug concentration over the range used. The weak bases amiodarone, amitriptyline and nortriptyline bound extensively to the microsomal membrane. The dependence of nortriptyline fu(mic) on drug and microsomal protein concentration is shown in Figure 1. The fraction unbound increased as the drug concentration increased and as the microsomal protein concentration decreased. The results were similar for amitriptyline.
Table 2.
Physicochemical characteristics and nonspecific binding. The concentration range is that used in the dialysis experiments. Log D7.4 was determined as described in the Methods section. The concentration of microsomal protein was 1 mg ml−1.
| Compound | log D7.4 | pKa | % ionization (pH 7.4) | Reported mean Km (µm) [reference] | Concentration range (µm) | Observed fu(mic) |
|---|---|---|---|---|---|---|
| Acids | ||||||
| Caffeine | 0.3 | 13.9 | 0.0 | 180a [14] | 20,250 | 0.96,1.1 |
| S-Naproxen | −0.7 | 4.2 | 100.0 | 143b [18] | 20,500 | 0.99,1.0 |
| Phenytoin | 2.1 | 8.3 | 11.1 | 30 [20] | 20,200 | 0.85,0.89 |
| Tolbutamide | 0.5 | 5.3 | 99.1 | 120 [19] | 20,500 | 0.97,1.1 |
| Bases | ||||||
| Amiodarone | > 7.0 | 6.6 | 12.5 | 310 [17] | 100 | < 0.01 |
| Amitriptyline | 1.6 | 9.4 | 99.0 | 67 [15] | 20–1000 | 0.35–0.73 |
| Nortriptyline | 1.1 | 9.7 | 99.3 | 21 [16] | 20–1000 | 0.35–0.70 |
High affinity component of caffeine N3-demethylation
O-Demethylation pathway.
Figure 1.

Nonspecific binding of nortriptyline to human liver microsomes. a) Dependence of fu(mic) on nortriptyline concentration at 1 mg ml−1 microsomal protein. b). Dependence of fu(mic) on microsomal protein concentration at 100 µm nortriptyline. Each point is the mean ± s.d. of triplicate determinations.
Saturable binding of amitriptyline and nortriptyline
The increase in free fractions of amitriptyline and nortriptyline with increase in total drug concentration indicated saturable binding. Figure 2 shows a standard binding plot and Scatchard plot for nortriptyline. The Scatchard plot is linear indicating saturable binding with a single binding component. Initial estimates of the maximum binding capacity, Bmax, and the dissociation constant, KD, were calculated from the Scatchard plot, and these values were then used in MK model to calculate final values by nonlinear least squares regression analysis. Binding parameters for nortriptyline and amitriptyline at varying microsomal protein concentrations are shown in Table 3. The binding capacity increased in an almost linear fashion as the microsomal protein concentration increased, whereas the Kd remained approximately constant. The binding parameters for amitriptyline and nortriptyline were similar, as would be expected from the similar physicochemical characteristics of the two drugs.
Figure 2.

Binding of nortriptyline to human liver microsomes 1 mg ml−1. a) Binding plot according to equation 1. b) Scatchard plot.
Table 3.
Nonspecific binding parameters for nortriptyline and amitriptyline at varying microsome concentrations.
| Microsomal protein (mg ml−1) | Bmax (µm) | KD1 (µm) |
|---|---|---|
| Nortriptyline | ||
| 0.5 | 220 ± 32 | 151 ± 42 |
| 1.0 | 382 ± 54 | 147 ± 44 |
| 2.0 | 707 ± 54 | 111 ± 3 |
| Amitriptyline | ||
| 1.0 | 375 ± 23 | 178 ± 33 |
Values are mean ± s.d. of three determinations.
Simulations of effects of nonspecific binding on in vitro enzyme kinetics
If the drug concentration range used in an in vitro determination of drug metabolism kinetics is similar to or above the KD for the nonspecific binding of the drug to the microsomal membrane, the fu(mic) will vary with the substrate concentration. A simulation of the impact of saturable nonspecific binding on reaction kinetics is shown in Figures 3 and 4. Figure 3 shows the effect of varying the binding KD whilst keeping the Km, Vmax, and Bmax constant. As the KD decreases relative to the Km for the reaction, the reaction velocity vs substrate concentration curve becomes increasingly sigmoidal. In the extreme case (a KD of 1 µm vs a Km of 200 µm) the predicted reaction velocity (metabolite formation) is negligible until saturation of nonspecific binding occurs at an added substrate concentration about that of the Bmax. Also shown are the Eadie-Hofstee plots, which become increasingly curvilinear as the ratio of KD to Km decreases.
Figure 3.

Simulation showing the effect of nonspecific binding with varying KD on Michaelis-Menten kinetics (a), and on an Eadie Hofstee plot (b). The plots use total (added) substrate concentration, whereas the reaction velocities were calculated using free (unbound) substrate concentrations.
Figure 4.

Simulation showing the effect of varying microsomal protein concentration on Michaelis-Menten kinetics (a), and on an Eadie Hofstee plot (b). Bmax was assumed to be proportional to microsomal protein concentration. Substrate concentration ranges used in kinetic studies are commonly 0.2 Km −3 Km.
A simulation of the effect of varying protein concentration whilst keeping Km, KD, Vmax, and Bmax constant, with KD approximately equal to Km, is illustrated in Figure 4. The Michaelis-Menten curve shifts to the right and becomes sigmoidal as the protein concentration and the degree of nonspecific binding increases. The Eadie-Hofstee plot shifts to the left and becomes nonlinear.
Discussion
The nonspecific binding of seven drugs with differing pharmacokinetics and physicochemical characteristics have been analysed in this study. Of the seven drugs, amiodarone, amitriptyline and nortriptyline showed marked nonspecific binding to microsomes. All three are lipophilic weak bases. Phenytoin, a lipophilic weak acid with a high pKa so that it is mostly unionized at pH 7.4, bound to microsomes to a minor extent. Tolbutamide and naproxen, which are less lipophilic and have lower pKa values than phenytoin, did not show measurable nonspecific binding. Caffeine also was not bound. These binding characteristics are as would be expected from the net negative charge on the microsomal membrane. The fu(mic) measured for phenytoin here was 0.88 and was independent of drug concentration. This is consistent with that found by Ludden et al. [21] who reported a fraction unbound of 0.77 using 1 mg ml−1 human liver microsomes and Tris buffer. Carlile et al. [22] have, however, recently reported a phenytoin fraction unbound of 0.36 with 1 mg ml−1 human liver microsomes and phosphate buffer. The reason for this discrepancy is unclear except that three different methods were used in the three studies (dialysis – this study; ultracentrifugation [21]; and microfiltration [22]). Warfarin which, like phenytoin, is a weak acid with a high pKa has been reported to have an fu(mic) of 0.85 at a microsomal protein concentration of 1.0 mg ml−1 and a warfarin concentration of 10 µm.
Amiodarone, which is extremely lipophilic, was bound so extensively that the free concentration, although detectable, was below the limit of quantification for the assay, but the unbound fraction was approximately 0.01 or less (at 100 µm amiodarone and 1 mg microsomal protein ml−1). The very extensive binding of amiodarone observed here is in agreement with the high nonspecific binding (fu(mic) < 0.07) of another highly lipophilic weak base, felodipine, reported previously [23].
The free fractions of the tricyclic antidepressants, amitriptyline and nortriptyline, were dependent on the microsome concentration (Figure 1). Obach [7] similarly showed that the binding of imipramine was dependent on the microsome concentration. At a microsomal protein concentration of 1 mg ml−1 and an imipramine concentration of 10 µm, the reported fu(mic) was 0.16. The corresponding values which we have found for amitriptyline and nortriptyline at 20 µm drug concentration were 0.35 and 0.33, respectively. Obach [7] examined the drug concentration dependence of imipramine binding over the range 1–100 µm and found fu(mic) increased from 0.14 to 0.22 over this range. We have examined higher concentrations of nortriptyline and amitriptyline in the present study and have found a much greater degree of variation in fu(mic) over the wider drug concentration range used.
Where drugs are subject to nonspecific membrane binding in a microsomal incubation, the free and available substrate concentration will be less than the added concentration. The apparent Km determined on the basis of the added concentration will then be higher than the ‘true’ Km, although the Vmax should not be affected. This will result in a falsely low value for the in vitro intrinsic clearance (Vmax/Km), and therefore for the predicted in vivo clearance. Two groups have suggested various corrections for nonspecific binding during the extrapolation to in vivo clearance [7, 8, 24, 25].
von Moltke et al. [24, 25] used mouse liver homogenate to determine a liver : water partition coefficient which was then used to correct for in vitro binding. However, the free drug concentration at the liver enzyme site in vivo is determined theoretically by the free drug concentration in the blood. Drug binding in the liver cell is a determinant of the overall liver:blood partition ratio, but not of the free drug concentration in the cytosol. Drug binding in vivo is accounted for in the clearance model as the fraction of drug unbound in plasma, fu. The in vitro correlate of free drug in the hepatocyte cytosol is the free drug concentration in the actual microsomal incubation used for the in vitro kinetic studies.
A nonspecific binding model
The nonspecific binding of amitriptyline and nortriptyline observed in the current study was saturable, and the data could be fitted to a standard binding model to determine the maximum binding capacity Bmax and the dissociation constant KD. This saturable binding behaviour is likely to apply at least to other lipophilic bases. The Bmax and KD values for nortriptyline were determined at varying microsome concentrations (Table 4). As might be expected, Bmax varied approximately proportionately with microsome concentration, whereas the KD remained constant.
Two situations may then occur in relation to correction of the Km to account for nonspecific binding. These are illustrated in Figure 5.
Figure 5.

Relationship between Km and KD. At the substrate concentration range used in kinetic studies when Km ≪ KD, the fu(mic) does not vary with substrate concentration (insert). When Km ≥ KD, the fu(mic) varies with substrate concentration over the range used in the kinetic study.
The first is where the Km, and therefore the substrate concentration range used in an in vitro kinetic study, is much less than the KD for nonspecific binding of the substrate. In this situation, fu(mic) is independent of the substrate concentration (Figure 5 insert), but will still depend on the microsomal protein concentration. The apparent Km (Km(app)) can then be corrected to the ‘true’ Km by multiplying by the fraction of unbound drug at the microsomal protein concentration used in the in vitro study.
| (4) |
This is analogous to linear pharmacokinetics where the drug concentration is much lower than the Km for a saturable elimination process such as metabolism or secretion. It is also analogous to linear plasma protein binding when the plasma drug concentration is much less than the concentration of protein binding sites. The fraction unbound then does not vary with drug concentration but does vary with the concentration of the binding proteins.
The second situation is where the substrate concentration range used in an in vitro study (determined by the apparent Km) is similar to or higher than the KD for nonspecific binding. The fraction of substrate unbound in the incubation mixture will then vary across the substrate concentration range used (Figure 5). This precludes a simple proportional correction of apparent Km for nonspecific substrate binding. This is analogous to zero order kinetics in vivo, or nonlinear plasma protein binding when the plasma drug concentration is similar to or higher than the concentration of the binding protein. In this situation, fu(mic) varies with both substrate and microsome concentrations, and complex effects on the in vitro kinetics are predicted. In both cases, sigmoidal kinetics and curvi-linear Eadie Hofstee plots result (see Figures 3 and 4). The sigmoidicity becomes greater as the Km (and therefore the substrate concentration range used in the in vitro kinetic study) increases relative to the KD for nonspecific binding.
Sigmoidal in vitro kinetics have been reported, particularly with CYP3A4 [15, 26–28]. This has been interpreted as autoactivation due to substrate binding at more than one site or to the simultaneous binding of two molecules of substrate at the active site [28, 29]. CYP3A4 is also activated by a number of flavonoids and steroids (heterotropic activators) which reduce the degree of cooperativity and at least partly restore hyperbolic kinetics [26, 28]. With CYP3A4, these effects can be substrate regioselective [30], and altered by changes in critical amino acids [31]. Recently, nonhyperbolic kinetics have been reported with a range of other CYP isoforms. These include: CYP1A2 [32]; CYP2B6 and CYP2E1 [33]; CYP2B6, CYP2C8, CYP2C9 and CYP3A5 [29]; and CYP1A2 and CYP2C9 [34].
We have shown in the current study that sigmoidal kinetics can be predicted to occur in some circumstances as a result of nonspecific binding of the substrate in the microsomal membrane. It is therefore possible that at least some cases of apparent substrate autoactivation are due to nonspecific substrate binding. We are currently exploring this possibility.
The nonspecific binding observed in this study of the weak bases nortriptyline, amitriptyline and amiodarone would increase the in vivo metabolic clearance predicted from in vitro data. This would tend to bring predicted and actual clearances closer, particularly for amiodarone where the correction factor is of the order of 100-fold or more. By contrast, the minor or absent nonspecific binding with the weak acids used in this study indicates that corrections for nonspecific binding will not need to be made for drugs with these physicochemical characteristics.
Acknowledgments
This work was supported by grant 981248 from the National Health and Medical Research Council of Australia.
References
- 1.Birkett DJ, Mackenzie PI, Veronese ME, Miners JO. In vitro approaches can predict human drug metabolism. Trends Pharmacol Sci. 1993;14:292–294. doi: 10.1016/0165-6147(93)90043-j. [DOI] [PubMed] [Google Scholar]
- 2.Miners JO, Veronese ME, Birkett DJ. In vitro approaches for the prediction of human drug metabolism. Ann Reports Med Chem. 1994;29:307–315. [Google Scholar]
- 3.Lin JH, Lu AYH. Inhibition and induction of cytochrome P450 and the clinical implications. Clin Pharmacokin. 1998;35:361–390. doi: 10.2165/00003088-199835050-00003. [DOI] [PubMed] [Google Scholar]
- 4.Houston BJ. Utility of in vitro drug metabolism data in predicting in vivo metabolic clearance. Biochem Pharmacol. 1994;49:1469–1479. doi: 10.1016/0006-2952(94)90520-7. [DOI] [PubMed] [Google Scholar]
- 5.Iwatsubo T, Hirota N, Ooie T, et al. Prediction of in vivo drug disposition from in vitro data based on physiological pharmacokinetics. Biopharm Drug Dispos. 1996;17:273–310. doi: 10.1002/(SICI)1099-081X(199605)17:4<273::AID-BDD961>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 6.Tucker GT. The rational selection of drug interaction studies: implications of recent advances in drug metabolism. Int J Clin Pharm Ther Toxicol. 1992;11:550–553. [PubMed] [Google Scholar]
- 7.Obach RS. Nonspecific binding to microsomes: impact on scale-up of in vitro intrinsic clearance to hepatic clearance as assessed through examination of warfarin, imipramine and propranolol. Drug Metab Dispos. 1997;25:1359–1369. [PubMed] [Google Scholar]
- 8.Obach RS. The importance of nonspecific binding in in vitro matrices, its impact on enzyme kinetic studies of drug metabolism reactions, and implications for in vitro-in vivo correlations. Drug Metab Dispos. 1996;24:1047–1049. [PubMed] [Google Scholar]
- 9.Chiba M, Fujita S, Suzuki T. Pharmacokinetic correlation between in vitro hepatic microsomal enzyme kinetics and in vivo metabolism of imipramine and desipramine in rats. J Pharm Sci. 1990;79:281–287. doi: 10.1002/jps.2600790402. [DOI] [PubMed] [Google Scholar]
- 10.Robson RA, Matthews AP, Miners JO, et al. Characterisation of theophylline metabolism in human liver microsomes. Br J Clin Pharmacol. 1987;24:293–300. doi: 10.1111/j.1365-2125.1987.tb03172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lowry OH, Rosenberg NJ, Farr AL, Randall RJ. Protein measurement with the Folin Phenol Reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 12.The Pharmaceutical Codex. 11th. London: The Pharmaceutical Press; Anonymous; pp. 646–648. [Google Scholar]
- 13.Holford NHG. MK Model. A tool for microcomputers. Pharmacokinetic evalution and comparison with standard computer programs. Clin Exp Pharmacol Physiol. 1985;9:95. [Google Scholar]
- 14.Tassaneeyakul W, Mohamed Z, Birkett DJ, et al. Caffeine as a probe for human cytochromes P450: validation using cDNA expression, immunoinhibition and microsomal kinetic and inhibitor techniques. Pharmacogenetics. 1992;2:173–183. doi: 10.1097/00008571-199208000-00004. [DOI] [PubMed] [Google Scholar]
- 15.Schmider J, Greenblatt DJ, Harmatz JS, von Moltke LL, Shader RI. N-demethylation of amitriptyline in vitro: Role of cytochrome P450, 3A (CYP3A) isoforms and effect of metabolic inhibitors. J Pharmacol Exp Ther. 1995. pp. 592–597. [PubMed]
- 16.Mellström B, Bertilsson C, Birgersson M, Göransson M, von Bahr C. E- and Z-10-hydroxylation of nortriptyline by human liver microsomes – methods and characterization. Drug Metab Dispos. 1983;11:115–119. [PubMed] [Google Scholar]
- 17.Fabre F, Julian B, Saint-aubert B, Joyeux H, Berger Y. Evidence for CYP3A-mediated N-deethylation of amiodarone in human liver microsomal fractions. Drug Metab Dispos. 1993;21:978–985. [PubMed] [Google Scholar]
- 18.Miners JO, Coulter S, Tukey RH, Veronese ME, Birkett DJ. Cytochromes P450 1A2 and 2C9 are responsible for the hepatic O-demethylation of R- and S-naproxen. Biochem Pharmacol. 1996;51:1003–1008. doi: 10.1016/0006-2952(96)85085-4. [DOI] [PubMed] [Google Scholar]
- 19.Miners JO, Smith KJ, Robson RA, McManus ME, Veronese ME, Birkett DJ. Tolbutamide hydroxylation by human liver microsomes. Kinetic characterisation and relationship to other cytochrome P-450 dependent xenobiotic oxidations. Biochem Pharmacol. 1988;37:1137–1144. doi: 10.1016/0006-2952(88)90522-9. [DOI] [PubMed] [Google Scholar]
- 20.Doecke CJ, Veronese ME, Pond SM, et al. Relationship between phenytoin and tolbutamide hydroxylations in human liver microsomes. Br J Clin Pharmacol. 1991;31:125–130. doi: 10.1111/j.1365-2125.1991.tb05499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ludden IK, Ludden TM, Collins JM, Pentikis HS, Strong JM. Effect of albumin on the estimation, in vitro, of phenytoin Vmax and Km values: implications for clinical correlation. J Pharmacol Exp Ther. 1997;282:391–396. [PubMed] [Google Scholar]
- 22.Carlile DJ, Hakooz N, Bayliss MK, Houston JB. Microsomal prediction of in vivo clearance of CYP2C9 substrates in humans. Br J Clin Pharmacol. 1999;47:625–635. doi: 10.1046/j.1365-2125.1999.00935.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baarnhielm C, Dahlback H, Skanberg I. In vivo pharmacokinetics of felodipine predicted from in vitro studies in rat, dog and man. Acta Pharmacol Toxicol. 1986;59:113–122. doi: 10.1111/j.1600-0773.1986.tb00142.x. [DOI] [PubMed] [Google Scholar]
- 24.von Moltke LL, Greenblatt DJ, Cotreau-bibbo MM, Duan SX, Harmatz JS, Shader RI. Inhibition of desipramine hydroxylation in vitro by serotonin-reuptake-inhibitor antidepressants, and by quinidine and ketoconazole: a model system to predict drug interactions in vivo. J Pharmacol Exp Ther. 1993;268:1278–1283. [PubMed] [Google Scholar]
- 25.von Moltke LL, Greenblatt DJ, Court MH, Duan SX, Harmatz JS, Shader RI. Inhibition of alprazolam and desipramine hydroxylation in vitro by paroxetine and fluvoxamine: comparison with other selective serotonin reuptake inhibitor antidepressants. J Clin Psychopharmacol. 1995;15:125–131. doi: 10.1097/00004714-199504000-00008. [DOI] [PubMed] [Google Scholar]
- 26.Andersson T, Miners JO, Veronese ME, Birkett DJ. Diazepam metabolism by human liver microsomes is mediated by S-mephenytoin hydroxylase and CYP3A isoforms. Br J Clin Pharmacol. 1994;38:131–137. doi: 10.1111/j.1365-2125.1994.tb04336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schwab GE, Raucy JL, Johnson EF. Modulation of rabbit and human hepatic cytochrome P-450-catalyzed steroid hydroxylations by α-naphthoflavone. Mol Pharmacol. 1988;33:493–499. [PubMed] [Google Scholar]
- 28.Ueng YF, Kuwabara T, Chun YJ, Guengerich FP. Cooperativity in oxidations catalyzed by cytochrome P450 3A4. Biochemistry. 1997;36:370–381. doi: 10.1021/bi962359z. [DOI] [PubMed] [Google Scholar]
- 29.Korzekwa KR, Krishnamachary N, Shou M, et al. Evaluation of atypical cytochrome P450 kinetics with two-substrate models: evidence that multiple substrates can simultaneously bind to cytochrome P450 active sites. Biochemistry. 1998;37:4137–4147. doi: 10.1021/bi9715627. [DOI] [PubMed] [Google Scholar]
- 30.Mäenpää J, Hall SD, Ring BJ, Strom SC, Wrighton SA. Human cytochrome P4503A (CYP3A) mediated midazolam metabolism – the effect of assay conditions and regioselective stimulation by α–naphthoflavone, terfenadine and testosterone. Pharmacogenetics. 1998;8:137–155. [PubMed] [Google Scholar]
- 31.Domanski TL, Liu J, Harlow GR, Halpert JR. Analysis of four residues within substrate recognition site 4 of human cytochrome P450 3A4: role in steroid hydroxylase activity and α-naphthoflavone stimulation. Arch Biochem Biophys. 1998;350:223–232. doi: 10.1006/abbi.1997.0525. [DOI] [PubMed] [Google Scholar]
- 32.Ekins S, Ring BJ, Binkley SN, Hall SD, Wrighton SA. Autoactivation and activation of the cytochrome P450s. Int J Clin Pharmacol Ther. 1998;36:642–651. [PubMed] [Google Scholar]
- 33.Ekins S, vandenBranden M, Ring SJ, Wrighton SA. Examination of purported probes of human CYP2B6. Pharmacogenetics. 1997;7:165–179. doi: 10.1097/00008571-199706000-00001. [DOI] [PubMed] [Google Scholar]
- 34.Venkatakrishnan K, Greenblatt DJ, von Moltke LL, Schmider J, Harmatz JS, Shader RI. Five distinct human cytochromes mediate amitriptyline N-demethylation in vitro: dominance of CYP 2C19 and 3A4. J Clin Pharmacol. 1998;38:112–121. doi: 10.1002/j.1552-4604.1998.tb04399.x. [DOI] [PubMed] [Google Scholar]