

Abstract
Aims
Inflammation reduces hepatic clearance of many drugs with unknown therapeutic consequences. This study was carried out to examine the effect of rheumatoid arthritis (RA) on the pharmacokinetics and pharmacodynamics of verapamil.
Methods
Eight RA patients were age- and sex-matched with eight healthy volunteers. The disease severity was assessed, and ECG, blood pressure and verapamil enantiomers concentrations were measured for 12 h post 80 mg oral verapamil. Serum interleukin-6 (IL-6) and nitrite (NO2−) were measured in predose samples.
Results
IL-6 and NO2− concentrations were significantly increased in parallel with disease severity. Oral clearance of both S- and R-verapamil was significantly decreased by RA. While the unbound fraction of S- and R-verapamil decreased by 5 and 7-fold, respectively, the unbound AUC remained unchanged for the more potent enantiomer, S-verapamil. AUC of norverapamil enantiomers was increased 2–3-fold. Despite elevated serum drug concentrations in RA, the potential to prolong the PR-interval was significantly reduced by one fold and the effect on the heart rate and blood pressure did not increase.
Conclusions
RA results in increased verapamil concentrations due likely to changes in protein binding, decreased clearance and/or altered hepatic blood flow. A significant decrease in dromotropic effect, despite increased serum drug concentrations, may be attributed to receptor down regulation caused by pro-inflammatory cytokines and/or NO.
Keywords: dromotropic response, rheumatoid arthritis, verapamil
Introduction
Inflammatory conditions such as rheumatoid arthritis (RA) and Crohn's disease have been shown to reduce hepatic clearance of several highly cleared drugs [1–5]. The therapeutic consequence of the effect is unknown. For drugs intended to treat inflammation, it is suggested that patients with RA are more predisposed to adverse drug reactions than the general population [6, 7] e.g. a higher frequency of allergic reactions to d-penicillamine [8, 9]. Since drugs used to treat inflammatory conditions typically exhibit a low hepatic extraction ratio, an increased reaction is unlikely to be due to altered pharmacokinetics induced by inflammation. On the other hand, the pharmacokinetics of drugs intended for the treatment of other conditions may be altered in patients with inflammatory disease. For example, clearance of many cardiovascular drugs is highly dependent on hepatic function [1–4]. This is important since it is estimated that about 40% of patients with rheumatic diseases take antihypertensive drugs [10, 11]. Very recently, inflammation has been suggested as the main determinant outcome of therapy failure in patients with unstable angina [12]. Moreover, the possibility of multiple diseases increases with ageing population.
Inflammation causes increased plasma concentration of α1-acid glycoprotein (AAG), thereby increasing protein binding and reducing unbound fraction. This, in turn, may result in reduced clearance of the total drug due to a diminished concentration of the unbound portion [13]. Both latter changes have been suggested as the underlying causes of reduced clearance of many drugs [1–5].
Verapamil, a phenylalkylamine calcium channel blocker, is widely used in the treatment of hypertension. It is bound to plasma proteins including AAG and is extensively metabolized upon the first-pass through the liver [14–16]. Following therapeutic doses, verapamil causes negative dromotropic effects reflected as a plasma concentration-dependent prolongation of PR interval and AV node block [17]. The PR interval prolongation is conveniently detectable even after small single doses [18].
The purpose of this study was to determine if rheumatoid arthritis influences the pharmacokinetics of verapamil, and whether this brings about changes in pharmacodynamics of the drug.
Methods
Materials
Rac-verapamil hydrochloride and norverapamil were gifts from G.D. Searle (Skokie, Ill). (+)-Glaucine and heptafluorobutanol were purchased from Aldrich (Milwaukee, WI). H.p.l.c. grade hexane (Caledon Laboratories, Georgetown, Canada), and propan-2-ol (BDH Inc., Toronto, Canada) and heptane (Mallinckrodt, Paris, KT), and 98% anhydrous ethanol (Stanley, Vancouver, Canada) were used. Asperigillus nitrate reductase 10 U ml−1, 0.1 m FAD, 1 mm NADPH, 1500 U ml−1 LDH, 100 mm pyruvic acid and triethylamine (TEA) were purchased from Sigma Chemical Co. (St Louis, MO).
Subjects and study protocol
The study was performed in accordance with the declaration of Helsinki. The protocol was approved by the University of Alberta Hospital Research and Ethics Committee. All participants provided written informed consent. Eight RA patients with no other diseases were age-and sex-matched with eight healthy volunteers (Table 1). They had maintained the same therapeutic regimen for at least 4 months. To avoid pharmacodynamic interactions [10, 11 19], patients under treatment with nonsteroidal antinflammatory drugs were excluded. All patients underwent physical and laboratory examination. A small but statistically significant difference in weight was observed between the two groups. They were no signs of hepatic or renal disease. The subjects fasted on the evening prior to the study. On the study day, they reported to the clinical investigation unit at 07.30 h. They were weighed and temperature recorded. An intravenous line was established in the antecubital vein for serial blood sampling. Surface electrodes were placed to record a standard lead I and aV5 ECG. The patients were then monitored for a minimum of 1 h or until a stable baseline was established for heart rate, PR interval and blood pressure. At time 0 h, 80 mg of rac-verapamil (Isoptin®, lot: 6C384, Searle Oakville, Canada) was administered with 200 ml water. Serial blood samples were drawn at 0, 20 and 40 min, and 1, 1.5, 3, 4, 6, 8 and 12 h and serum separated. Blood pressure and ECG measurements were recorded 1 min prior to each blood sample. Subjects ate a standard breakfast and lunch after the 1.5 and 6 h samples, respectively. Serum nitrite and IL-6 concentrations were measured in the time zero blood samples.
Table 1.
Patients characteristics.
| Parameter | Control | RA |
|---|---|---|
| Sex, Male/Female | 5/3 | 5/3 |
| Age (years) | 43.0 ± 5.1 | 43.0 ± 5.1 |
| Height (cm) | 174 ± 1.4 | 174 ± 1.7 |
| Weight (kg) | 76.7 ± 2.9 | 63.5 ± 3.4* |
| Number of joints involved | 0 | 16.3 ± 8.0* |
| Arthritic index | 0 | 3.9 ± 2.0* |
| Other medications | 1 on paracetamol prn | 2 on hydroxychloroquine |
| 1 on loratidine prn | 4 on methotrexate | |
| 1 on sulphasalazine | ||
| 1 on cyclosporin |
Significantly different from controls.
Disease severity (Table 1) was clinically assessed according to the American Rheumatism Association 1987 revised criteria [20]. An arthritic index was calculated using the number of joints involved and the severity as follows: 0, no joints involved; 1, 1–4 joints; 2, 5–9 joints; 3, > 10 joints involved. Swelling was assessed as 0: none; 1, mild; 2, moderate; 3, severe. Thus a maximum score of 6 would indicate severe disease.
Assay of verapamil and norverapamil
A previously published stereospecific assay was used [21] Briefly, to 100 μl of serum in a glass test tube were added 75 μl of 400 ng ml−1 (+)glaucine (internal standard), 100 μl 2m NaOH, 0.4 ml sodium phosphate buffer (pH 7.0, ionic strength 0.1), and 6 ml heptane. The sample was vortexed for 1 min and centrifuged at 2000 g for 10 min. The organic layer was transferred to a glass tube and evaporated to dryness in a vacuum centrifuge at 60°C. The resulting residue was reconstituted in 200 μl of mobile phase (hexane-isopropanol-ethanol-TEA, 85:7.5:7.5:1.0, v/v), and 100 μl injected into the h.plc.
A Waters (Millipore-Waters, Missassuaga, Canada) h.p.l.c. apparatus was used consisting of a twin piston pump, a WISP 710B autosampler, a column oven (31°C) and a 470 fluorescence detector set at excitation of 272 emission of 317 nm and bandwidth at 18 nm. The integrator was a Hewlett-Packard (Avondale,PA) 3390 A model. An achiral column (5 cm × 4.6 mm ID Supelcosil® LCSi column, Supelco Inc., Bellefonte PA) was serially attached to a chiral column (250 mm × 4.6 mm i.d. 10 μm Chiralpak AD, Daicel Chemical Ind., Tokyo, Japan). Standard curves were linear over a 2.5–200 ng ml−1 range. Sensitivity for R and S-verapamil was 2.5 ng ml−1 (CV <5%) and 7.5 ng ml−1 for R and S-norverapamil (CV <13%). The intra-and interday precisions were greater than 90%.
Electrocardiogram and haemodynamic analysis
A standard lead I and aV5 electrocardiogram was recorded using a Holter monitor (Hewlett-Packard, Avondale, PA) with full disclosure. Blood pressure was determined using a Dynamap (IVAC Instruments, Toronto, Canada) automated cuff syphgmomanometer. Mean arterial pressure (MAP) was calculated from
 |
where SBP and DBP were systolic and diastolic blood pressure, respectively. Subjects were in the supine state for 10 min prior and during ECG and blood pressure measurement. The mean of five PR intervals and heart rate measurements recorded during the minute before blood samples collection for the pharmacokinetic analysis was calculated.
Serum nitrite analysis
Serum nitrite (NO2−1) was measured using a previously published method [22]. Briefly, 100 μl of serum was incubated with 10 Units of Asperigillus nitrate reductase in the presence of 0.1 M flavine adenine dinucleotide, 1 mm nicotinamide adenine dinucleotide phosphate dehydro-genase to reduce all nitrate (NO3) to nitrite (NO2−). The reaction was quenched with 1500 U lactate dehydrogen-ase and 100 mm pyruvic acid. This was then treated with the Griess reagent, an equal mixture of 0.2% naphthyle-nethylendiene and 2% sulphanilamide in 5% ortho-phosphoric acid. Absorbance was measured at 540 nm using a Vmax microtitre plate reader (Molecular Devices Corp., Fisher Scientific, Edmonton, Canada). Calibration was performed using standard solutions of NaNO2 and NaNO3 to evaluate the dehydrogenase efficiency. The assay was linear within the examined range of 5–200 μm (CV <5%). None of the drugs used by our subjects (Table 1) including verapamil interfered with the assay. Due to an inadvertent destruction of samples, data from six controls and six RA patients were generated.
Interleukin 6 assay
Serum interleukin (IL)-6 concentrations were measured by ELISA (Medgenix Diagnostics, Fleurus, Belgium). This allowed for the quantification of IL-6–2.0 pg ml− (CV <8%). Due to an accidental destruction of samples, data from four controls and four RA patients were generated.
Protein binding
Serum for the protein binding study was pooled from the time zero h blood sample of both groups (n = 8/group). The pooled serum was adjusted to pH 7.4 with 0.1 m HCl. The serum was then fortified with 100 ng ml−1 of S-verapamil and 200 ng ml−1 R-verapamil to approximate in vivo drug concentrations. The sample was incubated at 37°C for 1 h then transferred to microparti-tion chambers (Amicon Division of W.R. Grace & Co, Danvers MA) for ultrafiltration. The chambers were centrifuged at 2000 g for 1 h. In addition, four chambers were loaded with phosphate buffer, pH 7.4, to determine the presence of any nonspecific binding or adsorption to the micropartition system. Both filtrate and nonfiltrate concentrations were measured and the fraction unbound (fu) determined as the unbound concentration divided by total concentration. To ensure concentrations were above the minimum quantifiable limit for the h.p.l.c. assay, 3 micropartitions of the 12 chambers were pooled allowing for a total of 4 measurements per group.
Treatment of data
Model independent analysis of serum verapamil enantio-mers was performed. The terminal elimination rate constant (λz) was calculated using a nonweighted nonlinear least-squares regression. AUC(0,∞) was calculated using the linear trapezoidal rule until the last experimental data point (Clast) plus Clast/λz. AUC of the unbound fraction was calculated from AUCu(0,∞) = AUC(0,∞) × fu where fu is the fraction unbound and it is assumed that verapamil protein binding is concentration independent [23]. Oral clearance (CL/F) was estimated for each patient from
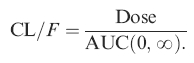 |
Percent changes in pharmacodynamic responses were calculated from the differences observed between the baseline and pos-treatment values. The area under the percent effect-time curve was calculated using the trapezoidal rule.
The unpaired and paired Student's t-test was used to compare the two groups and between the enantiomers, respectively. To test the significance of correlation between parameters, a least-squares linear regression was performed using a Pearson's r to determine goodness of fit. A multivariate discriminate analysis was used to assess serum IL-6 and NO2− as indicators of disease severity. The relative importance of each independent variable within the discriminant function was evaluated from the correlation between the independent variable and the canonical function [24].
Statistical significance was set at P < 0.05. The data are expressed as the mean ± s.e.mean.
Results
Serum IL-6 and NO2− were significantly elevated in the RA group in comparison with the control subjects (Figure 1). In addition, serum nitrite (r = 0.95, P < 0.00001, n = 12, Figure 2), IL-6 (r = 0.56, P < 0.05, n = 8) and S-verapamil AUC (r = −0.46, P < 0.05, n = 16). correlated with disease severity. Using a discriminant analysis, a subject could be correctly identified as control or rheumatoid 100% of the time based on the serum nitrite concentration (P < 0.04).
Figure 1.

Mean serum nitrite (NO2) (n = 6) and interleukin-6 (IL-6) (n = 4) concentrations just before oral administration of a single dose of 80 mg verapamil to control and RA patients. Error bars represent standard error of the mean. *Significantly different from control.
Figure 2.

Serum NO2 vs arthritic index.
RA caused a significant and substantial rise in serum concentrations of both enantiomers (Figure 3) as reflected in an approximately four and three fold increase in AUC(0,∞) of S-and R-verapamil, respectively (Table 2). Greater variability in serum verapamil concentrations was noticed in RA patients when compared with the control group. A similar numerical increase was observed for Cmax that reached significance for R-verapamil but not for S-verapamil. Terminal elimination half-life, was not significantly altered for either enantiomer. RA resulted in 6 and 5 fold decreased protein binding for R-and S-verapamil, respectively. However, calculation of AUCu(0,∞) (μg ml− min) revealed that RA significantly decreased R-verapamil but did not affect S-verapamil (Table 2).
Figure 3.

Serum verapamil enantiomers concentration-time profile in healthy control subjects (^) and patients with rheumatoid arthritis (•) after receiving a single 80 mg oral dose of verapamil., Error bars represent s.e. mean (n = 8/group).
Table 2.
Effect of rheumatoid arthritis (RA) on the pharmacokinetics of verapamil and norverapamil (NOR).
| Parameter | Control (Mean ± s.e.mean) | RA (Mean ± s.e.mean)) | ||
|---|---|---|---|---|
| tmax (h) | S | 1 (1–2.5) | 1 (1–3) | |
| 95% CI | −2.8 ∼ 2.8 | |||
| R | 1 (1,2.5) | 1 (1,2.5) | ||
| 95% CI | −3.0,−2.6 | |||
| Cmax (ng ml−1) | S | 29.2 ± 4.1 | 95.3 ± 41.1 | |
| 95% CI | −78.8,−53.4 | |||
| R | 144 ± 27 | 528 ± 27* | ||
| 95% CI | −410,−359 | |||
| t½ (h−1) | S | 4.1 ± 1.2 | 8.5 ± 0.7 | |
| 95% CI | −9.7,0.94 | |||
| R | 4.9 ± 0.7 | 3.6 ± 0.7 | ||
| 95% CI | −2.8,5.5 | |||
| AUC(0,∞) (μg ml−1 min) | S | 8.1 ± 1.2 | 33.6 ± 6.8* | |
| 95% CI | −31.7,−19.3 | |||
| R | 39.1±4.6 | 125 ± 34* | ||
| 95% CI | −98.6,−73.2 | |||
| Fraction unbound | S | 0.138 ± 0.007 | 0.028 ± 0.005* | |
| 95% CI | 0.061,0.16 | |||
| R | 0.073 ± 0.0008 | 0.011 ± 0.0008* | ||
| 95% CI | 0.018,0.107 | |||
| AUC(0,∞) R/S ratio | 5.6 ± 0.8 | 5.1±0.9 | ||
| 95% CI | −3.0,4.0 | |||
| AUC(0,∞) unbound (μg. ml−1 h) | S | 1.2 ± 0.12 | 1.1±0.61 | |
| 95% CI | −1.4,1.3 | |||
| R | 1.2 ± 0.6 | 2.7 ± 0.30* | ||
| 95% CI | −2.1,−1.2 | |||
| CL/F (1 h−1) | S | 361±69 | 162 ± 18* | |
| 95% CI | 159,239 | |||
| R | 67 ± 6.9 | 43 ± 8.7* | ||
| 95% CI | 10.9,37.1 | |||
| NOR AUC(0,12 h) (μg .ml−1 min) | S | 30.8 ± 12.5 | 68.7 ± 9.8* | |
| 95% CI | −55.2,−20.1 | |||
| R | 669 ± 121 | 1893 ± 254* | ||
| 95% CI | 1280,1168 | |||
| NOR/verapamil AUC(0,12 h) ratio | S | 4.5 ± 1.9 | 4.4 ± 1.3 | |
| 95% CI | −6.7,6.8 | |||
| R | 2.3 ± 0.3 | 2.2 ± 0.6 | ||
| 95% CI | 2.7,2.9 |
Significantly different from control; 95% CI, confidence interval of individual differences between control and rheumatoid arthritis; Data are mean ± s.e.mean except to tmax values which are median (range).
AUC(0,12 h) of S- and R-norverapamil was also significantly increased by RA. The terminal t½ of norverapamil was not estimated due to excessive fluctuations. Norverapamil/verapamil AUC ratios were not altered by the disease.
Despite the observed substantial elevation in serum total verapamil and norverapamil concentrations and no change in unbound S-verapamil, less dromotropic effect as measured by PR-interval prolongation was observed in RA (Figure 4, Table 3). Furthermore, AV blocks were noticed only in controls and not in RA patients. Similarly, other cardiac indices (i.e. blood pressure and heart rate), that were expected to be affected by higher verapamil concentrations observed in RA, remained at the level of control subjects (Table 3).
Figure 4.

Effect of rheumatoid arthritis on verapamil prolongation of PR interval following administration of a single oral dose 0f 80 mg kg−1 to healthy subject (^) and RA patients (•). Error bars represent s.e. mean (n = 8); *P<0.05, **P<0.01.
Table 3.
Effect of rheumatoid arthritis (RA) on various cardiac indices.
| Parameter | Control | RA | |
|---|---|---|---|
| Maximum percentage PR prolongation1 | 20.3 ± 2.9 | 11.8 ± 1.62 | |
| 95% CI | 0.58,14.6 | ||
| Area under percentage PR prolongation-time curve (% h) | 117.0 ± 11.6 | 60.0 ± 12.02 | |
| 95% CI | 60.5,111 | ||
| Maximum percentage reduction in heart rate1 | 22.6 ± 3.6 | 24.3 ± 7.7 | |
| 95% CI | 114,8.0 | ||
| Maximum percentage reduction blood pressure1 | Systolic | 10.3 ± 1.9 | 8.7 ± 2.3 |
| 95% CI | −5.3,8.5 | ||
| Diastolic | 8.1 ± 1.2 | 12.4 ± 1.8 | |
| 95% CI | −11.2,2.6 | ||
| Maximum percentage reduction MAP1 | 10.2 ± 1.6 | 9.6 ± 1.8 | |
| 95% CI | −6.6,7.3 | ||
| First degree AV node block | 2 out of 8 | Nil |
Mean (± s.e.mean) maximum percent reduction from baseline;
Significantly different from control; 95% CI, confidence interval of individual differences between control and rheumatoid.
In all subjects, the relationship between the PR interval and serum S-verapamil concentration was linear and significant (r = 0.71–0.98, P = 0.001–0.05, n = 13) except for three subjects in the RA group. These three demonstrated pronounced counter-clockwise hystereses. In all three cases the hysteresis collapsed to concave curves when the data were fitted to a one-compartment model with oral input linked to a theoretical effect compartment [25]. Pharmacokinetics-pharmacodynamics data for R-verapamil are not presented since the pharmacological activity of verapamil is mainly ascribed to the S enantiomer [26].
Discussion
This study demonstrates clearly that RA causes a decrease in verapamil oral clearance resulting in a substantially increased drug AUC (Figure 3, Table 2). This was not unexpected since, in the rat, endotoxin-induced inflammation causes similar changes in verapamil disposition [2]. In addition, reduced oral clearance of drugs with intermediate to high hepatic extraction ratios has been reported by many authors in both humans and animals with various inflammatory conditions [e.g. 1]. For an orally administered dose, AUC is affected by the unbound plasma concentration and enzyme activity [27, 28]. Inflammation increases AAG concentrations [29] and reduces intrinsic hepatic metabolism of many drugs [30–32]. Verapamil enantiomers are extensively bound to plasma AAG and albumin [33, 34]. They are also extensively metabolized by the liver [14] and to a lesser extent by the gut [35]. S- and R-verapamil have oral bioavailabilties of approximately 0.25 and 0.65, respectively [36], typical of a relatively highly and an intermediately extracted drug. The difference in the hepatic extraction ratio influences each enantiomer's sensitivity to these inflammation-induced changes [28]. Nevertheless, inflammation may increase plasma concentrations of drugs with both intermediate (e.g. hydroxy-chloroquine [37]), and high hepatic extraction (e.g. propranolol [38]). We did not examine the effect of route of administration on the inflammation-induced altered pharmacokinetics of verapamil. In the inflamed rat, however, increased plasma concentrations of propranolol, a drug with high hepatic extraction, are observed only after oral and not i.v. administration [4], suggesting reduced first pass metabolism.
Inflammatory diseases and individual proinflammatory cytokines can depress cytochrome (CYP) P450 isozyme [30, 32, 39, 40]. While the mechanism of this suppression is not clearly known, formation of NO, which may inhibit CYP450, is a plausible explanation [41]. RA is associated with an increased concentration of pro-inflammatory cytokines such as IL-1ß, tumour necrosis factor α and interferon γ [42] and NO [43, 44]. Our results (Figures 1 and 2) also demonstrate an average seven-fold increase in serum IL-6 and 2-fold increase in serum NO2−, a stable breakdown product of NO.
RA could also change hepatic blood flow due to involvement of prostanoids, NO and endothelins [45, 46]. Nevertheless, at least in the rat adjuvant arthritis, the hepatic blood flow remains unchanged [47].
The AUC(0,12 h) of norverapamil was also elevated in the presence of RA. However, no changes in metabolite to parent drug (norverapamil/verapamil) ratios were observed (Table 2). This may indicate that the rise in the concentration of norverapamil is a reflection of that of the parent drug. The present data, however, do not permit a clear interpretation of the effect of RA on norverapamil.
Our patients were taking other medications during the study (Table 1). Four severely afflicted patients with relatively higher AUC values were receiving methotrexate during the study. Three patients with moderate RA and consequently AUC values closer to normal were receiving hydroxychloroquine and sulphasalazine. The RA patient with the lowest measured AUC was on cyclosporin. Nevertheless, we noticed a significant association between the disease activity and oral clearance of verapamil similar to what has been reported for propranolol in the rat [38]. We also observed a much greater variability in the pharmacokinetics of verapamil in RA patients than in healthy subjects (Figure 3). This may be explained by the fact that patients with a wide range of disease activity were studied.
Regardless of the exact underlying mechanism, substantial increases in the total verapamil concentrations were observed in RA patients. This, however, did not result in increased response. Quite the contrary, the dromotropic effect of verapamil decreased and its hypotensive potency remained unchanged in RA. The percent prolongation in PR interval significantly decreases to approximately 50% in RA when compared with healthy controls (Table 3, Figure 4). In addition, 1° AV-block, a side-effect associated with elevated concentrations of verapamil [17], occurred in two healthy controls but not in any of the RA patients. Two explanations may be plausible for our observation; (1) reduced response due to reduced plasma unbound verapamil concentrations secondary to elevation of plasma AAG levels in RA, and (2) reduced sensitivity of calcium channel receptors. The former explanation can be ruled out since the AUC of the unbound S-verapamil did not change in the presence of RA (Table 2]. The unbound AUC of R-verapamil did decrease. R-verapamil, however, has 10–20 fold less dromotropic activity than its antipode [48]. It has been reported that norverapamil, which possesses negligible dromotropic effect compared with verapamil [49], can displace verapamil from plasma proteins [50]. However, displacement of verapamil by norverapamil should lead to an increase in fu and an increased rather than decreased effect. Consistent with our observation in RA, Abernethy et al. [36] observed reduced verapamil dromotropism with no changes in protein binding in the elderly. Therefore, it appears that RA results in alterations in normal cardiac responsiveness to drugs, which are independent of pharmacokinetic changes. This may indicate a down-regulation of the l-type Ca2+ channels in the heart secondary, perhaps, to the elevation of NO in RA. NO is a powerful activator of guanylyl cyclase [51] and has been shown to have negative inotropic effects on the heart [52]. In our study, a strong negative correlation was observed between serum NO2− and percentagePR interval prolongation. Therefore, there exists the possibility of a significant link between a RA-induced rise in NO and reduced dromotropic effect of verapamil. However, this does not preclude a direct effect of the pro-inflammatory cytokines on cardiac function. For example, binding of IL-2 to cardiac IL-2 receptors causes an increase in Ca2+ ion flux [53]. Such alterations in Ca2+ currents could alter the normal inotropic and dromotropic responsiveness of the myocardium to drugs such as verapamil.
Reduced,-adrenergic receptor density in neutrophil of RA patients has been observed [54]. In addition, down-regulation of α-adrenergic receptors by pro-inflammatory cytokines and inflammatory conditions has also been observed in asthma [55] as well as in congestive heart failure [56]. Changes in receptor function due to decoupling of β-receptors from guaninine nucleotide binding protein [G-protein), and altered intracellular protein kinase C activity may be involved [57]. The same mechanism may operate for verapamil since it also alters the intracellular function of protein kinase C [58].
Our study had two limitations. Firstly it was conducted after a single dose, and secondly the RA patients were normotensive. Secondly, the effect of inflammatory conditions on the blood pressure controlling effect of verapamil in hypertensive patients remains unknown. However, it is known that inflammation may directly determine therapeutic failure in patients with unstable angina [12].
In conclusion, RA results in increased concentrations of verapamil and norverapamil. Regardless of the mechanism involved in the altered pharmacokinetics of verapamil, the elevation in drug concentrations was accompanied by a significant decrease in dromotropic activity. This may be attributable to a receptor down-regulation caused by increased expression of pro-inflammatory cytokines and/ or NO. Implications of this observation may reach beyond verapamil and RA since other inflammatory conditions (e.g. arthritis, infection, asthma) and other receptors may be involved. Recently, the rate of therapy failure [12] and mortality [59] following myocardial infarction have been shown to be associated with the elevation of serum C-reactive protein, an indicator of inflammation. With increasing age, multiple disease states are more likely to occur. Treatment of cardiovascular diseases in a patient with rheumatoid arthritis may therefore require closer attention to prevent therapeutic failure.
Acknowledgments
This study was supported in part by Medical Research Council of Canada Grant 983587. The authors wish to thank Ann Gilianus, RN and the Misericordia Hospital Cardiac Monitoring Unit for the use of the digital Holter monitors, Lori Zuk, RN and the nurses of the Clinical Investigation Unit for their help in patient care and Dr Reza Mehvar of Texas Tech University for his assistance with setting up of the h.p.l.c. assay.
References
- 1.Schneider RE, Bishop H, Kendall MJ, Quarterman CP. Effect of inflammatory disease on plasma concentrations of three β-adrenoreceptor blocking agents. Int J Clin Pharmacol Ther Tox. 1981;19:158–162. [PubMed] [Google Scholar]
- 2.Laethem ME, Belpaire FM, Wijnant P, Rosseel MT, Bogaert MC. Influence of endotoxin on the steroselective pharmacokinetics of oxprenolol, propranolol, and verapamil in the rat. Chirality. 1994;6:405–410. doi: 10.1002/chir.530060508. [DOI] [PubMed] [Google Scholar]
- 3.Belparie F, De Smet B, Chindavijal B, Fraeyman N, Bogaert MG. Effect of turpentine-induced inflammation on the disposition kinetics of propranolol, metoprolol, and antipyrine in the rat. Fund Clin Pharmacol. 1989;3:79–88. doi: 10.1111/j.1472-8206.1989.tb00667.x. [DOI] [PubMed] [Google Scholar]
- 4.Piquette-Miller M, Jamali F. Selective effect of adjuvant arthritis on the disposition of propranolol enantiomers in rats detected using a stereospecific HPLC assay. Pharm Res. 1993;10:294–299. doi: 10.1023/a:1018907431893. [DOI] [PubMed] [Google Scholar]
- 5.Piquette-Miller M, Jamali F. Effect of adjuvant arthritis on the disposition of acebutolol enantiomers in rats. Agents Actions. 1992;37:290–296. doi: 10.1007/BF02028122. [DOI] [PubMed] [Google Scholar]
- 6.Wolfe F. Adverse drug reactions of DMARR and DC-ARTs in rhematoid arthritis. Clin Exp Rheum. 1997;15:S75–S81. [PubMed] [Google Scholar]
- 7.Poulton K, Griffith S, Thomson W, et al. Adverse drug reactions in patients with rheumatoid arthritis and HLA-DR3. Eur J Immunogen. 1998;25:62–66. [Google Scholar]
- 8.Lyle WH. Penicillamine. Clin Rheum Dis. 1979;5:569–601. [Google Scholar]
- 9.Walshe JM., Chairman Round Table Discussion-Proper use of penicillamine. Postgrad Med J. 1974;50:80–83. [Google Scholar]
- 10.Oates J. Antagonism of antihypertensive drug therapy by nonsteroidal anti-inflammatory drugs. Hypertension. 1988;11:114–116. doi: 10.1161/01.hyp.11.3_pt_2.ii4. [DOI] [PubMed] [Google Scholar]
- 11.Houston MC. Nonsteroidal anti-inflammatory drugs and antihypertensives. Am J Med. 1991;17:42S–47S. doi: 10.1016/0002-9343(91)90485-g. [DOI] [PubMed] [Google Scholar]
- 12.Verheggen PW, de Maat MP, Cats VM, et al. Inflammatory status as a main determinant of outcome in patients with unstable angina, independent of coagulation activation and endothelial cell function. Eur Heart J. 1999;20:567–574. doi: 10.1053/euhj.1998.1312. [DOI] [PubMed] [Google Scholar]
- 13.Belpaire F, Bogaert M, Rosseneu M. Binding of β-adrenoreceptor blocking drugs to human serum albumin, to α1-acid-glycoprotein and to human serum. Eur J Clin Pharmacol. 1982;23:246–2533. doi: 10.1007/BF00545224. [DOI] [PubMed] [Google Scholar]
- 14.Eichelbaum M, Ende M, Remberg G, Schomerus M, Dengler HJ. The metabolism of DL-[14C]verapamil in man. Drug Metab Dispos. 1979;7:145–148. [PubMed] [Google Scholar]
- 15.Kroemer H, Gautier J, Beaune P, Henderson C, Wolf C, Eichelbaum M. Identification of P450 enzymes involved in metabolism of verapamil in humans. Naunyn-Schmiedeberg's Arch Pharmacol. 1993;348:332–337. doi: 10.1007/BF00169164. [DOI] [PubMed] [Google Scholar]
- 16.Tracy TS, Korzekwa KR, Gonzalez FJ, Wainer IW. Cytochrome P450 isoforms involved in metabolism of the enantiomers of verapamil and norverapamil. Br J Clin Pharmacol. 1999;47:545–552. doi: 10.1046/j.1365-2125.1999.00923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thomas S, Stone C, Koury S. Cardiac dysrhythmias in severe verapamil overdose: characterization with a canine model. Eur J Emerg Med. 1996;3:9–13. doi: 10.1097/00063110-199603000-00002. [DOI] [PubMed] [Google Scholar]
- 18.Johnston A, Burgess C, Hamer J. Systemic availability of oral verapamil and effect on PR interval in man. Br J Clin Pharmacol. 1981;12:397–400. doi: 10.1111/j.1365-2125.1981.tb01233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson AG. NSAIDs and blood pressure. Clinical importance for older patients. Drugs Aging. 1998;12:17–27. doi: 10.2165/00002512-199812010-00003. [DOI] [PubMed] [Google Scholar]
- 20.Schumacher H, Klippel J, Koopman W, editors. Primer of the rheumatic diseases. 10. William Byrd Press Richmond VA: Arthritis Foundation; 1993. p. 328. [Google Scholar]
- 21.Shibukawa A, Wainer I. Simultaneous direct determination of the enantiomers of verapamil and norverapamil in plasma using a derivatized amylose HPLC chiral stationary phase. J Chromatogr. 1992;574:85–92. doi: 10.1016/0378-4347(92)80101-u. [DOI] [PubMed] [Google Scholar]
- 22.Grisham M, Johnson G, Gautreaux M, Berg R. Measurement of nitrate and nitrite in extracellular fluids: a Window to systemic nitric oxide metabolism. Meth Comp Meth Enzym. 1990;7:84–90. [Google Scholar]
- 23.Keefe D, Yin-Gail Y, Kates R. Verapamil protein binding in patients and normal subjects. Clin Pharmacol Ther. 1981;29:21–26. doi: 10.1038/clpt.1981.4. [DOI] [PubMed] [Google Scholar]
- 24.Tatsuoka M. Selected topics in advanced statistics: An elementary approach 6. Discriminant analysis: The study of group differences. Champaign Ill.: Institute for Personality and Ability Testing; 1970. pp. 48–56. [Google Scholar]
- 25.Holford NH, Sheiner LB. Understanding the dose-effect relationship: clinical application of pharmacokinetic-pharmacodynamic models. Clin Pharmacokin. 1981;6:429–453. doi: 10.2165/00003088-198106060-00002. [DOI] [PubMed] [Google Scholar]
- 26.Echizen H, Brecht T, Niedergesäss S, Vogelgesang B, Eichelbaum M. The effect of dextro-, levo-, and racemic verapamil on atrioventricular conduction in humans. Am Heart J. 1985a;109:210–217. doi: 10.1016/0002-8703(85)90585-x. [DOI] [PubMed] [Google Scholar]
- 27.Wilkinson G. Clearance approaches in pharmacology. Pharmacol Rev. 1987;39:1–47. [PubMed] [Google Scholar]
- 28.Rowland M, Tozer NT. Chapter 12. 3. Baltimore MD: Williams & Wilkins; 1985. Clinical pharmacokinetics, concepts and applications. [Google Scholar]
- 29.Fey G, Fuller G. Regulation of acute phase gene expression by inflammatory mediators. Mol Biol Med. 1987;4:323–338. [PubMed] [Google Scholar]
- 30.Cawthorne M, Palmer E, Green J. Adjuvant induced arthritis and drug-metabolizing enzymes. Biochem Pharmacol. 1976;25:2683–2688. doi: 10.1016/0006-2952(76)90257-4. [DOI] [PubMed] [Google Scholar]
- 31.Descotes J. Immunomodulating agents and hepetic drug metabolizing enzymges. Drug Metab Rev. 1985;16:175–185. doi: 10.3109/03602538508991434. [DOI] [PubMed] [Google Scholar]
- 32.Peterson TC, Renton KW. Kupfer cell factor mediated depression of hepatic parenchymal cell cytochrome P-450. Biochem Pharmacol. 1986;35:1491–1497. doi: 10.1016/0006-2952(86)90114-0. [DOI] [PubMed] [Google Scholar]
- 33.McGowan F, Reiter M, Pritchett E, Shand D. Verapamil plasma binding: Relationship to α1-acid glycoprotein and drug efficacy. Clin Pharmacol Ther. 1993;33:485–490. doi: 10.1038/clpt.1983.66. [DOI] [PubMed] [Google Scholar]
- 34.Gross A, Heuer B, Eichelbaum M. Stereoselective protein binding of verapamil enantiomers. Biochem Pharmacol. 1988;37:4623–4627. doi: 10.1016/0006-2952(88)90330-9. [DOI] [PubMed] [Google Scholar]
- 35.Fromm M, Dilger K, Busse D, Kroemer H, Eichelbaum M, Klotz U. Gut wall metabolism of verapamil in older people: Effects of rifampicin-mediated enzyme induction. Br J Clin Pharmacol. 1998;45:247–255. doi: 10.1046/j.1365-2125.1998.00669.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abernethy D, Wainer I, Longstreth J, Adrawis N. Stereoselective verapamil disposition and dynamics in aging during racemic verapamil administration. J Pharmacol Exp Ther. 1993;266:904–911. [PubMed] [Google Scholar]
- 37.Emami J, Pasutto FM, Jamali F. Effect of experimental diabetes mellitus and arthritis on the pharmacokinetics of hydroxy-chloroquine enantiomers in rats. Pharm Res. 1998;15:897–903. doi: 10.1023/a:1011928732588. [DOI] [PubMed] [Google Scholar]
- 38.Piquette-Miller M, Jamali F. Influence of severity of inflammation on the disposition kinetics of propranolol enantiomers in ketoprofen-treated and untreated adjuvant arthritis. Drug Metab Dispos. 1995;23:240–245. [PubMed] [Google Scholar]
- 39.Chen YL, Vraux VL, Leneveua A. Acute-phase reponse, IL-6 and alteration of cyclosporin pharmacokinetics. Clin Pharmacol Ther. 1994;55:649–660. doi: 10.1038/clpt.1994.82. [DOI] [PubMed] [Google Scholar]
- 40.Abdel-Rassak Z, Loyer P, Fautrel A, et al. Cytokines dow-regulate expression of major cytochrome P450 enzymes in adult human hepatocytes in primary culture. Mol Pharmacol. 1993;44:707–715. [PubMed] [Google Scholar]
- 41.Khatsenko O, Gross S, Rifkind A, Vane J. Nitric oxide is a mediator of the decrease in CYP450-dependent metabolism caused by immunostimulants. Proc Nat Acad Sci USA. 1993;90:11147–11151. doi: 10.1073/pnas.90.23.11147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Arend W, Dayer J. Cytokines and cytokine inhibitors oral antagonists in rheumatoid arthritis. Arth Rheumol. 1990;33:305–315. doi: 10.1002/art.1780330302. [DOI] [PubMed] [Google Scholar]
- 43.Ueki Y, Miyake S, Tominaga Y, Eguchi K. Increased nitric oxide levels in patients with rheumatoid arthritis. J Rheumatol. 1996;23:230–236. [PubMed] [Google Scholar]
- 44.Robinson D. Lipid mediators, active oxygen, amines, nitric oxide, kinins and clotting factors in rheumatoid arthritis. In: Schmacher H, Klippel J, Koopman W, editors. Primer on the Rheumatic Diseases. 10. William Byrd Press Richmond VA: Arthritis Foundation; 1993. pp. 46–49. [Google Scholar]
- 45.Robotham J, Clemens M, Geiger K. Regulation of hepatic blood flow during resuscitation from hemorrhagic shock. Role of NO and endothelins. Am J Physiol. 1997;272:H2736–H2745. doi: 10.1152/ajpheart.1997.272.6.H2736. [DOI] [PubMed] [Google Scholar]
- 46.Myers S, Turnage R, Hernandez R, Castenada A, Valentine R. Autoregulation of renal and splanchnic blood flow following infra-renal aortic clamping is mediated by nitric oxide and vasodilator prostanoids. J Cardiovasc Surg. 1996;37:97–103. [PubMed] [Google Scholar]
- 47.Walker K, Barber H, Hawksworth G. Mechanism responsible for altered propranolol disposition in adjuvant-induced arthritis in the rat. Drug Metab Dispos. 1986;14:482–486. [PubMed] [Google Scholar]
- 48.Echizen H, Vogelgesang B, Eichelbaum M. Effects of d,l-verapamil on atrioventricular conduction in relation to its stereoselective first-pass metabolism. Clin Pharmacol Ther. 1985;38:71–76. doi: 10.1038/clpt.1985.137. [DOI] [PubMed] [Google Scholar]
- 49.Johnson K, Balderston S, Piper J, Mann D, Reiter M. Electrophysiological effects of verapamil metabolites in the isolated heart. J Cardiovasc Pharmacol. 1991;17:830–837. doi: 10.1097/00005344-199105000-00020. [DOI] [PubMed] [Google Scholar]
- 50.Johnson J, Akers W. Influence of metabolites on protein binding of verapamil. Br J Clin Pharmacol. 1995;39:536–538. doi: 10.1111/j.1365-2125.1995.tb04492.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Murad F, Forstermann U, Nakane M, et al. The nitric oxide-cGMP signal transduction system for intracellular and intercellular communication. Adv Second Messenger Phosphoprotein Res. 1993;28:101–109. [PubMed] [Google Scholar]
- 52.Finkel M, Oddis C, Jacob T, Watkins S, Hattler B, Simmons R. Negative inotropic effects of cytokines on the heart mediated by nitric oxide. Science. 1992;257:387–389. doi: 10.1126/science.1631560. [DOI] [PubMed] [Google Scholar]
- 53.Gross G. Curtis MJ. Inflammatory Mediators and the Stunned Myocardium Immunopharmacology of the Heart. San Diego: Academic Press; 1993. pp. 98–124. Chapter 8. [Google Scholar]
- 54.Baerwald C, Graefe C, Von Wichert P, Krause A. Decreased density of beta-adrenergic receptors on peripheral blood mononuclear cells in patients with rheumatoid arthritis. J Rheumatol. 1992;19:204–210. [PubMed] [Google Scholar]
- 55.Meurs H, Kauffman HF, Koeter GH, Timmermans A, de Vries K. Regulation of the beta-receptor-adenylate cyclase system in lymphocytes of allergic patients with asthma: possible role for protein kinase C in allergen-induced nonspecific refractoriness of adenylate cyclase. J Allergy Clin Immunol. 1987;80:326–339. doi: 10.1016/0091-6749(87)90039-x. [DOI] [PubMed] [Google Scholar]
- 56.Bavendiek U, Brixius K, Frank K. Altered inotropism in the failing human myocardium. Basic Res Cardiol. 1996;91:9–16. doi: 10.1007/BF00795356. [DOI] [PubMed] [Google Scholar]
- 57.Strasser R, Benovic J, Lefkowitz R, Caron M. The beta-adrenergic receptor kinase: role in homologous desensitization in S49 lymphoma cells. Adv Exp Med Biol. 1988;232:503–517. doi: 10.1007/978-1-4684-9042-8_43. [DOI] [PubMed] [Google Scholar]
- 58.DePitrillo P, Abernethy D, Wainer I, Andrawis N. Verapamil decreases lymphocyte protein kinase C activity in humans. Clin Pharmacol Ther. 1994;55:44–50. doi: 10.1038/clpt.1994.8. [DOI] [PubMed] [Google Scholar]
- 59.Pietila KO, Harmoinen AP, Jokiniitty J, Pasternack AI. Serum C-reactive protein concentration in acute myocardial infarction and its relationship to mortality during 24 months of follow-up in patients under thrombolytic treatment. Eur Heart J. 1996;17:1345–1349. doi: 10.1093/oxfordjournals.eurheartj.a015068. [DOI] [PubMed] [Google Scholar]