

Abstract
Aims
To evaluate the effects of cimetidine and Maalox® (aluminium hydroxide 1.35 g and magnesium hydroxide 1.2 g) on the pharmacokinetics of ziprasidone.
Methods
Eleven healthy young subjects aged 18–45 years were given single oral doses of ziprasidone 40 mg on three occasions at least 7 days apart. On one occasion ziprasidone was administered alone, on another occasion ziprasidone was co-administered with oral cimetidine 800 mg and on a third occasion ziprasidone was co-administered with oral Maalox®.
Results
The administration of cimetidine increased the ziprasidone AUC(0, ∞) by 6% but there were no statistically significant differences in Cmax, tmax or λz between the ziprasidone + cimetidine group and the ziprasidone group. The administration of Maalox® did not produce any statistically significant differences in AUC(0, ∞), Cmax, tmax or λz between the ziprasidone + Maalox® group and the ziprasidone group.
Conclusions
The pharmacokinetics of ziprasidone are not affected by concurrent administration of cimetidine or Maalox®. This suggests that other nonspecific inhibitors of cytochrome P450 and antacids are unlikely to alter the pharmacokinetics of ziprasidone.
Keywords: ziprasidone, aluminium, cimetidine, interaction, magnesium, pharmacokinetics
Introduction
Ziprasidone has a pharmacokinetic profile unaffected by age and gender [1]. It is extensively metabolized to inactive derivatives in humans, and in vitro studies using human liver microsomes have shown that the oxidative metabolism of ziprasidone is primarily mediated by the 3A4 isoform of cytochrome P450 (CYP3A4) [2]. The absorption of orally administered ziprasidone is enhanced when it is taken immediately after eating, perhaps as a consequence of improved drug solubilization in the presence of food [3].
Cimetidine is an H2 receptor antagonist which is commonly used to treat duodenal ulcers, benign gastric ulcers and hypersecretory states. It inhibits several isoforms of cytochrome P450, including CYP3A4 [4]. This inhibition can result in alterations in the elimination of co-administered drugs which are also oxidatively metabolized by CYP3A4. In addition, cimetidine can alter gastric pH and therefore has the potential to affect the absorption of co-administered drugs.
Antacids are commonly used in the symptomatic relief of dyspeptic symptoms and, usually in combination with H2 receptor antagonists, in the treatment of duodenal ulcers. As they function by raising gastric pH, they have the potential to affect the absorption of co-administered drugs. Furthermore, they can influence the pharmacokinetics of co-administered drugs through absorption or chelation.
This study was designed to determine whether or not multiple-dose cimetidine or antacid administration alters the pharmacokinetics of ziprasidone.
Methods
Subjects
The subjects were healthy, young (18–45 years) adults of either gender. All subjects weighed ≤91 kg and were within 10% of their ideal body weight for age, height, sex and frame [5]. None was pregnant or lactating and none was a smoker. All subjects provided written informed consent.
Protocol
This was an open-label, randomized, three-way crossover study designed to determine whether multiple doses of cimetidine and Maalox® alter the pharmacokinetics of ziprasidone. The study protocol was approved by an independent institutional review board.
The study comprised three 1 day treatment periods separated by intervals of at least 7 days. During each treatment period each subject received one of three different treatments in one of six sequences according to a computer-generated randomization. These three treatments were: ziprasidone 40 mg alone; ziprasidone 40 mg + cimetidine 800 mg; and ziprasidone 40 mg + Maalox® 30 ml (aluminium hydroxide 1.35 g and magnesium hydroxide 1.2 g).
Ziprasidone hydrochloride capsules were administered orally, after a standard breakfast at approximately 09.00 h. Cimetidine tablets were administered once daily, at approximately 07.00h, commencing 2 days before ziprasidone administration and continuing until 1 day after ziprasidone dosing. Maalox® suspension was administered on the evening before the administration of ziprasidone (approximately 23.00 h), immediately before the ziprasidone dosing (approximately 09.00 h), and after lunch (approximately 13.00 h) and dinner (approximately 18.00 h) on the day that ziprasidone was taken.
Pharmacokinetic sampling
Blood samples for the determination of serum ziprasidone concentrations were collected immediately before dosing and at fixed intervals up to 36 h after each dose of ziprasidone.
Pharmacokinetic analysis
Serum concentrations of ziprasidone were determined using high performance liquid chromatography involving solid-phase extraction and u.v. detection. The assay had a dynamic range of 1.0–250.0 ng ml−1[6]. Ziprasidone concentrations below the lower limit of quantification were assigned a value of 0 ng ml−1 in pharmacokinetic calculations.
The maximum serum concentration of ziprasidone (Cmax) and the earliest time at which Cmax occurred (tmax) were estimated directly from the experimental data. The terminal elimination rate constant for ziprasidone (λz) was estimated using least-squares regression analysis of the ziprasidone concentration–time data obtained during the terminal log-linear phase. The terminal half-life of ziprasidone (t½,z) was calculated as ln 2/λz, and the mean half-life of ziprasidone was estimated as ln 2/mean λz. The area under the serum concentration–time curve from time zero to the last time (t) serum ziprasidone was measurable (AUC(0, t)) was estimated by the linear trapezoidal approximation. The area under the serum concentration–time curve from the time t to infinity (AUC(t, ∞)) was calculated as Cpest/λz, where Cpest is the estimated concentration at the last time serum ziprasidone was measurable based on the regression analysis of the terminal log-linear phase. The total area under the serum concentration–time curve (AUC(0, ∞)) was estimated as the sum of the AUC(0, t) and AUC(t, ∞) values.
Statistical analysis
Natural log-transformed AUC(0, ∞) and Cmax values, and untransformed tmax and λz values, were analysed in a two-way analysis of variance (anova) model containing sequence, subject within sequence, period and treatment effects (PROC GLM of SAS®). The sequence effect was tested using the subject within sequence mean square as the error term and the period effect was tested using the within subject mean square error as the error term.
Geometric means and standard deviations were calculated for AUC(0, ∞) and Cmax. Arithmetic means and standard deviations were calculated for tmax and λz. Adjusted means, and their variances and covariances, were then calculated using the least-squares means statement from SAS®. These were then used to estimate the difference between the treatment effects.
Student’s t-tests with 95% confidence limits were used to examine pair-wise differences between the groups. Values of P< 0.05 were considered to be statistically significant.
Results
Subjects
Twelve subjects entered the study (Table 1). Ten completed all three treatment periods and were included in the pharmacokinetic analysis (n = 10 for all three treatment periods).
Table 1.
Demographic characteristics.
| Characteristic | Men (n =3) | Women (n =9) | Total (n =12) |
|---|---|---|---|
| Age (years) | |||
| Mean | 23 | 32 | 30 |
| Range | 21–25 | 21–44 | 21–44 |
| Weight (kg) | |||
| Mean | 64.6 | 58.5 | – |
| Range | 62–68 | 46–69 | – |
| Ethnic origin | |||
| White | 3 | 8 | 11 |
| Black | 0 | 1 | 1 |
One subject discontinued the study after experiencing adverse events and only completed the ziprasidone treatment period. Another subject discontinued the study for personal reasons after completing the ziprasidone + Maalox® and ziprasidone + cimetidine treatment periods.
Pharmacokinetics
The mean serum ziprasidone concentration–time curves for the three treatment groups were broadly similar, but suggested a small but not statistically significant delay in the occurrence of Cmax in the ziprasidone + Maalox® group compared with the ziprasidone alone group (mean ratio 108.4%; 95% CI: 82.8,134) (Figure 1).
Figure 1.
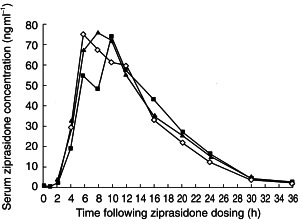
Mean serum ziprasidone concentrations by treatment group (ziprasidone ◊; ziprasidone + Maalox®▪; ziprasidone + cimetidine ▴).
The pharmacokinetic parameters for ziprasidone across the three treatment groups were generally similar (Table 2). Individual AUC(0, ∞) and Cmax values are shown in Figures 2 and 3. The only statistically significant difference between the treatment groups, for any of the pharmacokinetic parameters, occurred in the comparison of mean AUC(0, ∞) values in the ziprasidone + cimetidine group with the ziprasidone alone group. This parameter was 6.2% greater in the ziprasidone + cimetidine group than in the ziprasidone alone group (95% CI: 100.6,113.5). None of the differences in Cmax, tmaxand λz between the ziprasidone + cimetidine group and the ziprasidone alone group was statistically significant. The t½,z of ziprasidone in the ziprasidone + cimetidine group was 3.1% longer than that in the ziprasidone alone group.
Table 2.
Summary of pharmacokinetic parameters of ziprasidone.
| Pharmacokinetic parameter | Ziprasidone (n =10) | Ziprasidone + cimetidine (n =10) | Ziprasidone + Maalox® (n =10) | Ziprasidone + cimetidine vs ziprasidone Ratio of (AUC(0, ∞) and Cmax) and difference in (tmax and λz) means (95% CI) | Ziprasidone + Maalox® vs ziprasidone Ratio of (AUC(0, ∞) and Cmax) and difference in (tmax and λz) means (95% CI) |
|---|---|---|---|---|---|
| AUC(0,∞)a (ng ml−1 h) | 939±178 | 998±210 | 952±190 | 106.9% (100.6, 113.5) | 101.4% (95.6, 107.5) |
| Cmaxa (ng ml−1) | 91±27 | 92±30 | 96±51 | 102.1% (80, 130.4) | 105.3% (82.8, 134) |
| tmaxb (h) | 8±2 | 9±3 | 11±3 | 1.05 (−1.4751, 3.5751) | 2.6 (0.1134, 5.0866) |
| λzb (h−1) | 0.182±0.02 | 0.176±0.02 | 0.181±0.03 | −0.033 (−0.0183, 0.0118) | −0.0005 (−0.0153, 0.0143) |
| t1/2,zc (h) | 3.81 | 3.93 | 3.82 | – | – |
Geometric means and standard deviations.
Arithmetic means and standard deviations.
Calculated as ln2/mean λz.
CI – confidence interval.
Figure 2.
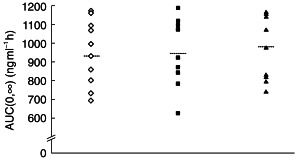
Individual AUC(0,∞) values by treatment group (ziprasidone ◊; ziprasidone + Maalox®▪; ziprasidone + cimetidine ▴). Group means are shown as horizontal dashed lines.
Figure 3.
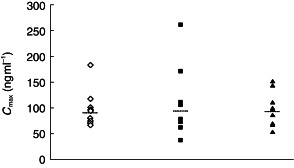
Individual Cmax values by treatment group (ziprasidone ◊; ziprasidone + Maalox®▪; ziprasidone + cimetidine ▴). Group means are shown as horizontal dashed lines.
The mean AUC(0,∞), Cmax, tmax and λz values in the ziprasidone + Maalox® group were not significantly different from those in the ziprasidone alone group. The t½,z of ziprasidone in the ziprasidone + Maalox® group was almost identical to that in the group receiving ziprasidone alone.
Discussion
Findings from this study indicate that concomitant administration of cimetidine produces a small increase in systemic exposure to ziprasidone as measured by the AUC(0,∞). Although this increase was statistically significant, it was small (6%) and is not considered to be clinically meaningful. None of the other pharmacokinetic parameters that were measured showed any statistically or clinically significant differences between the ziprasidone + cimetidine and ziprasidone alone groups. Similarly, no statistically significant differences between the ziprasidone + Maalox® and ziprasidone alone groups were observed for any of the measured pharmacokinetic parameters. The delay in reaching Cmax is unlikely to be of clinical relevance.
The lack of clinically significant effects of cimetidine on the pharmacokinetics of ziprasidone was observed despite the fact that oxidative ziprasidone metabolism is primarily mediated by CYP3A4 [2]. As cimetidine is known to be a non-specific inhibitor of CYP3A4 [4], the findings of this study suggest that ziprasidone could also be metabolized via alternative pathways. In view of the in vitro activity of ziprasidone for the inhibition of CYP3A4 [4], however, a single dose of ziprasidone would seem unlikely to inhibit this isoenzyme substantially in vivo. Nevertheless, the modest increase in the AUC(0,∞) is in accord with this hypothesis. Alternatively, ziprasidone may be excreted unchanged to a greater degree than normal in the presence of cimetidine.
In conclusion, the findings of this study indicate that the administration of multiple doses of cimetidine or of Maalox® do not exert any clinically significant influence on the single-dose pharmacokinetics of ziprasidone 40 mg.
References
- 1.Wilner KD, Tensfeldt TG, Baris B, et al. Single- and multiple-dose pharmacokinetics of ziprasidone in healthy young and elderly volunteers. Br J Clin Pharmacol. 2000;49(Suppl. 1):15S–20S. doi: 10.1046/j.1365-2125.2000.00148.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prakesh C, Kamel A, Cui D, et al. Identification of the major human liver cytochrome P450 isoform responsible for the primary metabolites of ziprasidone and prediction of possible drug interactions. Br J Clin Pharmacol. 2000;49(Suppl. 1):35S–42S. doi: 10.1046/j.1365-2125.2000.00151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lebel M, Proulx MJ, Allard S, et al. Influence of a high fat breakfast on the absorption and pharmacokinetics of CP-88,059, a new antipsychotic. Clin Invest Med. 1993;16:B18. Abstract 117. [Google Scholar]
- 4.Knodell RG, Browne DG, Gwozdz GP, et al. Differential inhibition of individual human liver cytochrome P450 by cimetidine. Gastroenterology. 1991;101:1680–1691. doi: 10.1016/0016-5085(91)90408-d. [DOI] [PubMed] [Google Scholar]
- 5.Metropolitan height and weight tables Stat Bull Metrop Life Found. 1983;64:3–9. [PubMed] [Google Scholar]
- 6.Janiszewski JS, Fouda HG, Cole RO. Development and validation of a high-sensitivity assay for an antipsychotic agent, CP-88,059, with solid-phase extraction and narrow-bore high-performance liquid chromatography. J Chromatogr. 1995;668:133–139. doi: 10.1016/0378-4347(95)00071-p. [DOI] [PubMed] [Google Scholar]