

Abstract
Aims
To identify the cytochrome P450 (CYP) isoform(s) responsible for the formation of the primary metabolite of ziprasidone (ziprasidone sulphoxide), to determine the kinetics of its formation and to predict possible drug interactions by investigating CYP isoform inhibition in an in vitro study.
Methods
In vitro metabolism of [14C]-ziprasidone was studied using human liver microsomes. The metabolites were identified using mass spectrometry. The kinetics of metabolite formation were determined using [14C]-ziprasidone (10–200 μm) over 5 min, and Km and Vmax were estimated from Lineweaver-Burk plots. IC50 values for the inhibition of specific probe substrates for CYP1A2, CYP2C9, CYP2C19, CYP2D6 and CYP3A4, by ziprasidone, risperidone and 9-hydroxyrisperidone were also determined using human liver microsomes from three subjects. Mean Ki values were calculated.
Results
Three CYP-mediated metabolites – ziprasidone sulphoxide, ziprasidone sulphone and oxindole acetic acid – were identified. The apparent Km and Vmax values for the formation of the major metabolite, ziprasidone sulphoxide (measured as the sum of sulphoxide and sulphone) were 235 μm and 1.14 nmol mg−1 protein min−1, respectively. Isoform-selective inhibitors and recombinant enzymes indicated that CYP3A4 is responsible for the formation of ziprasidone metabolites. Ziprasidone was not a substrate for the other isoforms studied. Similar in vitro inhibition of CYP2D6 (Ki 6.9–16 μm) and CYP3A4 (Ki 64–80 μm) was obtained with ziprasidone, risperidone and 9-hydroxyrisperidone. The in vivo free drug concentrations associated with clinically effective doses of ziprasidone are at least 1500-fold lower than the mean Ki for either CYP2D6 inhibition or CYP3A4 inhibition.
Conclusions
Ziprasidone is predominantly metabolized by CYP3A4 in human liver microsomes and is not expected to mediate drug interactions with coadministered CYP substrates, at clinically effective doses.
Keywords: cytochrome P450, drug interactions, enzyme kinetics, human liver microsomes, ziprasidone
Introduction
The primary oxidative metabolites in normal human volunteers, following a single 20 mg oral dose of radiolabelled ziprasidone, were identified as ziprasidone sulphoxide, ziprasidone sulphone, benzisothiazole piperazine (BITP) and oxindole acetic acid. These metabolites were also detected following in vitro incubation of ziprasidone with human liver microsomes [1]. Cytochromes P450 (CYP) are involved in the oxidative metabolism of many drugs, environmental pollutants, procarcinogens and other xenobiotics [2, 3]. Changes in the CYP-dependent metabolism of a particular drug may occur when a co-administered drug acts as an inducer or inhibitor of the same CYP isoform. This can alter the systemic exposure of the drug and thereby affect its pharmacological activity. To characterize and predict possible drug interactions in humans, it is important to determine which CYPs are involved in the oxidative metabolism of a new drug [4–7].
This report presents the results of in vitro studies which aimed to: (1) identify the CYP isoforms responsible for the formation of the primary metabolite of ziprasidone (ziprasidone sulphoxide); (2) elucidate the kinetics of ziprasidone sulphoxide formation following incubation of ziprasidone with human liver microsomes; and (3) predict possible drug–drug interactions by determining Ki values for in vitro inhibition of the major human CYP isoforms.
Methods
Chemicals
Ziprasidone: 5-[2-[4-(1,2-benzisothiazole-3-yl)-1-piperazin-1-yl}ethyl]-6-chloro-1,3-dihydro-2H-indole-2-one hydrochloride hydrate (Figure 1). [14C]-Ziprasidone: ziprasidone labelled at the C-2 carbon of the ethyl side chain attached to the piperazine (Figure 1). [3H]-Ziprasidone: ziprasidone labelled at the C-7 position of the benzisothiazole ring (Figure 1). Ziprasidone sulphoxide: 6-chloro-5-(2-[4-(1-oxo-1,2-benzisothiazol-3-yl)-piperazin-1-yl]ethyl)-1,3-dihydro-2H-indole-2-one (Figure 1). Ziprasidone sulphone: 6-chloro-5-(2-[4-(1,1-dioxo-1,2-benzisothiazol-3-yl)-piperazin-1-yl]ethyl)-1,3-dihydro-2H-indole-2-one (Figure 1). Oxindole acetic acid: (6-chloro-2-oxo-2,3-dihydro-1H-indol-5-yl)acetic acid (Figure 1). BITP: 1,2-{benzisothiazole-3-yl}-1-piperazine (Figure 1). [14C]-CP-118, 954 (internal standard): 5,7-dihydro-3-[2–1-(phenylmethyl)ethyl]-6H-pyrolo [4, 5-f]1,2-benzisoxazol-6-one. The compounds listed above were prepared at Pfizer Central Research, Groton, CT, USA, using published procedures [8, 9]. NADP+, (±)-isocitric acid, isocitric dehydrogenase, 1-aminobenzotriazole (ABT) and quinidine were purchased from Sigma Chemical Co. (St Louis, MO, USA). Ketoconazole was obtained from Research Diagnostics Inc. (Flanders, NJ, USA). Sulphaphenazole was obtained from Ultrafine Chemicals (Salford, Manchester, UK). Furafylline was obtained from Research Biochemicals Inc. (Natick, MA, USA). All other reagents and solvents were of analytical grade.
Figure 1.
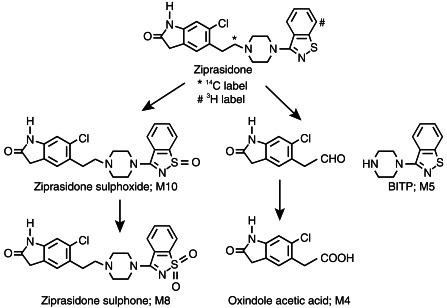
Major in vitro metabolic pathways of ziprasidone by human liver microsomes.
Preparation of microsomes
Human liver samples were obtained from organ donors. Liver and microsome samples were stored at −70oC until used. Microsomes and liver homogenates (S-9) were prepared by differential centrifugation using standard procedures [10]. Protein concentrations were measured by the bicinchoninic acid assay [11] using crystalline bovine serum albumin as standard and CYP concentrations were determined by the method of Omura and Sato [12]. Each microsomal preparation was characterized for five major drug metabolizing CYP isoforms: CYP1A2; CYP2C9; CYP2C19; CYP2D6 and CYP3A4. The activities of CYP1A2, CYP2C9, CYP2C19, CYP2D6 and CYP3A4 were assessed using phenacetin [13], tolbutamide [14], S-mephenytoin [15], bufuralol [16], and testosterone [17], respectively, as probe substrates.
Microsomes prepared from human B-lymphoblastoid cells, containing cDNA-expressed CYP1A2 were purchased from Gentest Corporation (Woburn, MA, USA). Recombinant CYPs expressed in insect cells (CYP2C9, CYP2C19, CYP2D6 and CYP3A4) were generated in the Molecular Sciences Department at Pfizer Inc. and microsomal subcellular fractions were prepared by standard centrifugation procedures [10]. The activities of expressed CYP1A2 (7-ethoxyresorufin deethylation), CYP2C9 (tolbutamide hydroxylation), CYP2C19 (S-mephenytoin hydroxylation), CYP2D6 (bufuralol 1′-hydroxylation), and CYP3A4 (testosterone 6-β-hydroxylation), were 0.1, 1.8, 0.11, 0.91, and 9.0 nmol mg−1 microsomal protein min−1, respectively.
Incubations
The incubation mixture (final volume 1 ml) contained liver microsomes (0.5 μm CYP), [14C]-ziprasidone (50 μm, 0.2% DMSO), 100 mm potassium phosphate buffer (pH 7.4) and an NADPH-generating system (9 mm MgCl2, 0.54 mm NADP+, 6.2 mm (±)-isocitric acid, and 0.5 U ml−1 isocitric dehydrogenase). The reactions were initiated by addition of an NADPH regenerating system after a 5 min preincubation period. Reactions were incubated at 37oC for 30 min and were stopped by the addition of methanol (2 ml). Incubations with S-9 fraction contained protein equivalent to 32 mg ml−1 of liver and incubations with recombinant CYP contained 1 mg ml−1 of microsomal protein.
Time course for metabolite formation
The time course of ziprasidone sulphone and ziprasidone sulphoxide metabolite formation with human liver microsomes was studied at 10 μm and 100 μm concentrations of [14C]-ziprasidone. Reactions were carried out as described above, were terminated at 0, 5, 15, 30, 45, and 60 min, and analysed by high pressure liquid chromatography (h.p.l.c.) using a radioactivity monitor (described below).
Kinetics of ziprasidone metabolism
Nine concentrations of [14C]-ziprasidone, ranging from 10 to 200 μm, were studied in the kinetic experiments using human liver (HL-1011) microsomes. Incubations were carried out for 5 min. Reaction rates were linear with respect to protein concentration and time of incubation. Apparent Km and Vmax values for the formation of ziprasidone sulphoxide (sum of ziprasidone sulphoxide and ziprasidone sulphone) were estimated from Lineweaver-Burk plots using linear regression analysis [18].
Correlation studies
Formation rates of ziprasidone sulphoxide and sulphone were also measured using microsomes from eight different human livers (HL-H5, HL-H6–2, HL-1003–3, HL-1008, HL-1009, HL-1011, HL-1013, HL-1014) at a substrate concentration of 50 μm as described above. Formation rates were correlated with activities of isoform-selective probe substrates to assess the possible involvement of individual isoforms.
Inhibition studies
Inhibitors of CYP3A4, ketoconazole (10 μm); CYP2C9, sulphaphenazole (50 μm); CYP1A2, furafylline (20 μm); CYP2D6, quinidine (5 μm) and a general inhibitor of CYP, 1-aminobenzotriazole (ABT, 5 mm) were incubated with [14C]-ziprasidone and S-9. The inhibitors were dissolved in water or methanol. The concentration of organic solvent in the incubates was maintained at no greater that 0.5% of the incubation volume to avoid an effect of solvent on metabolism. Incubations were performed in duplicate and the pairs of resulting values averaged.
Quantification of metabolites formed with human liver microsomes
[14C]-ziprasidone metabolites formed in microsomal incubations (described above) were quantified by h.p.l.c. with radioactivity monitoring (β-RAM). A 100 μl aliquot of internal standard (IS) [14C]-CP-118, 954 (0.01 mg ml−1) was added to the terminated reaction and the mixture was evaporated to dryness on a Speed-Vac evaporator. The residue was reconstituted in 300 μl of mobile phase (described below; solvent A: solvent B, 50:50, v/v), sonicated and centrifuged to remove protein. A 60 μl portion of the supernatant was analysed by h.p.l.c. A standard curve was constructed by plotting the peak area (counts min−1) ratio of [14C]-ziprasidone to IS against the concentration of [14C]-ziprasidone added to the mixture. Unknown concentrations of metabolites were estimated by interpolation from the standard curve.
The h.p.l.c. system consisted of a Rheodyne injector, LDC/Milton Roy constametric CM4000 gradient pump, Waters Lambda-Max model 481 u.v. detector, radioactivity monitor (β-RAM) and an SP 4200 computing integrator. Chromatography was carried out on a YMC basic h.p.l.c. column (4.6 mm× 250 mm, 5 μm) with a binary mixture of methanol (solvent A) and 20 mm ammonium acetate (pH 5.0, solvent B). The mobile phase initially consisted of solvent A: solvent B (30:70, v/v) for 5 min, which was then linearly programmed to a solvent ratio of 50:50 over 35 min, maintained for 1 min and then linearly increased to a ratio of 90:10 over 9 min. The system was allowed to re-equilibrate for approximately 10 min before making the next injection. Retention times of metabolites were compared with those of synthetic standards to confirm identity.
Inhibition of isoform-selective substrates by ziprasidone, risperidone and 9-hydroxyrisperidone
IC50 values (50% inhibition of the activity observed in the absence of the inhibitor) for the inhibition of probe substrates specific for the activities of CYP1A2, CYP2C9, CYP2C19, CYP2D6 and CYP3A4, by ziprasidone, risperidone and 9-hydroxyrisperidone, were determined using individual preparations of human liver microsomes from three subjects. Incubate concentrations of ziprasidone, risperidone and 9-hydroxyrisperidone ranged from 0.1 μm to 100 μm. Final incubate concentrations of the solvent DMSO were kept constant at 0.2% (v/v). Probe substrate concentrations were 20 μm for phenacetin, 200 μm for tolbutamide, 100 μm for S-mephenytoin, 10 μm for bufuralol and 20 μm for testosterone. IC50 values < 100 μm were determined using the equation Y = 100 –[100X/(X+ B)], where X=inhibitor concentration and B= IC50 value. Ki values were experimentally determined for interactions with CYP2D6 by Dixon plot. Bufuralol concentrations were 5, 10, and 20 μm, and inhibitor concentrations were 0, 5, 10, and 20 μm. Individual Ki values were calculated as the mean of each intercept. Ki values for the interaction with CYP3A4 were calculated from IC50 values using the equation IC50= (1+[S]/Km)Ki as defined by Segel [19] for a competitive inhibitor.
Results
CYP-dependent metabolism of ziprasidone
A representative chromatogram of the supernatant following incubation of [14C]-ziprasidone with human liver microsomes is shown in Figure 2. As seen in Figure 2, five radioactive peaks were observed in microsomal incubations. Only three peaks with retention times of 20.5, 39.6 and 40.5 min, were identified by mass spectrometry as oxindole acetic acid (M4), ziprasidone sulphone (M8) and ziprasidone sulphoxide (M10), respectively. These structures were confirmed by comparing respective retention times with synthetic standards onh.p.l.c. [8]. Metabolite formation was NADPH-dependent and was largely abolished in the presence of 1-aminobenzotriazole (ABT, 5 mm) indicating that CYP was responsible (data not shown).
Figure 2.
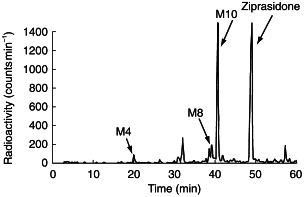
H.p.l.c. radiochromatograms of metabolites of ziprasidone in human liver microsomes. M4: oxindole acetic acid; M8: ziprasidone sulphone; M10: ziprasidone sulphoxide.
Time course for the formation of metabolites
The time course for the combined formation of ziprasidone sulphoxide and ziprasidone sulphone metabolites at two ziprasidone concentrations (10 μm and 100 μm) is shown in Figure 3. Formation rates of the two metabolites were linear for the first 5 min.
Figure 3.
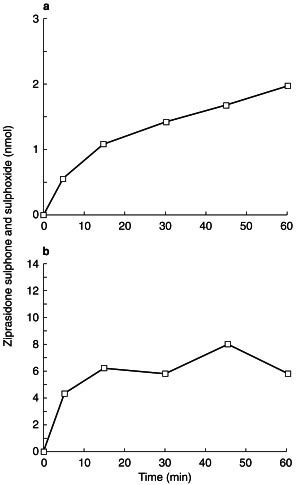
Time course for the formation of ziprasidone sulphoxide and sulphone at a concentration of 10 µm (a) and 100 µm (b).
Kinetics of the formation of metabolites
A typical reaction velocity/substrate concentration curve for the combined formation of the metabolites, ziprasidone sulphoxide and ziprasidone sulphone, is shown in Figure 4. The apparent Vmax and Km values for the formation of ziprasidone sulphoxide (sum of sulphoxide and sulphone) were calculated as 1.14 nmol mg−1 protein min−1 and 235 μm, respectively, which represents an intrinsic clearance value of 0.0048 ml mg−1 protein min−1.
Figure 4.
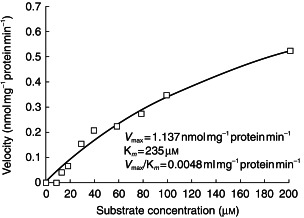
Typical velocity/concentration (Michaelis-Menten) plot for the formation of ziprasidone sulphoxide (measured as combined sulphoxide and sulphone) by human liver microsomes.
Correlation studies
Rates of formation of ziprasidone sulphoxide (sum of sulphoxide and sulphone) with liver microsomes from eight human individuals were measured at a substrate concentration of 50 μm. Formation of ziprasidone sulphoxide (sum of sulphoxide and sulphone) correlated (r = 0.83) significantly with testosterone 6-β-hydroxylation activity (CYP3A4, Table 1). The formation of ziprasidone sulphoxide (sum of sulphoxide and sulphone) also correlated (r =0.70) with S-mephenytoin hydroxylase activity (CYP2C19). No significant correlations were found between the sulphoxidation of ziprasidone and the other CYP specific substrates.
Table 1.
Correlation of various CYP isoform-selective monoxygenase activities with the sulphoxidation (sulphoxide+sulphone) of ziprasidone in human liver microsomes.
| Reaction | CYP isoform | Correlationcoefficient (r)a |
|---|---|---|
| Phenacetin O-deethylation | CYP1A2 | 0.291 |
| Tolbutamide hydroxylation | CYP2C9 | 0.438 |
| S-mephenytoin 4′-hydroxylation | CYP2C19 | 0.699 |
| Bufuralol-1′-hydroxylation | CYP2D6 | 0.149 |
| Testosterone 6β-hydroxylation | CYP3A4 | 0.825 |
Derived from a panel of liver samples from eight individuals.
Inhibition studies
Inhibition of metabolite formation by each inhibitor is summarized in Table 2. Ketoconazole (10 μm) inhibited the formation of ziprasidone sulphoxide (sulphoxide and sulphone) by 79% and oxindole acetic acid (N-dealkylated product) completely. Sulphaphenazole and furafylline did not inhibit the formation of ziprasidone sulphoxide. The formation of oxindole acetic acid was inhibited (∼50%) by furafylline. The formation of ziprasidone metabolites was not inhibited by quinidine but rather increased.
Table 2.
Effects of isoform-specific inhibitors of human CYP on the metabolism of ziprasidone.
| % Inhibitiona | ||||
|---|---|---|---|---|
| Inhibitors | Concentration (μm) | Target CYP | M4b (N-dealkylation) | M8 + M10 (sulphone + sulphoxide) |
| Ketoconazole | 10 | 3A4 | 100 | 79 |
| Sulphaphenazole | 50 | 2C9 | 9.5 | 8.0 |
| Furafylline | 20 | 1A2 | 50 | 0 |
| Quinidine | 5 | 2D6 | −62c | −91c |
| ABTd+ ketoconazole | 5000 + 10 | Non-specific | 100 | 88 |
Duplicate determinations.
Oxindole acetic acid.
Stimulation.
ABT–1-Aminobenzotriazole.
Metabolism by recombinant CYP isoforms
The ability of specific CYP isoforms to metabolize ziprasidone was determined using recombinant CYP1A2, CYP2C9, CYP2C19, CYP2D6 and CYP3A4. H.p.l.c.-radioactivity profiles of ziprasidone metabolites following incubation of [14C]-ziprasidone or [3H]-ziprasidone with CYP3A4 are shown in Figure 5(a) and (b), respectively. CYP3A4 catalysed the formation of the N-dealkylated metabolite (M5), as well as ziprasidone sulphone (M8) and ziprasidone sulphoxide (M10). These oxidative metabolites were not detected in incubations containing recombinant CYP1A2, CYP2C9, CYP2C19 or CYP2D6. As the enzyme activities of the recombinant CYPs were selected to reflect activities typical of a human liver microsomal preparation (see Methods), it can be concluded that these latter isoforms do not contribute significantly to ziprasidone metabolism.
Figure 5.
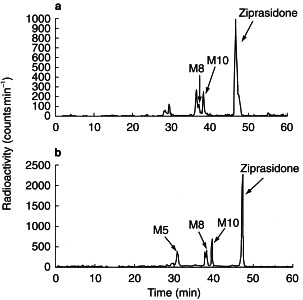
H.p.l.c. radiochromatograms of metabolites of [14C]-ziprasidone (a) and [3H]-ziprasidone (b) by recombinant CYP3A4. M8: ziprasidone sulphone; M10: ziprasidone sulphoxide.
In vitro inhibition of CYP isoforms by ziprasidone
Interactions with CYP2D6 exhibited the lowest mean IC50 values of 31 μm for ziprasidone, 14 μm for risperidone, and 31 μm for 9-hydroxyrisperidone (Table 3). IC50 values for interaction with CYP3A4 were higher at 72, 75 and 90 μm, respectively. All of the other IC50 values were greater than 100 μm except that for the interaction between 9-hydroxyrisperidone and CYP2C19 (52 μm). Mean Ki values for interaction with CYP2D6 were similar for all three compounds; 11 μm for ziprasidone, 6.9 μm for risperidone and 16 μm for 9-hydroxyrisperidone (Table 4). Mean Ki values for interaction with CYP3A4 were calculated from IC50 values assuming competitive inhibition and an average Km value of 100 μm. These were 64 μm for ziprasidone, 67 μm for risperidone, and 80 μm for 9-hydroxyrisperidone.
Table 3.
Interaction of ziprasidone, risperidone and 9-hydroxyrisperidone with human liver cytochromes P450: IC50values for inhibition of probe substrate activities.
| IC50 value (μm)a | |||
|---|---|---|---|
| CYPb | Ziprasidone | Risperidone | 9-Hydroxy-risperidone |
| CYP1A2 | >100 | >100 | >100 |
| CYP2C9 | >100 | >100 | >100 |
| CYP2C19 | >100 | >100 | 52c |
| CYP2D6 | 31 ± 37 | 14 ± 9 | 31 ± 11 |
| CYP3A4 | 72 ± 29 | 75 ± 16 | 90 ± 5 |
IC50 values <100 μm were determined from Y=100–(100X/(X+B)), where X=inhibitor concentration and B = IC50. Values aremean± s.d. (n = 3).
CYP=cytochrome P450.
Mean of two values. One set of human liver microsomes hadlow CYP2C19 activity.
Table 4.
Ki values for inhibition of bufuralol 1′-hydroxylase activity (CYP2D6) by ziprasidone, risperidone, and 9-hydroxyrisperidone.a
| Ki value (μm)a | |||
|---|---|---|---|
| Human liver IDb | Ziprasidone | Risperidone | 9-Hydroxy-risperidone |
| HL 1010–2 | 3.2 | 3.0 | ND |
| HL H35–2 | 2.9 | 2.7 | ND |
| HL 1011 | 11 | 7.8 | ND |
| HL 1018 | 9.5 | 8.0 | 19 |
| HL 1020 | 15 | 6.1 | 14 |
| HL 1021 | 23 | 14 | 16 |
| Mean | 11 | 6.9 | 16 |
| ± s.d. | 7.6 | 4.1 | 2.5 |
Determined from Dixon plots.
Identification of human liver microsomes.
ND – not determined.
Discussion
The present work describes the in vitro metabolism of ziprasidone using human liver microsomes, S-9 and recombinant human CYP isoforms. The oxidative metabolites produced from ziprasidone by human liver microsomes were ziprasidone sulphoxide, ziprasidone sulphone, oxindole acetic acid and BITP. These metabolites were excreted in vivo[20]. Ziprasidone sulphoxide and sulphone were the major circulating metabolites in all species [20–22]. Kinetic analysis of ziprasidone sulphoxide formation (measured as the sum of sulphoxide and sulphone) produced a monophasic Lineweaver-Burk plot suggesting that a single CYP isoform was responsible for sulphoxide formation. Based on CYP isoform-selective chemical inhibitors, correlation studies and metabolism by recombinant human CYP enzymes, the work presented here demonstrates that human liver CYP3A4 has a major role in the oxidative metabolism of ziprasidone.
Ketoconazole, a selective inhibitor of CYP3A4 [23], markedly inhibited the formation of the major metabolites of ziprasidone. The formation of ziprasidone sulphoxide (sum of ziprasidone sulphoxide and ziprasidone sulphone) significantly correlated with CYP3A4 and CYP2C19 specific monooxygenase activities, whereas no correlation was observed between ziprasidone metabolite formation and bufuralol 1′-hydroxylation (CYP2D6), 7-ethoxyresorufin de-ethylation (CYP1A2), or tolbutamide hydroxylation (CYP2C9). The formation of ziprasidone sulphoxide and sulphone correlated with CYP2C19 activity (r = 0.70) but these metabolites were not detected following incubation of [14C]-ziprasidone with recombinant CYP2C19. As the probe substrate activities of CYP2C19 correlated with CYP3A4 (r = 0.52), this correlation may be fortuitous, indicating that CYP2C19 was probably not involved in the formation of ziprasidone sulphoxide and sulphone. Whilst CYP3A4 is primarily responsible for ziprasidone metabolism in human liver microsomes, studies in healthy men have shown that cimetidine (a CYP3A4 inhibitor) does not affect ziprasidone metabolism [24].
Consistent with the results obtained from correlation and inhibition studies, microsomes containing human recombinant CYP3A4 catalysed the conversion of [14C]-ziprasidone to ziprasidone sulphoxide and ziprasidone sulphone and [3H]-ziprasidone to BITP, ziprasidone sulphoxide and ziprasidone sulphone. The N-dealkylated metabolite, oxindole acetic acid, was not detected following incubation of [14C]-ziprasidone with CYP3A4. N-dealkylation involves initial hydrogen abstraction with subsequent hydroxylation at an alpha carbon to form an unstable carbinolamine intermediate. This intermediate then decomposes to form an aldehyde which can be oxidized by aldehyde oxidase or CYP to form an acid metabolite [25]. It is possible that oxindole acetaldehyde was formed following incubation of [14C]-ziprasidone with CYP3A4. However, no attempt to identify the aldehyde intermediate could be made because a synthetic standard was not available. As the 14C label was located on the C-2 carbon of the ethyl side chain attached to the piperazine, the counterpart of the N-dealkylated product, BITP, could not be detected either. The formation of BITP was confirmed by incubating [3H]-ziprasidone, labelled at the C-7 position of the benzisothiazole, with recombinant CYP3A4.
The interaction of ziprasidone, risperidone and the major in vivo metabolite of risperidone, 9-hydroxyrisperidone, with human CYPs was determined by investigating the ability of these compounds to inhibit the metabolism of isoform-specific metabolic pathways for each of the five major human hepatic drug-metabolizing CYPs; CYP1A2, CYP2C9, CYP2C19, CYP2D6 and CYP3A4. The most potent interaction was observed with CYP2D6. Similar mean Ki values of 11, 6.9 and 16 μm were obtained for inhibition of CYP2D6-mediated bufuralol 1-hydroxylation by ziprasidone, risperidone, and 9-hydroxyrisperidone, respectively. As demonstrated in this report, ziprasidone is not a substrate for CYP2D6 but does bind as a potential inhibitor. On the other hand, risperidone demonstrates a polymorphic metabolism typical of a CYP2D6 substrate [26], an interaction which is consistent with the low Ki value observed in this study. A weaker interaction with CYP3A4 was observed for each of these compounds, with Ki values ranging from 3- to 10-fold higher than for CYP2D6. IC50 values for all other interactions were generally greater than 100 μm and were considered not to be clinically significant.
At the upper end of the therapeutic dose range for ziprasidone (80 mg twice daily), the mean serum Cmax achieved in humans is approximately 326 ng ml−1. Plasma protein binding of ziprasidone has been determined in a clinical study to range from 99.65 to 99.94%[T. Smolarek, personal communication]. Considering only the higher free fraction value, an unbound drug Cmax was calculated, which is equivalent to a free drug Cmax of 0.007 μm. To determine the potential for in vivo drug interactions, this serum Cmax is compared with the in vitro inhibition constant, Ki. At the therapeutic dose, the free drug concentration of ziprasidone is approximately 1500-fold lower than the mean Ki exhibited for CYP2D6 and more than 9000-fold lower than the mean Ki calculated for CYP3A4. Thus, ziprasidone is not expected to affect significantly the metabolism of coadministered drugs that are metabolized by these isoforms. Interactions with the other major drug-metabolizing CYP isoforms were less potent and therefore drug interactions with these isoforms are less likely.
Estimated steady-state plasma concentrations for risperidone and its active metabolite, 9-hydroxyrisperidone, in schizophrenic patients receiving risperidone, 8 mg twice daily, were approximately 20 and 110 ng ml-1, respectively [27]. Assuming a peak-to-trough ratio of 5 for risperidone (t½,z of 3 h in extensive metabolisers) and a peak-to-trough ratio of 1 for its metabolite (t½,z of 20 h), the anticipated steady-state Cmax concentrations for risperidone and its metabolite would be 100 and 110 ng ml−1, respectively. The unbound (fraction unbound of 0.1) and total plasma Cmax for risperidone would then be 0.024 and 0.24 μm, respectively. These concentrations are 300- and 30-fold lower than their respective Ki values for CYP2D6. Thus, although the unbound Cmax for risperidone is greater than that for ziprasidone, based on our best estimates of therapeutic concentrations of risperidone at clinically efficacious doses, inhibition of coadministered substrates for CYP2D6 would also not be anticipated for risperidone, even with the additional contribution from its active metabolite, 9-hydroxyrisperidone.
The results of this in vitro study indicate that ziprasidone can be administered together with substrates of CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4, without the expectation of pharmacokinetic interactions. Although ziprasidone interacts with CYP2D6 and CYP3A4, it is not expected to inhibit these isozymes at clinically effective doses. There is some uncertainty as to the appropriate in vivo drug concentration with which to compare the in vitro-derived inhibitory constants [28]. However, even using a conservative estimate of relevant in vivo concentrations, total ziprasidone Cmax, the anticipated inhibition by ziprasidone, acting as a competitive inhibitor of CYP2D6, would only be approximately 4%. The lack of clinically significant inhibition of CYP2D6 by ziprasidone has been confirmed in vivo in a study in healthy volunteers. In this study, treatment with ziprasidone (80 mg) did not change the urinary dextromethorphan/dextrorphan ratio, an indicator of CYP2D6 activity, whereas treatment with paroxetine (20 mg), which has been shown to inhibit coadministered substrates of CYP2D6 [29], increased the ratio 10-fold [30].
Acknowledgments
We would like to thank Dr Keith McCarthy and Ms Cynthia Morrency for synthesizing radiolabelled ziprasidone, Dr Larry Tremaine for helpful suggestions and Ms Dayna Mankowski for technical assistance.
References
- 1.Kamel A, Prakash C. Identification of in vitro metabolites of CP-88,059 in humans: characterization of metabolites by LC-MS/MS. Proceedings of the 43rd ASMS Conference on Mass Spectrometry and Allied Topics 887.
- 2.Nelson DR, Kamataki T, Waxman DJ, et al. The P450 superfamily: update on new sequences, gene mapping, accession numbers, early trivial names and nomenclature. DNA Cell Biol. 1993;12:1–51. doi: 10.1089/dna.1993.12.1. [DOI] [PubMed] [Google Scholar]
- 3.Gonzalez FJ. The molecular biology of cytochrome P450s. Pharmacol Rev. 1989;40:243–288. [PubMed] [Google Scholar]
- 4.Peck CC, Temple R, Collins JM. Understanding consequences of concurrent therapies. JAMA. 1993;269:1550–1552. [PubMed] [Google Scholar]
- 5.Tucker GT. The rational selection of drug interaction studies: implications of recent advances in drug metabolism. Int J Clin Pharmacol Ther Toxicol. 1992;30:550–553. [PubMed] [Google Scholar]
- 6.Wrighton SA, Vandenbranden M, Stevens J, et al. In vitro methods for assessing human hepatic drug metabolism: their use in drug development. Drug Metab Rev. 1993;25:453–484. doi: 10.3109/03602539308993982. [DOI] [PubMed] [Google Scholar]
- 7.Rodriguez AD. Use of in vitro metabolism studies in drug development. An industrial perspective. Biochem Pharmacol. 1994;48:2147–2156. doi: 10.1016/0006-2952(94)00312-2. [DOI] [PubMed] [Google Scholar]
- 8.Prakash C, Kamel A, Anderson W, Howard H. Metabolism and excretion of an antipsychotic drug ziprasidone in rat following oral administration of a mixture of 14C and 3H labelled ziprasidone. Drug Metab Dispos. 1997;25:206–218. [PubMed] [Google Scholar]
- 9.Howard HR, Shenk KD, Smolarek TA, et al. Synthesis of 3H- and 14C-labeled CP-88,059: a potent atypical antipsychotic agent. J Labelled Compd Radiopharm. 1994;34:117–125. [Google Scholar]
- 10.Tweedie DJ, Burke MD. Differential effects of phenobarbitone and 3-methylcholanthrene induction on the hepatic microsomal metabolism of the beta-carbolines harmine and harmol. Biochem Pharmacol. 1983;32:653–663. doi: 10.1016/0006-2952(83)90490-2. [DOI] [PubMed] [Google Scholar]
- 11.Smith PK, Krohn RI, Hermanson GT, et al. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 12.Omura T, Sato R. The carbon monoxide binding pigment of liver microsomes, I. Evidence of its hemoprotein nature. J Biol Chem. 1964;239:2370–2378. [PubMed] [Google Scholar]
- 13.Tassaneeyakul W, Birkett DJ, Veronese ME, et al. Specificity of substrate and inhibitor probes for human cytochromes P450 1A1 and 1A2. J Pharmacol Exp Ther. 1993;265:401–407. [PubMed] [Google Scholar]
- 14.Miners JO, Smith KJ, Robson RA, et al. Tolbutamide hydroxylation by human liver microsomes. Biochem Pharmacol. 1988;37:137–144. doi: 10.1016/0006-2952(88)90522-9. [DOI] [PubMed] [Google Scholar]
- 15.Meier UT, Kronbach T, Meyer UA. Assay of mephenytoin metabolism in human liver microsomes by high performance liquid chromatography. Anal Biochem. 1985;151:286–291. doi: 10.1016/0003-2697(85)90177-0. [DOI] [PubMed] [Google Scholar]
- 16.Kronbach T. Bufuralol, dextromethorphan, and debrisoquine as prototype substrates for human P450IID6. Meth Enzymol. 1991;206:509–517. doi: 10.1016/0076-6879(91)06120-r. [DOI] [PubMed] [Google Scholar]
- 17.Sonderfan AJ, Arlotto MP, Dutton DR, et al. Regulation of testosterone hydroxylation by rat liver microsomal cytochrome P-450. Arch Biochem Biophys. 1987;255:27–41. doi: 10.1016/0003-9861(87)90291-8. [DOI] [PubMed] [Google Scholar]
- 18.Orsi BA, Tipton KF. Kinetic analysis of progress curves. Meth Enzymol. 1979;63:159–183. doi: 10.1016/0076-6879(79)63010-0. [DOI] [PubMed] [Google Scholar]
- 19.Segel IH. Enzyme Kinetics: Behaviours and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems. New York: John Wiley and Sons; 1975. pp. 100–226. [Google Scholar]
- 20.Prakash C, Kamel A, Anderson W, Wilner K. Characterization of metabolites of ziprasidone in human by LC-MS/MS. Proceedings of the 44th ASMS Conference on Mass Spectrometry and Allied Topics 563.
- 21.Prakash C, Kamel A, Anderson W, Fouda H. Characterization of metabolites of CP-88,059 in rat using HPLC/RAM/ESI/MS/MS. Proceedings of the 42nd ASMS Conference on Mass Spectrometry and Allied Topics 355.
- 22.Prakash C, Kamel A, Anderson W. Biotransformation of antipsychotic drug ziprasidone in dogs. Proceedings of the 4th International ISSX Meeting 312.
- 23.Newton DJ, Wang RW, Lu AY. Cytochrome P450 inhibitors. Evaluation of specificities in the in vitro metabolism of therapeutic agents by human liver microsomes. Drug Metab Dispos. 1995;23:154–158. [PubMed] [Google Scholar]
- 24.Wilner KD, Hansen RA, Folger CJ, Geoffroy P. The pharmacokinetics of ziprasidone in healthy volunteers treated with cimetidine or antacid. Br J Clin Pharmacol. 2000;49(Suppl. 1):57S–60S. doi: 10.1046/j.1365-2125.2000.00154.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Testa B. In: The Metabolism of Drugs and Other Xenobiotics. Testa B, Caldwell J, editors. New York, Academic Press.; pp. 203–234. [Google Scholar]
- 26.Huang ML, Van Peer A, Woestenborghs R, et al. Pharmacokinetics of the novel antipsychotic agent risperidone and the prolactin response in healthy subjects. Clin Pharmacol Ther. 1993;54:257–268. doi: 10.1038/clpt.1993.146. [DOI] [PubMed] [Google Scholar]
- 27.Heykants J. The pharmacokinetics of risperidone in humans. The Ist International Risperidone Investigators Meeting; :19–20. [Google Scholar]
- 28.Ring BJ, Binkley SN, Roskos L, Wrighton SA. Effect of fluoxetine, norfluoxetine, sertraline and desmethylsertraline on human CYP3A catalyzed 1(-hydroxy midazolam formation in vitro. J Pharmacol Exp Ther. 1995;275:131–135. [PubMed] [Google Scholar]
- 29.Brosen K, Hansen JG, Nielsen KK, Sindrup SH, Gram LF. Inhibition by paroxetine of desipramine metabolism in extensive but not in poor metabolizers of sparteine. Eur J Clin Pharmacol. 1993;44:349–355. doi: 10.1007/BF00316471. [DOI] [PubMed] [Google Scholar]
- 30.Wilner KD, Demattos SB, Anziano RJ, Apseloff G, Gerber N. Ziprasidone and the activity of cytochrome P450 2D6 in healthy extensive metabolisers. Br J Clin Pharmacol. 2000;49(Suppl. 1):43S–47S. doi: 10.1046/j.1365-2125.2000.00152.x. [DOI] [PMC free article] [PubMed] [Google Scholar]