

Abstract
Aims
To assess the effects of multiple oral doses of ketoconazole on the single-dose pharmacokinetics of oral ziprasidone HCl.
Methods
This was a 14-day, open-label, randomized, crossover study in 14 healthy subjects aged 18–31 years. Group 1 received oral ketoconazole 400 mg once daily for 6 days, followed by a 2 day wash-out period and 6 days of placebo administration. Group 2 received placebo followed by ketoconazole. Single oral doses of ziprasidone HCl 40 mg were administered on days 5 and 13 in both groups. Ziprasidone pharmacokinetic parameters were compared between placebo and ketoconazole administration periods.
Results
Co-administration of ziprasidone with ketoconazole was associated with a modest increase in ziprasidone exposure; mean ziprasidone AUC(0,∞) increased by 33%, from 899 ng ml−1 h with placebo to 1199 ngml−1 h with ketoconazole. Mean Cmax increased by 34%, from 89 ng ml−1 to 119 ng ml−1, respectively. The treatment effect on both of these parameters was statistically significant (P< 0.02). Most adverse events were of mild intensity. There were no serious adverse events, laboratory abnormalities, abnormal ECGs, or clinically significant alterations in vital signs throughout the study.
Conclusions
The concurrent administration of ketoconazole and ziprasidone led to modest, statistically significant increases in ziprasidone exposure, although the changes seen were not considered clinically relevant. This suggests that other inhibitors of CYP3A4 are unlikely to significantly affect the pharmacokinetics of ziprasidone.
Keywords: interaction, ketoconazole, pharmacokinetics, ziprasidone
Introduction
Studies conducted in vitro have shown that the oxidative metabolism of ziprasidone is mediated primarily by isoform 3A4 of the hepatic cytochrome P450 system (CYP3A4) [1, 2], and that ziprasidone does not inhibit cytochromes CYP1A2, CYP2C9, CYP2C19, CYP2D6, or CYP3A4 at clinically relevant concentrations [2, 3]. Ziprasidone is therefore unlikely to interfere with the cytochrome-dependent metabolism and the pharmaco-kinetics of a large number of widely used drugs such as antiarrhythmics, antihistamines, tricyclic antidepressants, β-adrenoceptor blockers, and calcium channel antagonists.
Phase I studies in healthy subjects have also demonstrated no clinically significant pharmacokinetic interactions between ziprasidone and cimetidine (which inhibits several isoforms of CYP including CYP3A4), ethinyloestradiol (which is metabolized by CYP3A4), or carbamazepine (a substrate for, and potent inducer of, CYP3A4), suggesting that co-administration with other CYP3A4 inhibitors or inducers will not present a problem [4–6].
Ketoconazole, an imidazole derivative and broad-spectrum antifungal treatment for superficial and systemic fungal infections, is a potent inhibitor of CYP3A4. It has been shown to reduce the oxidative metabolism of drugs such as warfarin and some benzodiazapines [7], as well as antihistamines and antacids that share the CYP3A4 pathway [8, 9].
The following open-label, randomized, placebo-controlled, crossover study in healthy volunteers evaluated the effect of ketoconazole inhibition of CYP3A4 on ziprasidone pharmacokinetics.
Methods
Subjects
Fourteen healthy individuals (men or women) 18–45 years old, weighing no more than 91 kg and within 10% of their ideal body weight for age, height, gender, and frame [10] were enrolled into the study. Subjects with evidence or a history of clinically significant allergic (except for untreated asymptomatic seasonal allergies), haematological, renal, endocrine, pulmonary, gastrointestinal, cardiovascular, hepatic, psychiatric, or neurological disease (including all forms of epilepsy) were excluded. Smokers, subjects with any condition that could affect drug absorption, and subjects with known drug or alcohol dependence or drug allergies were also excluded. Women were required to have been surgically sterilized, or at least 2 years postmenopausal, or to have been using reliable contraception for at least 3 months.
Subjects were excluded if they had taken any pre-scription medication (except contraceptives), over-the-counter, or recreational drugs within 2 weeks, or any investigational drug within 4 weeks of study entry. Alcohol and concomitant medications were not allowed during the study.
The study had institutional review board approval. All subjects gave informed written consent.
Protocol
This was a randomized, open-label, placebo-controlled, crossover study. Following screening, subjects were randomized to two groups. Subjects in group 1 received once-daily oral ketoconazole 200 mg after food on the morning of day 1, which was increased to 400 mg on days 2–6. After a 2 day wash-out period on days 7 and 8, they then received placebo on days 9–14. Subjects in group 2 were treated exactly as in group 1 except that they received placebo on days 1–6 and ketoconazole on days 9–14. Single oral doses of ziprasidone HCl 40 mg were administered on days 5 and 13. During the 14-day study period, subjects received ketoconazole or placebo on an outpatient basis. However, subjects were housed in the research facility under continuous medical supervision for at least 12 h prior to, and 36 h after dosing with ziprasidone on days 5 and 13.
Ziprasidone was administered with 100 ml water immediately after consumption of a standard high-fat breakfast. Ketoconazole and placebo were taken with 50 ml water after a light breakfast, except on day 5 (group 1) or day 13 (group 2) when they were co-administered with ziprasidone. When ziprasidone was administered, subjects did not eat, lie down (except for vital sign measurements), or drink caffeinated or decaffeinated beverages for up to 4 h after dosing.
Pharmacokinetic sampling and analysis
Blood samples were collected before morning dosing on days 1, 4, 5, 6, 12, 13, and 14, in tubes containing EDTA, for the determination of trough plasma concentrations of ketoconazole. Plasma was separated from whole blood samples by centrifugation within 1 h of collection. On days 5 and 13 (ziprasidone dosing days), blood samples were collected in plain tubes (no preservative, anticoagulant, or serum separator) just before ziprasidone dosing and then 1, 2, 4, 6, 8, 12, 18, 24, and 36 h after the morning ziprasidone dose. These samples were allowed to clot at room temperature to extract serum for the measurement of ziprasidone concentration. All samples were stored at −20 °C until analysed for either ziprasidone or ketoconazole concentrations.
Serum ziprasidone concentrations were determined using h.p.l.c.) assay using a solid phase extraction procedure and ultraviolet (u.v.) detection [11]. This assay had a dynamic range of 1–250 ng ml−1 with concentrations below 1 ng ml−1 reported as 0 ng ml−1. Trough plasma ketoconazole concentrations were also assayed using a validated h.p.l.c. method with u.v. detection. This assay had a dynamic range of 50–5000 ng ml−1.
Data analysis
The effect of ketoconazole on ziprasidone pharmacokinetics was assessed by comparing pharmacokinetic parameters on days 5 and 13 between both treatment groups. Maximum observed serum concentrations of ziprasidone (Cmax), and the time at which Cmax occurred (tmax) were determined directly from the experimental data. The terminal elimination phase rate constant (λz) was estimated from individual concentration–time curves using least-squares regression analysis of data obtained during the log-linear phase. The terminal phase half-life of ziprasidone (t½,z) was calculated as ln 2/λz and the mean half-life of ziprasidone was estimated as ln 2/mean λz.
Total area under the concentration–time curve, AUC(0,∞), was determined as the sum of AUC(0, t) and AUC(t,∞). AUC(0, t) was estimated using linear trapezoidal approximation, where t is the time of the last sample with quantifiable concentrations of ziprasidone; AUC(t, ∞) was estimated as Cpest /λz, where Cpest was the estimated concentration at time t, based on regression analysis from the log-concentration–time curve.
An estimated 14 subjects (seven to receive ketoconazole and seven to receive placebo) were required to detect a 50% difference in the change in AUC(0,∞) between the two groups with 80% power and a 5% significance level. An analysis of variance (anova) model containing sequence, period, and treatment effects was used to analyse natural log-transformed AUC(0,∞) and Cmax in PROC GLM of SAS® at the 5% significance level. Differences between ziprasidone pharmacokinetic parameters with concomitant placebo and ketoconazole treatments were estimated by comparing adjusted means for groups 1 and 2, their variances, covariances and 95% confidence limits, calculated using the SAS® LSMEANS function.
Tolerability assessments
Tolerability assessments during the study were clinical laboratory tests including liver function tests (alkaline phosphatase, aspartate aminotransferase [SGOT], alanine aminotransferase [SGPT], and total bilirubin) 24 h before day 1 and on days 5 and 13, and vital sign measurements (blood pressure and pulse rate) on days 1–6 and 9–14. Physical examinations were performed at screening, on completion, or discontinuation from the study.
Adverse events occurring during study treatment and up to 6 days after the last day of dosing were recorded using standard COSTART definitions and classified according to the investigator’s assessment of intensity (mild, moderate, severe) and causal relationship to study drug.
Results
Subjects
A total of 14 subjects (6 men and 8 women) were enrolled in the study. Mean age and body weights of the men were 25.5 years (range 18–31 years) and 73.8 kg (range 68–83 kg), respectively. All six men were White. Mean age and weight of the women were 29.3 years (range 22–44 years) and 58.8 kg (range 49–68 kg), respectively. Six of the women were White, one was Black and one was Hispanic. Thirteen subjects completed the study. One subject discontinued from the study for personal reasons, after taking placebo for 4 days in the second treatment phase.
Pharmacokinetics
Figure 1 shows the mean serum ziprasidone concentration–time curves on days 5 and 13 for both groups. Mean ziprasidone pharmacokinetic parameters for both groups on days 5 and 13 are summarized in Table 1. Mean pharmacokinetic parameters were similar in both groups following administration of ziprasidone with ketoconazole. Following administration of ziprasidone with placebo, tmax occurred 3 h later in group 2 compared with group 1; all other parameters were similar in both groups. anova showed that there were no significant effects on AUC(0,∞) and Cmax of either the sequence or the period in which the study drugs were given (Table 1). Thus, data from corresponding treatment phases of the two groups were combined for analysis of treatment effects.
Figure 1.
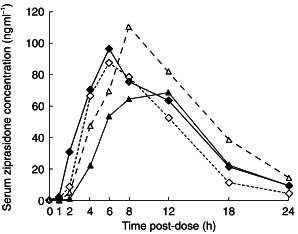
Mean serum ziprasidone concentrations on days 5 and 13 for groups 1 and 2. Group 1, day 5 ketoconazole (♦), group 1 day 13 placebo (◊); group 2, day 5 placebo (▴), and group 2 day 13 ketoconazole (▵).
Table 1.
Mean±s.d. pharmacokinetic parameters for ziprasidone on days 5 and 13 during placebo and ketoconazole administration.
| Group 1 | Group 2 | ||||
|---|---|---|---|---|---|
| Day 5 Ziprasidone + ketoconazole (n = 6) | Day 13 Ziprasidone + placebo (n = 6) | Day 5 Ziprasidone + placebo (n = 7) | Day 13 Ziprasidone + ketoconazole (n = 7) | Pd (S, P effects) | |
| AUC(0,∞)a (ng ml−1 h) | 1107 ± 232 | 917 ± 165 | 882 ± 335 | 1299 ± 351 | 0.66, 0.23 |
| Cmaxa (ng ml−1) | 117 ± 36 | 101 ± 28 | 79 ± 31 | 121 ± 57 | 0.57, 0.19 |
| tmaxb (h) | 8.3 ± 2.9 | 7.0 ± 2.8 | 10.0 ± 2.6 | 10.0 ± 2.6 | 0.49, 0.55 |
| λzb (h−1) | 0.198 ± 0.067 | 0.177 ± 0.05 | 0.151 ± 0.029 | 0.169 ± 0.051 | 0.28, 0.88 |
| t½,zc (h) | 3.5 | 3.9 | 4.6 | 4.1 | – |
Geometric means and s.d.
Arithmetic means and s.d.
Calculated as ln 2/mean λz.
Significance for sequence (S) and period (P) effects, in that order.
Co-administration of ziprasidone with ketoconazole was associated with an increase in overall ziprasidone exposure compared with placebo (Table 2). Adjusted mean values for ziprasidone AUC(0,∞) increased by 33%, from 899 ng ml−1 h with placebo to 1199 ngml−1 h with ketoconazole. Corresponding adjusted mean Cmax values increased by 34%, from 89 ng ml−1 with placebo to 119 ng ml−1 with ketoconazole (Table 2). These changes were associated with a small increase in ziprasidone λz from 0.164 h−1 (placebo) to 0.183 h−1 (ketoconazole), corresponding to a modest decrease in ziprasidone t½,z from 4.2 h (placebo) to 3.8 h (ketoconazole). Adjusted mean values for ziprasidone tmax were similar with values of 8.5 h for placebo and 9.2 h for ketoconazole.
Table 2.
Summary of adjusted (anova) mean pharmacokinetic parameters for ziprasidone on days 5 and 13 during placebo and ketoconazole administration.
| Ziprasidone + placebo (n =13) | Ziprasidone + ketoconazole (n =13) | Ratio (AUC and Cmax) or difference (tmax, λz) | 95% CI | Pd (T effect) | |
|---|---|---|---|---|---|
| AUC(0,∞)a(ng ml−1 h) | 899 | 1199 | 133.3% | 112.4–158.1% | 0.003 |
| Cmaxa (ng ml−1) | 89 | 119 | 133.5% | 106.9–166.7% | 0.015 |
| tmaxb (h) | 8.5 | 9.2 | 0.7 | −1.7–3.0 | 0.549 |
| λzb (h−1) | 0.164 | 0.183 | 0.019 | −0.012–0.051 | 0.197 |
| t½,zc (h) | 4.2 | 3.8 | 0.45 | – | – |
Adjusted geometric mean.
Adjusted arithmetic mean.
Calculated as ln 2/mean λz.
Significance for treatment (T) effect.
anova showed that the effects of drug administration (i.e. ziprasidone plus placebo vs ziprasidone plus ketoconazole) on ziprasidone AUC(0,∞) and Cmax were statistically significant (P< 0.02) (Table 2). In contrast, anova showed no significant effect of either period or treatment on tmax, but a slight influence of sequence on tmax was shown to be statistically significant (P = 0.05; Table 1). There was no apparent difference in ziprasidone pharmacokinetics between men and women (data not shown).
Mean trough plasma ketoconazole concentrations determined 1 day before and just before ziprasidone administration were similar in the ketoconazole–placebo sequence (group 1; 236–286 ng ml−1) and the placebo–ketoconazole sequence (group 2; 278–302 ng ml−1). One day after ziprasidone administration, mean trough ketoconazole concentrations were approximately 2-fold higher compared with those before ziprasidone dosing.
Tolerability
The majority of adverse events experienced by patients in groups 1 and 2 were of mild severity. Two adverse events of moderate intensity were recorded; one in a subject given ketoconazole alone (headache) and one in a subject given ketoconazole and ziprasidone (dizziness).
Mild to moderate adverse events were recorded in 2/14 (14%) subjects given placebo alone, 4/14 (29%) subjects given ketoconazole alone, 4/13 (30%) subjects given ziprasidone with placebo, and 10/14 (71%) subjects given ziprasidone and ketoconazole together. The most frequent treatment-emergent adverse events seen in subjects given ziprasidone with ketoconazole were mild or moderate dizziness (5/14 subjects), asthenia (4/14 subjects) and somnolence (3/14 subjects). While taking ziprasidone with placebo, subjects experienced asthenia (2/13 subjects), dizziness (1/13 subject), and somnolence (1/13 subject). There were no serious adverse events, treatment-emergent laboratory abnormalities or significant changes in vital signs throughout the study, or within a 6 day period after study completion.
One subject receiving ziprasidone with placebo (group 1) experienced a single, brief episode of mild syncope and dizziness on day 13 that was judged by the investigator to be related to study medication. One subject receiving ziprasidone with ketoconazole (group 2) experienced a single, brief episode of mild syncope 8 h after ziprasidone administration on day 13. This episode resolved spontaneously within 1 min, and was judged to be unrelated to study medication.
Discussion
Ketoconazole is a potent inhibitor of the P450 isozyme CYP3A4. Numerous reports have demonstrated that the concurrent administration of ketoconazole reduces the metabolism of other drugs metabolized by this isozyme [7, 12–14]. The primary route of the oxidative metabolism of ziprasidone is mediated by CYP3A4 and it is important to investigate whether co-administration with ketoconazole would produce clinically relevant changes in ziprasidone exposure and safety. Dhis study examined the pharmacokinetics and safety of ziprasidone coadministered with ketoconazole (under steady-state conditions) or placebo in healthy volunteers.
Concurrent administration of a single dose of ziprasidone (40 mg) with multiple doses of ketoconazole (400 mg day−1), given to attain steady-state conditions, resulted in a modest but statistically significant increase in ziprasidone exposure compared with concurrent administration of ziprasidone and placebo. This change, observed as a 34% increase in Cmax and a 33% increase in AUC(0,∞) (P< 0.02), is considered clinically insignifi-cant.
There were no statistically significant changes in tmax, t½,z, and λz when ziprasidone was given with ketoconazole. The difference in tmax between the placebo phases of group 1 and group 2 (7.0 h vs 10.0 h) represented a statistically significant sequence effect on tmax (P =0.05). However, there were no significant treatment or period effects on tmax and no significant sequence, treatment, or period effects on the other pharmacokinetic parameters.
In a single-dose study involving healthy volunteers, a small (6%) increase in ziprasidone exposure was observed following co-administration with cimetidine 800 mg (a nonspecific CYP inhibitor), as measured by AUC(0,∞) [4]. No statistically significant changes in Cmax, tmax, or λz were observed. A study on the effect of CYP3A4 induction on ziprasidone, using carbamazepine, showed a statistically significant but modest reduction in steady-state ziprasidone exposure; AUC(0,12 h) and Cmax decreased by 36% and 27%, respectively [6]. However, these results suggest that neither CYP3A4 inhibitors nor CYP3A4 inducers have a clinically significant effect on the pharmacokinetics of ziprasidone. The clinically insignificant effect of ketoconazole on ziprasidone pharmacokinetics contrasts with the interaction between ketoconazole and model CYP3A4 substrates, such as triazolobenzodiazepines. For example, administration of ketoconazole 400 mg day−1 for 4 days increases the AUC of a single dose of triazolam by > 2000%[15].
Trough plasma ketoconazole concentrations were elevated during concurrent administration with ziprasidone. This increase is believed to be related to the consumption of the standardized high-fat breakfast prior to ketoconazole dosing on the days when ziprasidone was administered, leading to enhanced drug solubilization, absorption, and bioavailability [16, 17].
The modest increase in ziprasidone exposure following ketoconazole administration is consistent with inhibition of CYP3A4 by ketoconazole and the results of in vitro studies, which show that CYP3A4 is the major CYP isoform involved in ziprasidone metabolism. Other CYP isoforms (CYP1A2, CYP2C9, CYP2C19, and CYP2D6) are unlikely to be implicated in ziprasidone metabolism based on the evidence of human liver microsome studies [3]. Isoform-selective substrate studies conducted in human hepatic microsomes determined that ziprasidone concentrations required to inhibit CYP3A4 and CYP2D6 would be at least 1500-fold higher than those achievable clinically [3]. The Ki for ziprasidone at CYP3A4 is 64 µm[3]. Thus, ziprasidone is not expected to inhibit the oxidative metabolism of CYP3A4 substrates.
The majority of adverse events reported were of mild intensity. Adverse events most often reported in subjects receiving ziprasidone and ketoconazole were dizziness, asthenia, and somnolence. There were no treatment-related laboratory abnormalities or abnormal vital signs during the study and 6 days after study completion.
In conclusion, ziprasidone is oxidatively metabolized by CYP3A4, and the potent CYP3A4 inhibitor, ketoconazole, does not cause clinically significant increases in ziprasidone exposure following single oral doses of 40 mg. This suggests that ziprasidone dose modifications are unlikely to be necessary in patients receiving ketoconazole or other potent CYP3A4 inhibitors.
References
- 1.Prakash C, Kamel A, Anderson W, Fouda H. Characterization of metabolites of CP-88 059 in rat using HPLC/RAM/ESI/MS/MS. Proceedings of the 42nd ASMS Conference on Mass Spectrometry and Allied Topics. Chicago, USA.
- 2.Prakash C, Kamel A, Cui D, Whalen RD, Miceli JJ, Tweedie DJ. Ziprasidone metabolism and cytochrome P450 isoforms. Biol Psychiatry. 1997;42:40S. [Google Scholar]
- 3.Prakash C, Kamel A, Cui D, Whalen RD, Miceli JJ, Tweedie D. Identification of the major human liver cytochrome P450 isoform (s) responsible for the formation of the primary metabolites of ziprasidone and prediction of possible drug interactions. Br J Clin Pharmacol. 2000;49(Suppl. 1):35S–42S. doi: 10.1046/j.1365-2125.2000.00151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilner KD, Hansen RA, Folger CJ, Geoffroy P. The pharmacokinetics of ziprasidone in healthy volunteers treated with cimetidine or antacid. Br J Clin Pharmacol. 2000;49(Suppl. 1):57S–60S. doi: 10.1046/j.1365-2125.2000.00154.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muirhead GJ, Harness J, Holt PR, Oliver S, Anziano RJ. Ziprasidone and the pharmacokinetics of a combined oral contraceptive. Br J Clin Pharmacol. 2000;49(Suppl. 1):49S–56S. doi: 10.1046/j.1365-2125.2000.00153.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miceli JJ, Laurent A. The effect of carbamazepine on the steady-state pharmacokinetics of ziprasidone in healthy volunteers. Br J Clin Pharmacol. 2000;49(Suppl. 1):65S–70S. doi: 10.1046/j.1365-2125.2000.00157.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bennett JE. Antimicrobial agents: antifungal agents. In: Hardman JJG, Limbird LE, Molinoff PB, Ruddon RW, Goodman Gilman A, editors. The Pharmacological Basis of Therapeutics. 9. UAS: McGraw-Hill; 1996. pp. 1175–1190. [Google Scholar]
- 8.Ament PW, Paterson A. Drug interactions with the non-sedating antihistamines. Am Fam Physician. 1997;56:223–231. [PubMed] [Google Scholar]
- 9.Sadowski DC. Drug interactions with antacids. Mechanisms and clinical significance. Drug Safety. 1994;11:395–407. doi: 10.2165/00002018-199411060-00002. [DOI] [PubMed] [Google Scholar]
- 10.Metropolitan height and weight tables Stat Bull Metrop Life Found. 1983;64:3–9. [PubMed] [Google Scholar]
- 11.Janiszewski JS, Fouda HG, Cole RO. Development and validation of a high-sensitivity assay for an antipsychotic agent, CP-88,059, with solid-phase excavation and narrow-bore high-performance liquid chromatography. J Chromatogr. 1995;668:133–139. doi: 10.1016/0378-4347(95)00071-p. [DOI] [PubMed] [Google Scholar]
- 12.Babe KS, Serafin WE. Histamine, Bradykinin, and their atagonists. In: Hardman JJG, Limbird LE, Molinoff PB, Ruddon RW, Goodman Gilman A, editors. The Pharmacological Basis of Therapeutics. 9. USA: McGraw-Hill; 1996. pp. 581–600. [Google Scholar]
- 13.Ereshefsky L. Pharmacokinetics and drug interactions: update for new antipsychotics. J Clin Psychiatry. 1996;57:12–25. [PubMed] [Google Scholar]
- 14.Raeissi SD, Guo Z, Dobson GL, Artursson P, Hidalgo IJ. Comparison of CYP3A activities in a subclone of Caco-2 cells (TC7) and human Intestine. Pharm Res. 1997;14:1019–1025. doi: 10.1023/a:1012197110917. [DOI] [PubMed] [Google Scholar]
- 15.Preskorn SH. Clinical Pharmacology of Selective Serotonin Reuptake Inhibitors. Caddo, OK, USA: Professional Communications; 1996. [Google Scholar]
- 16.Daneshmend TK, Warnock DW, Ene MD, et al. Influence of food on the pharmacokinetics of ketoconazole. Antimicrob Agents Chemother. 1984;25:1–3. doi: 10.1128/aac.25.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lelawongs P, Barone AB, Colaizzi JL, et al. Effects of food and gastric acidity on absorption of orally administered ketoconazole. Clin Pharmacol. 1988;7:228–235. [PubMed] [Google Scholar]