

Abstract
Aims
To evaluate the effect of steady-state carbamazepine administration on the steady-state pharmacokinetics of ziprasidone in healthy young adults, in an open, randomised, parallel-group study.
Methods
Twenty-five subjects were randomized to one of two treatment groups. Group 1 received 20 mg ziprasidone twice daily on days 1 and 2, and a single dose on day 3. A single 100 mg dose of carbamazepine was given once daily on days 5 and 6 and twice daily on days 7 and 8, followed by 200 mg twice daily until day 28 and on the morning only on day 29. Ziprasidone 20 mg was also administered twice daily on days 26 and 27 and in the morning only on day 28. Group 2 received the same treatment regimen with carbamazepine replaced by placebo. Pharmacokinetic data were obtained on days 3 and 28.
Results
Nine subjects in group 1 and 10 in group 2 completed all three treatment periods (ziprasidone, carbamazepine or placebo; and ziprasidone plus carbamazepine or placebo). Carbamazepine administration to group 1 was associated with modest reductions in ziprasidone exposure, with mean decreases in ziprasidone AUC(0,12 h) and Cmax values of 36% and 27%, respectively, on day 28 compared with day 3 (P<0.03). The mean differences between day 28 and day 3 ziprasidone AUC(0,12 h) and Cmax values were also statistically significantly greater in the carbamazepine group than in the placebo group. The mean half-life of ziprasidone decreased by 1 h from day 3 to day 28 in the subjects receiving carbamazepine, compared with virtually no change in the placebo group. All adverse events were mild or moderate in severity and there were no serious adverse events, or clinically significant changes in ECGs and vital signs throughout the study.
Conclusions
Induction of CYP3A4 with carbamazepine led to a modest reduction (<36%) in steady-state exposure to ziprasidone that is believed to be clinically insignificant.
Keywords: carbamazepine, interaction, pharmacokinetics, ziprasidone
Introduction
Studies performed in vitro using human liver microsomes have shown that the oxidative metabolism of ziprasidone is mediated primarily by CYP3A4 [1]. In vivo studies have demonstrated no clinically significant pharmacokinetic interactions between ziprasidone and cimetidine (which inhibits several isoforms of CYP including CYP3A4), ethinyloestradiol (which is metabolized by CYP3A4), or ketoconazole (a potent inhibitor of CYP3A4), suggesting that co-administration with other CYP3A4 inhibitors or inducers will not require dose adjustment [2–4].
Treatment strategies for schizophrenia need to take into account the treatment of mood disorders because these are associated with a poor therapeutic outcome, an increased risk of relapse and a high rate of suicide [5–7]. While antidepressants may be beneficial, carbamazepine has been advocated as an adjunctive therapy for some patients with psychoses with mood disorders, particularly for schizoaffective disorder [8–11]. Carbamazepine is widely used in epilepsy, bipolar disorder and other affective disorders, in individuals with and without psychoses. It is oxidatively metabolized extensively by CYP3A4 [12, 13] and is a potent inducer of this isozyme, with plasma drug concentrations frequently decreasing during the first month of therapy. This induction has been shown to enhance the metabolism of other CYP3A4 substrates, including antiepileptics, tricyclic antidepressants, oral anticoagulants, calcium channel blockers, oral contraceptives, chemotherapeutic agents, and antipsychotics [14]. This can result in clinically relevant drug interactions [15], for example haloperidol levels are 50% lower when carbamazepine is co-administered [16, 17].
The following open-label, randomized, placebocontrolled, parallel-group study in healthy volunteers evaluated the effect of subchronic carbamazepine administration on the steady-state pharmacokinetics of ziprasidone.
Methods
Subjects
Twenty-five healthy young men and women (18–45 years) weighing no more than 91 kg and within 10% of their ideal body weight for age, height, gender, and frame [18] were enrolled into the study. Subjects with evidence or a history of clinically significant allergic (except for untreated asymptomatic seasonal allergies), haematological, renal, endocrine, pulmonary, gastrointestinal, cardiovascular, hepatic, psychiatric, or neurological disease (including all forms of epilepsy) were excluded. Smokers, subjects with any condition that could affect drug absorption, and subjects with known drug or alcohol dependence or drug allergies were also excluded. Women were required to have been surgically sterilized, or at least 2 years postmenopausal, or to have been using reliable contraception for at least 3 months.
Subjects were excluded if they had taken any prescription medication (except contraceptives), over-the-counter or recreational drugs within 2 weeks, or any investigational drug within 4 weeks of study entry. Alcohol and concomitant medications were not allowed during the study.
The study had institutional review board approval. All subjects gave informed written consent.
Protocol
This was an open-label, randomized, placebo-controlled, parallel-group study designed to evaluate the influence of subchronic administration of carbamazepine on the steady-state pharmacokinetics of ziprasidone in healthy subjects.
The study comprised two treatment groups and three treatment periods. On days 1–3 (period 1) both groups received ziprasidone to establish baseline steady-state ziprasidone pharmacokinetics. On days 5–25 (period 2) the subjects were given either carbamazepine (group 1) or placebo (group 2). On days 26–28 (period 3) the subjects received ziprasidone in combination with either carbamazepine (group 1) or placebo (group 2).
Ziprasidone was given orally to both groups as one 20 mg capsule twice daily throughout periods 1 and 3, except on days 3 and 28 when only the morning doses were administered. Carbamazepine (Tegretol XR®, Ciba-Geigy) was given orally to subjects in group 1 as one 100 mg tablet once daily on days 5 and 6, as one 100 mg tablet twice daily on days 7 and 8, and as one 200 mg tablet twice daily on days 9–28, as per instructions included in the package insert (Tegretol XR® dosage instructions, Ciba-Geigy). Placebo was given orally in the same dose regimen as carbamazepine to subjects in group 2. On the morning of day 29, subjects in groups 1 and 2 received either a single 200 mg carbamazepine tablet or placebo, respectively.
The interval between morning and evening doses was 12 h. All doses were administered with 50 ml water, immediately after eating a standard meal over 20 min.
Pharmacokinetic sampling and analysis
Blood samples (sufficient for 3 ml of serum) for the determination of serum ziprasidone concentrations were collected in plain tubes (no preservative, anticoagulant, or serum separator), from all subjects prior to and at 1, 2, 3, 4, 6, 8, 10, 12, 18, 24, and 36 h following the morning dose of ziprasidone on days 3 and 28. Blood samples for the determination of plasma carbamazepine were collected in heparinized tubes, prior to morning dosing on days 5, 10, 14, 18, 22, 25, and 28. Ziprasidone blood samples were allowed to clot at room temperature. Serum (for ziprasidone) and plasma (for carbamazepine) were separated from whole blood samples by centrifugation within 1 h of collection. Samples were stored at −20°C until analysed for ziprasidone and carbamazepine concentrations. Serum concentrations of ziprasidone were determined using a validated high-performance liquid chromatography assay involving solid-phase extraction and u.v. detection. The assay had a dynamic range of 1–250 ng ml−1[19]. Ziprasidone concentrations below the lower limit of quantification were assigned a value of 0 ng ml−1 in pharmacokinetic calculations. Plasma concentrations of carbamazepine were determined using a fluorescence polarization immunoassay.
Data analysis
Steady-state area under the ziprasidone serum concentration–time curve from time zero to 12 h postdose (AUC(0, 12 h)) was estimated using linear trapezoidal approximation. Maximum observed serum concentrations of ziprasidone (Cmax), and the time at which Cmax occurred (tmax) were determined directly from the experimental data. The terminal phase elimination rate constant for ziprasidone (λz) was estimated using least-squares regression analysis of the serum ziprasidone concentration–time data obtained during the log-linear phase. The terminal phase half-life of ziprasidone (t½,z) was calculated as ln 2 /λz and the mean half-life of ziprasidone was estimated as ln 2/mean λz.
An estimated 14 subjects were required to detect a 50% difference in the change (final from baseline) in AUC(0,12 h) between the two treatments with 80% power and a 5% significance level. The effect of carbamazepine on the steady-state pharmacokinetics of ziprasidone was assessed from the changes between day 3 and day 28 in the pharmacokinetic parameters. Parameters were analysed using a two-sample Student’s t-test with 95% confidence limits to compare between-group differences on days 3 and 28, and within-subject changes between day 3 and day 28. Values of P< 0.05 were considered to be statistically significant.
Results
Subjects
A total of 13 subjects were randomized to group 1. Their mean age was 28.8 years (range 23–35 years). Twelve subjects were men with mean body weight 74.6 kg (range 57–89 kg), and the woman weighed 61.7 kg. Mean age of the 12 subjects randomized to group 2 was 31.2 years (range 24–45 years). Eleven were men with mean body weight 76.7 kg (range 60–90 kg), and the woman weighed 60.1 kg. Nine subjects in group 1 and 10 in group 2 completed all three treatment periods and were included in the pharmacokinetic analysis. All adverse events were mild or moderate in severity and there were no serious adverse events, or clinically significant changes in ECGs and vital signs throughout the study.
Pharmacokinetics
Mean serum ziprasidone concentration–time curves for the two groups were similar on day 3 (Figure 1). However, on day 28, following the administration of carbamazepine to subjects in group 1, the serum concentrations of ziprasidone were lower than in the placebo group 2 (Figure 1).
Figure 1.
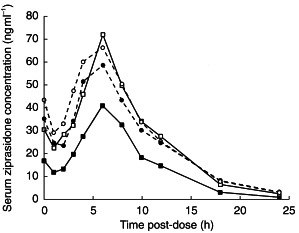
Mean serum ziprasidone concentrations in subjects receiving ziprasidone (40 mg day−1) before (day 3) and after (day 28) the administration of placebo or carbamazepine for 21 days. Day 3 group 1 – carbamazepine (•), day 3 group 2 – placebo (○), day 28 group 1 – carbamazepine (▪), day 28 Group 2 – placebo (□).
There were no statistically significant differences in mean ziprasidone pharmacokinetic parameters between the two groups on day 3. In contrast, there were statistically significant differences between the two groups in the mean steady-state AUC(0,12 h) and Cmax values on day 28 (both P< 0.001; Table 1), following administration of carbamazepine or placebo for 25 days. There were also statistically significant differences between placebo and carbamazepine groups in the mean difference between day 28 and day 3 values for AUC(0,12 h), Cmax and λz (all P< 0.03).
Table 1.
Mean±s. d. pharmacokinetic parameters of ziprasidone.
| Pharmacokinetic parameter | Group 1 (carbamazepine) (n = 9) | Group 2 (placebo) (n =10) | Difference (ratiod) Group 1 – Group 2 means (95% CI) | P value (Group 1 vs Group 2) |
|---|---|---|---|---|
| Day 3 | ||||
| AUC(0,12 h) (ng ml−1 h) | 445 ± 155 | 532 ± 146 | 83.7% (62, 113) | 0.23 |
| Cmaxa (ng ml−1) | 65 ± 25 | 76 ± 3 | 86.1% (61, 122) | 0.38 |
| tmaxb (h) | 5 ± 3 | 6 ± 2 | −1.10 (−3.4, 1.1) | 0.30 |
| λzb (h−1) | 0.160 ± 0.024 | 0.171 ± 0.026 | −0.011 (−0.035, 0.013) | 0.34 |
| t½,zc (h) | 4.3 | 4.1 | – | – |
| Day 28 | ||||
| AUC(0,12 h) (ng ml−1 h) | 285 ± 79 | 495 ± 131 | 57.7% (44, 75) | <0.001 |
| Cmaxa (ng ml−1) | 48 ± 13 | 79 ± 19 | 60.4% (47, 78) | <0.001 |
| tmaxb (h) | 6 ± 1 | 6 ± 2 | 0.40 (−1.1, 1.9) | 0.57 |
| λzb (h−1) | 0.211 ± 0.042 | 0.175 ± 0.046 | 0.037 (−0.006, 0.079) | 0.09 |
| t½,zc (h) | 3.3 | 4.0 | – | – |
| Ratio Day 28/Day3 | ||||
| AUC(0,12 h) (ng ml−1 h) | 0.64 ± 0.20 | 0.93 ± 0.12 | – | – |
| Cmaxa (ng ml−1) | 0.73 ± 0.20 | 1.04 ± 0.25 | – | – |
| t½,zc (h) | 0.77 ± 0.13 | 1.06 ± 0.43 | – | – |
Geometric means and s.d.
Arithmetic means and s.d.
Calculated as ln 2/mean λz.
AUC(0,12 h) and Cmax were analysed on the log scale and back-transformed for calculation of the group 1 – group 2 ratio and 95% confidence limits of differences between means.
Within group 1 (carbamazepine), a mean 27% decrease in Cmax (from 65 to 48 ng ml−1) and a mean 36% decrease in AUC(0,12 h) (from 445 to 285 ng ml −1h) were associated with carbamazepine administration between days 3 and 28. Consistent with these changes was a 32% increase in mean λz, which corresponded to a decrease in mean t½,z of 1 h (3.3 vs 4.3 h). Over the same period, the pharmacokinetic parameters for group 2 (placebo) remained essentially unchanged between days 3 and 28. Mean Cmax increased from 76 to 79 ng ml−1, mean AUC(0,12 h) decreased from 532 to 495 ng ml −1 h, λz increased from 0.171 to 0.175 h−1 and the t½,z decreased from 4.1 to 4.0 h. Figure 2 shows individual AUC(0,12 h), Cmax and t½,z values for carbamazepine and placebo on days 3 and 28.
Figure 2.
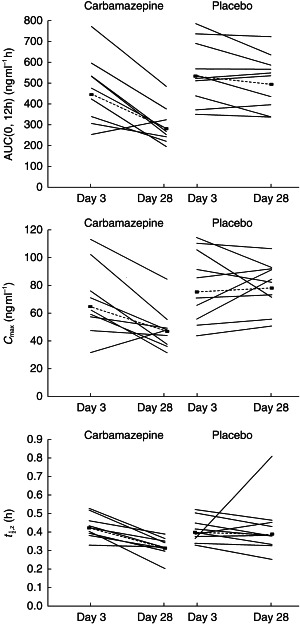
Mean AUC(0,12 h), Cmax and t½,z on days 3 and 28 in subjects receiving ziprasidone (40 mg day−1) before (day 3) and after (day 28) the administration of placebo or carbamazepine for 21 days. Solid lines (----) represent individual observed values, and dashed lines (▪––––▪) represent geometric (Cmax, AUC(0,12 h)) and harmonic (t1/2) mean values.
Discussion
After oral administration of ziprasidone to healthy subjects, ziprasidone is metabolized extensively with less than 5% excreted in the urine and faeces as unchanged drug. Based on the structures of the major metabolites, four routes of metabolism have been identified [20]. In vitro studies using human liver microsomes have demonstrated that CYP3A4 is the major CYP isoform responsible for the oxidative metabolism of ziprasidone [1].
Carbamazepine is a potent inducer of CYP3A4 and is known to influence the pharmacokinetics of numerous drugs that are metabolized by this isozyme [14]. The clinical relevance of such interactions depends on their magnitude and the therapeutic index in terms of the relative effects of drug concentrations on their therapeutic effects as compared with their adverse exposure [21]. The principal findings from the study demonstrate that no clinically significant interaction between carbamazepine and ziprasidone occurs.
Baseline characteristics of the two groups appeared to be well matched in age, weight, gender, and race. The similarity in steady-state pharmacokinetic parameters on day 3, prior to initiation of carbamazepine or placebo administration, also supports the good match between the patient groups. Plasma carbamazepine concentrations were within the clinically relevant concentration range throughout the observation period.
Pharmacokinetic assessments showed that ziprasidone exposure decreased in group 1 subjects on day 28 following concomitant carbamazepine administration from day 5. Within this group, statistically significant decreases in mean AUC(0,12 h) and Cmax, of 36% and 27%, respectively, occurred compared with day 3.
Between-group comparison revealed that mean ziprasidone AUC(0,12 h) and Cmax values in group 1 (carbamazepine) were statistically significantly lower than those in group 2 (placebo) at day 28. In addition, the changes between the day 28 and day 3 values were compared between groups to adjust for the small (but not statistically significant) differences in the ziprasidone pharmacokinetic variable values on day 3. In this calculation, the between-group differences persisted. Indeed, there were statistically significant differences between the carbamazepine and placebo groups in the mean day 28 to day 3 change in ziprasidone AUC(0,12 h), Cmax, and λz values.
The moderate reduction in ziprasidone exposure in the carbamazepine group is consistent with induction of CYP3A4 by carbamazepine [14] and supports the results of in vitro studies, which show that CYP3A4 is the major isoform involved in the oxidative metabolism of ziprasidone. Other CYP isoforms (1A2, 2C9, 2C19, and 2D6) are unlikely to be implicated in ziprasidone metabolism on the evidence of human liver microsome studies. In human hepatic microsomes the ziprasidone concentrations required to inhibit CYP3A4 and CYP2D6 would be at least 1500-fold higher than those achievable clinically. The Ki for ziprasidone at CYP3A4 is 64 µm[1]. Thus ziprasidone is unlikely to inhibit the oxidative metabolism of CYP3A4 substrates.
Changes in the pharmacokinetics of other antipsychotics that are metabolized by CYP3A4, including haloperidol, clozapine and quetiapine, have been observed during co-administration with carbamazepine. It has been shown that carbamazepine decreases plasma concentrations of haloperidol by 50–60%. The clinical implication of carbamazepine co-administration appears to be equivocal, as the psychotic symptoms of some patients receiving moderate doses of haloperidol have deteriorated as a consequence, while others receiving higher doses of haloperidol did not deteriorate [22, 23]. Clozapine is at least partly metabolized by CYP3A4 and its plasma levels have also been shown to decrease by approximately 50% during carbamazepine treatment [24]. The pharmacokinetics of some of the newer antipsychotics are affected by co-administration of carbamazepine. Quetiapine is metabolized by CYP3A4, and the CYP3A4 inducer, phenytoin, has been shown to increase its mean oral clearance by 5-fold, which corresponds to a reduction in quetiapine exposure of about 80%. Thus, dose adjustment may be needed when drugs such as phenytoin or carbamazepine are co-administered with quetiapine [25].
In a single-dose study involving healthy volunteers, a 6% increase in ziprasidone exposure was observed following co-administration with cimetidine (CYP3A4 inhibitor), as measured by AUC(0,∞) [2]. In a multiple-dose study involving healthy volunteers, a 33% increase in ziprasidone exposure (AUC(0, ∞)) was seen following co-administration with ketoconazole (a potent CYP3A4 inhibitor) [4]. Cmax also increase by 34%. These results, and the modest effects of carbamazepine on ziprasidone pharmacokinetics, suggest that neither CYP3A4-inhibitors nor CYP3A4 inducers have a clinically significant effect on the pharmacokinetics of ziprasidone, nor are they expected to necessitate dose adjustment of ziprasidone.
In conclusion, the findings of this study are consistent with CYP3A4 induction increasing the metabolism of ziprasidone. They indicate that the effects of concomitant carbamazepine therapy on the AUC(0,12 h) and Cmax of ziprasidone are unlikely to be clinically relevant.
References
- 1.Prakash C, Kamel A, Cui D, et al. Identification of the major human liver cytochrome P450 isoform responsible for the primary metabolites of ziprasidone and prediction of possible drug interactions. Br J Clin Pharmacol. 2000;49(Suppl. 1):35S–42S. doi: 10.1046/j.1365-2125.2000.00151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wilner KD, Hansen RA, Folger CJ, Geoffroy P. The pharmacokinetics of ziprasidone in healthy volunteers treated with cimetidine or antacid. Br J Clin Pharmacol. 2000;49(Suppl. 1):57S–60S. doi: 10.1046/j.1365-2125.2000.00154.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Muirhead GJ, Harness J, Holt PR, Oliver S, Anziano RJ. Ziprasidone and the pharmacokinetics of a combined oral contraceptive. Br J Clin Pharmacol. 2000;49(Suppl. 1):49S–56S. doi: 10.1046/j.1365-2125.2000.00153.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miceli JJ, Smith M, Robarge L, Morse T, Laurent A. The effect of ketoconazole on ziprasidone pharmacokinetics – a placebo-controlled crossover study in healthy volunteers. Br J Clin Pharmacol. 2000;49(Suppl. 1):71S–76S. doi: 10.1046/j.1365-2125.2000.00156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keefe RS, Mohs RC, Losonczy MF. Characteristics of a very poor outcome in schizophrenia. Am J Psychiatry. 1987;144:889–895. doi: 10.1176/ajp.144.7.889. [DOI] [PubMed] [Google Scholar]
- 6.Prasad AJ. Attempted suicide in hospitalised schizophrenics. Acta Psychiatr Scand. 1986;74:41–42. doi: 10.1111/j.1600-0447.1986.tb06224.x. [DOI] [PubMed] [Google Scholar]
- 7.Blak DW, Winokur G, Warrack G. Suicide in schizophrenia: the Iowa Record Linkage Study. J Clin Psychopharmacol. 1996;16:158–169. [PubMed] [Google Scholar]
- 8.Azorin JM. Long-term treatment of mood disorders in schizophrenia. Acta Psychiatr Scand. 1995;388:20–23. doi: 10.1111/j.1600-0447.1995.tb05940.x. [DOI] [PubMed] [Google Scholar]
- 9.Albani F, Riva R, Baruzzi A, et al. Carbamazepine clinical pharmacology: a review. Pharmacopsychiatry. 1995;28:235–244. doi: 10.1055/s-2007-979609. [DOI] [PubMed] [Google Scholar]
- 10.Pantelis C, Barnes TR. Drug strategies and treatment-resistant schizophrenia. Aust NZ J Psychiatry. 1996;30:20–37. doi: 10.3109/00048679609076070. [DOI] [PubMed] [Google Scholar]
- 11.Johns CA, Thompson JW. Adjunctive treatments in schizophrenia: pharmacotherapies and electroconvulsive therapy. Schizophr Bull. 1995;21:607–619. doi: 10.1093/schbul/21.4.607. [DOI] [PubMed] [Google Scholar]
- 12.Levy RH. Cytochrome P450 isozymes and antiepileptic drug interactions. Epilepsia. 1994;36(Suppl 5):S8–S13. doi: 10.1111/j.1528-1157.1995.tb06007.x. [DOI] [PubMed] [Google Scholar]
- 13.Spatzenegger M, Jaeger W. Clinical importance of hepatic cytochrome P450 in drug metabolism. Drug Metab Rev. 1995;27:397–417. doi: 10.3109/03602539508998329. [DOI] [PubMed] [Google Scholar]
- 14.Spina E, Pisani F, Perucca E. Clinically significant pharmacokinetic drug interactions with carbamazepine: an update. Clin Pharmacokinet. 1996;31:198–214. doi: 10.2165/00003088-199631030-00004. [DOI] [PubMed] [Google Scholar]
- 15.Ketter TA, Flockhart DA, Post M, et al. The emerging role of cytochrome P450 3A4 in psychopharmacology. J Clin Psychopharmacol. 1995;15:387–398. doi: 10.1097/00004714-199512000-00002. [DOI] [PubMed] [Google Scholar]
- 16.Ereshefsky L. Pharmacokinetics and drug interactions: update for new antipsychotics. J Clin Psychiatry. 1996;57(Suppl 11):12–25. [PubMed] [Google Scholar]
- 17.Kahn EM, Schulz SC, Perel JM, Alexander JE. Change in haloperidol level due to carbamazepine – a complicating factor in combined medication for schizophrenia. J Clin Psychopharmacol. 1990;10:54–57. doi: 10.1097/00004714-199002000-00011. [DOI] [PubMed] [Google Scholar]
- 18.Metropolitan height and weight tables. Stat Bull Metrop Life Found. 1983;64:3–9. [PubMed] [Google Scholar]
- 19.Janiszewski JS, Fouda HG, Cole RO. Development and validation of a high-sensitivity assay for an antipsychotic agent, CP-88,059, with solid-phase excavation and narrow-bore high-performance liquid chromatography. J Chromatogr. 1995;668:133–139. doi: 10.1016/0378-4347(95)00071-p. [DOI] [PubMed] [Google Scholar]
- 20.Prakash C, Kamel A, Gummerus J, et al. Metabolism and excretion of a new antipsychotic drug, ziprasidone, in humans. Drug Metab Dispos. 1997;25:863–872. [PubMed] [Google Scholar]
- 21.Preskorn SH. Caddo OK, editor. Clinical pharmacology of selective serotonin reuptake inhibitors. 1996. Professional Communications.
- 22.Meyer MC, Baldessarini RJ, Goff DC, et al. Clinically significant interactions of psychotropic agents with antipsychotic drugs. Drug Safety. 1996;15:333–346. doi: 10.2165/00002018-199615050-00004. [DOI] [PubMed] [Google Scholar]
- 23.Iwahashi K, Miyatake R, Suwaki H, et al. The drug-drug effects of haloperidol on plasma carbamazepine levels. Clin Neuropharmacol. 1995;18:233–266. doi: 10.1097/00002826-199506000-00003. [DOI] [PubMed] [Google Scholar]
- 24.Tiihonen J, Vartiainen H, Hakola P, et al. Carbamazepine-induced changes in plasma levels of neuroleptics. Pharmacopsychiatry. 1995;28:26–28. doi: 10.1055/s-2007-979584. [DOI] [PubMed] [Google Scholar]
- 25.Misra LK, Erpanbach JE, Hamlyn H, et al. Quetiapine: a new atypical antipsychotic. SDJ Med. 1998;51:189–193. [PubMed] [Google Scholar]