

Abstract
Aims
To determine whether ziprasidone alters the metabolizing activity of the 2D6 isoenzyme of cytochrome P450 (CYP2D6).
Methods
Twenty-four healthy young subjects aged 18–45 years were screened for CYP2D6 metabolizing activity and shown to be extensive metabolizers of dextromethorphan. These subjects were then randomized to receive a single dose of ziprasidone 80 mg, paroxetine 20 mg or placebo, 2 h before receiving a dose of dextromethorphan. Urine samples for the determination of dextromethorphan concentrations were collected over the 8 h period following dextromethorphan dosing, and used for the determination of dextromethorphan/dextrorphan ratios. Blood samples were collected immediately before and up to 10 h after the administration of ziprasidone or paroxetine, and used to derive pharmacokinetic parameters of ziprasidone and paroxetine.
Results
There were no statistically significant changes in the urinary dextromethorphan/dextrorphan ratio in the ziprasidone group or the placebo group. By contrast, there was a 10-fold increase in the urinary dextromethorphan/dextrorphan ratio in the paroxetine group and this differed significantly from those in the ziprasidone and placebo groups (P = 0.0001).
Conclusions
The findings of this study suggest that ziprasidone does not inhibit the clearance of drugs metabolized by CYP2D6.
Keywords: dextromethorphan, dextrorphan, metabolism, paroxetine, pharmacokinetics, ziprasidone
Introduction
Studies performed in vitro using human liver microsomes have shown that the metabolism of ziprasidone is mediated by the cytochrome P450 (CYP) oxidase system. By using selective inhibitors of each of the five major isoenzymes of CYP, it has been demonstrated that this hepatic metabolism is primarily mediated by CYP3A4 [1]. However, studies performed using specific probe substrates for each of the five major isoenzymes of CYP found that ziprasidone is a weak inhibitor of CYP2D6 (Ki= 11 µm) [1]. As ziprasidone binds extensively to plasma proteins (> 99%), the maximum concentration of unbound ziprasidone attained during treatment at the maximal therapeutic dose (160 mg day−1, in two divided daily doses) is estimated to be approximately 0.007 µm. As such, it is approximately 1500-fold lower than the Ki of ziprasidone for CYP2D6. An in vivo clinical study was used to confirm these in vitro findings.
CYP2D6 is clinically one of the most important CYP isoenzymes, known to be at least partly responsible for the oxidation of many drugs. These include 5-HT-selective re-uptake inhibitors (e.g. paroxetine, fluoxetine), tricyclic antidepressants (e.g. desipramine, amitriptyline), typical antipsychotics (e.g. haloperidol, thioridazine), new antipsychotics (e.g. clozapine, risperidone), β-adrenoceptor antagonists (alprenolol, propranolol), antiarrhythmics (encainide, flecainide), opiates (e.g. dextromethorphan, codeine) and various other drugs (e.g. debrisoquine, sparteine) [2–4]. Several of these drugs may be administered in combination with ziprasidone.
CYP2D6 activity is genetically controlled. In most races, the presence of a polymorphism in the nucleic acid sequence of the relevant gene gives rise to a bimodal distribution of CYP2D6-activity phenotypes within populations. Thus, a certain proportion of any given polymorphic population can be classified as poor metabolisers, with relatively low rates of metabolism for substrates of the isoenzyme, and extensive metabolizers, with relatively high rates of metabolism for substrates of the isoenzyme [4, 5].
The activity of CYP in vivo can be assessed by dosing subjects with a specific probe substrate for the isoenzyme of interest and monitoring changes in the relative concentrations of that substrate and its metabolites. Dextromethorphan is a model probe substrate for CYP2D6 because its metabolism to dextrorphan is primarily mediated by this isoenzyme [6]. One way to assess the in vivo activity of this isoenzyme is therefore to dose subjects with dextromethorphan and monitor urinary concentrations of unchanged dextromethorphan and dextrorphan [7]. Changes in the urinary dextromethorphan/dextrorphan ratio that occur during exposure to a particular drug of interest can then be attributed to the effects of that drug on CYP2D6. Paroxetine has been shown to be a potent inhibitor of CYP2D6, both in vitro and in vivo, using the dextromethorphan/dextrorphan ratio [2, 8, 9]. It can therefore be used as a positive control in studies designed to assess the nature of interactions between investigational drugs and CYP2D6.
Here we report the findings of a study designed to assess whether or not ziprasidone inhibits the in vivo activity of CYP2D6 activity by examining the urinary dextromethorphan/dextrorphan ratio in extensive metabolizers given ziprasidone, paroxetine or placebo.
Methods
Subjects
The subjects were healthy young (18–45 years) adults of either gender who were extensive metabolizers of dextromethorphan (mean urinary dextromethorphan/dextrorphan ratio ≤ 0.03 based on three separate determinations).
All subjects weighed ≤ 94 kg and were within 15% of their ideal weight for their age, height, sex and frame [10]. None of the subjects had received any of the investigational drugs for at least 4 weeks prior to the study nor any other drug therapy (except contraceptive medication) in the 2 weeks preceding entry into the study or during the study. The women had been surgically sterilized, or were at least 2 years postmenopausal or had been practising successful contraception for at least 3 months. Subjects who smoked, or who had any condition possibly affecting drug absorption were excluded. All subjects provided written informed consent.
Protocol
This was an open-label, placebo-controlled, randomized, parallel-group study designed to determine whether ziprasidone alters the CYP2D6 metabolizing activity of healthy volunteers who are extensive metabolizers of dextromethorphan, using paroxetine as a positive control. The study was approved by an independent institutional review board.
During screening, each subject received dextromethorphan HBr (Parke-Davis), at a dose of 30 mg with 240 ml water, on three separate occasions, separated by at least 1 day, to determine their urinary dextromethorphan/dextrorphan ratio. Individuals who had met the study criterion for an extensive metabolizer of dextromethorphan (dextromethorphan/dextrorphan ratio ≤ 0.03) and also met the other study entry criteria entered the active treatment phase of the study.
On the day of study drug administration, subjects were randomised to receive a single oral dose of ziprasidone HCl 80 mg (n = 8), paroxetine (Paxil®, SmithKline Beecham) 20 mg (n = 8), or placebo (n = 8). Ziprasidone was administered as two 40 mg capsules, paroxetine was administered as one 20 mg tablet and placebo was administered as two capsules. All study medications were given in the early morning after an overnight fast of at least 8 h. A 30 mg dose of dextromethorphan was then given 2 h after receiving the randomized study medication.
Pharmacokinetic sampling
Venous blood samples for the determination of serum ziprasidone and plasma paroxetine concentrations were collected immediately before dosing and 1, 2, 4, 6, 8 and 10 h after ziprasidone or paroxetine dosing.
Urine samples for the determination of dextromethorphan concentrations were collected over the 8 h period following dextromethorphan dosing. After the volume of each sample had been measured, a 40 ml aliquot was taken for determination of dextromethorphan/dextrorphan ratios.
Pharmacokinetic assessments
Serum concentrations of ziprasidone were determined using a validated high performance liquid chromatography (h.p.l.c.) assay with solid-phase extraction and ultraviolet detection. The assay had a dynamic range of 1.0–250.0 ng ml−1[11]. Ziprasidone concentrations below the lower limit of quantification were assigned a value of 0 ng ml−1 in pharmacokinetic calculations.
Plasma concentrations of paroxetine were determined using a validated h.p.l.c./fluorescence assay involving liquid–liquid extraction and derivatization with dansyl chloride [12]. Paroxetine concentrations below the lower limit of quantification (≤ 2 ng ml−1) were assigned a value of 0 ng ml−1 in pharmacokinetic calculations.
Urinary dextromethorphan/dextrorphan ratios were determined using liquid chromatography involving liquid–liquid extraction and liquid chromatography with fluorescent detection [13].
The maximum observed serum/plasma concentrations of ziprasidone/paroxetine (Cmax), and the earliest time at which Cmax occurred (tmax) were estimated directly from the experimental data. The area under the serum/plasma ziprasidone/paroxetine concentration–time curve from time zero to 10 h postdosing (AUC(0, 10 h)) was estimated using linear trapezoidal approximation.
Statistical evaluation
It was estimated, on the basis of previous experience (Pfizer Inc., data on file) that a sample size of eight subjects per treatment group would provide at least 80% power to detect a 100% difference in the dextromethorphan/dextrorphan ratio using a 5% significance level. A 100% increase in the dextromethorphan/dextrorphan ratio translated to an approximate 30% increase in the concentration of drugs which are known substrates for CYP2D6 (Ereshefsky L, personal communication). A Student’s t-test was performed to compare the change from baseline dextromethorphan/dextrorphan ratio for the ziprasidone group with that of the placebo group. Values of P< 0.05 were considered to be statistically significant.
The pharmacokinetic parameters were summarized using descriptive statistics. Geometric means and standard deviations were calculated for AUC(0,10 h) and Cmax. Arithmetic means and standard deviations were calculated for tmax. Postdose dextromethorphan/dextrorphan ratios were compared with the average of three screening ratios to detect any statistical differences from placebo. PROC GLM of SAS® was used to test whether there was a difference between the change from baseline dextromethorphan/dextrorphan ratio between treatments.
Results
Subjects
Twenty-four subjects entered the study (eight in each treatment group) (Table 1). All subjects completed the study. One subject who received paroxetine did not have detectable paroxetine concentrations during the 10 h postdose sampling period and was excluded from the summary pharmacokinetic analysis.
Table 1.
Demographic characteristics.
| Characteristic | Ziprasidone | Paroxetine | Placebo |
|---|---|---|---|
| Number of subjects | 8 | 8 | 8 |
| Men | 4 | 5 | 7 |
| Women | 4 | 3 | 1 |
| Mean age (range) (years) | 27.7 (21–39) | 26.6 (20–31) | 26.1 (20–39) |
| Men | 29.0 (22–33) | 28.0 (24–31) | 26.7 (20–39) |
| Women | 26.5 (21–39) | 24.3 (20–31) | 22.0 (22–22) |
| Mean weight (range) (kg) | |||
| Men | 82.8 (68–94) | 75.8 (60–85) | 74.9 (67–83) |
| Women | 55.9 (43–69) | 55.9 (48–65) | 59.4 (59–59) |
| Ethnic origin | |||
| White | 8 | 2 | 6 |
| Asian | 0 | 3 | 1 |
| Black | 0 | 2 | 0 |
| Hispanic | 0 | 1 | 1 |
Pharmacokinetics
The mean±s.d. AUC(0,10 h), Cmax and tmax values for ziprasidone were 311 ± 157 ng ml−1 h, 55 ± 35 ng ml−1 and 4 ± 1 h, respectively. The mean±s.d. AUC(0,10 h), Cmax and tmax values for paroxetine were 32.9 ± 17.1 ng ml−1 h, 6 ± 4 ng ml−1 and 6 ± 2 h, respectively (Table 2).
Table 2.
Mean± s.d. values of ziprasidone and paroxetine pharmacokinetic parameters.
| Treatment group | ||
|---|---|---|
| Pharmacokinetic parameter | Ziprasidone (n = 8) | Paroxetine (n = 7)a |
| AUC(0,10 h) (ng ml−1 h) | 311 ± 157 | 32.9 ± 17.1 |
| Cmax (ng ml−1) | 55 ± 35 | 6 ± 4 |
| tmax (h) | 4 ± 1 | 6 ± 2 |
Excludes one subject who received paroxetine but in whom the drug was not detectable in plasma during the 10 h postdose sampling period.
There were no appreciable changes in the mean urinary dextromethorphan/dextrorphan ratios from baseline to 10 h in either the ziprasidone group or the placebo group. By contrast, there was a 10-fold increase in the mean urinary dextromethorphan/dextrorphan ratio from baseline to 10 h in the paroxetine group (Table 3, Figure 1). The mean change in the urinary dextromethorphan/dextrorphan ratio in the ziprasidone group was not statistically significantly different from that in the placebo group (P =0.9), but was statistically significantly different from that in the paroxetine group (P = 0.0001). The mean change in the urinary dextromethorphan/dextrorphan ratio in the paroxetine group was also significantly different from that in the placebo group (P = 0.0001).
Table 3.
Summary of urinary dextromethorphan/dextrorphan ratios
| Parameter | Ziprasidone (n = 8) | Paroxetine (n = 7)a | Placebo (n = 8) |
|---|---|---|---|
| Mean baseline value | 0.009 | 0.007 | 0.005 |
| Mean value after treatment | 0.007 | 0.072 | 0.004 |
| Mean± s.d. change | −0.002 ± 0.004 | 0.065 ± 0.043 | −0.001 ± 0.001 |
| P value vs placebo | 0.9231 | 0.0001 | – |
| P value vs paroxetine | 0.0001 | – | – |
Excludes one subject who received paroxetine but in whom the drug was not detectable in plasma during the 10 h postdose sampling period.
Figure 1.
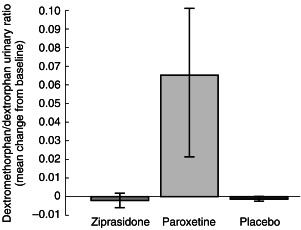
Mean±s.d. change from baseline in urinary dextromethorphan/dextrorphan.
Discussion
Findings from this study indicate that the administration of a single 80 mg dose of ziprasidone does not influence the activity of CYP2D6, as indicated by the urinary dextromethorphan/dextrorphan ratio. The mean urinary dextromethorphan/dextrorphan ratio in the ziprasidone group decreased by 22% (from 0.009 to 0.007) and that in the placebo group decreased by 20% (from 0.005 to 0.004). By contrast, there was a 10-fold increase (from 0.007 to 0.072) in the mean urinary dextromethorphan/dextrorphan ratio in the paroxetine group. This change in the paroxetine group served as a positive control for the ability to detect a change in CYP2D6 metabolizing activity in this study. As the urinary dextromethorphan/dextrorphan ratio is a reliable measure of CYP2D6 activity in vivo[6] these findings suggest that ziprasidone does not influence the activity of CYP2D6 in vivo when administered to extensive metabolizers as a single 80 mg dose. The results also suggest that clinically relevant concentrations of ziprasidone are unlikely to influence the CYP2D6-mediated metabolism of any of numerous drugs with which it may be concomitantly administered in clinical practice.
The serum concentrations of ziprasidone in the present study, obtained following the administration of a single 80 mg dose to healthy men and women under fasting conditions, were similar to those obtained in other studies in which a single 20 mg dose was given to healthy fed young (18–45 years) individuals [14, 15]. The pharmacokinetics of paroxetine in the present study were unremarkable and were similar to those previously reported [16]. The undetectable plasma concentrations of paroxetine in one subject may reflect delayed absorption and large intersubject variability of paroxetine following administration.
The findings of this in vivo study are in accord with those of an in vitro study which evaluated CYP2D6-mediated drug metabolism [1]. The highest steady-state Cmax found for ziprasidone (dose 160 mg day−1) was found to be 326 ng ml−1 (Pfizer Inc., data on file) and this equates to a free serum ziprasidone concentration of 0.007 µm (ziprasidone is highly bound (> 99%) by serum proteins [17, 18]). As the in vitro Ki for ziprasidone for CYP2D6 is 11 µm[1] the free concentration is approximately 1500-fold less than the in vitro Ki.
In conclusion, the findings of this study suggest that ziprasidone does not influence the oxidative metabolism of drugs by CYP2D6 at clinically relevant concentrations.
References
- 1.Prakash C, Kamel A, Cui D, et al. Identification of the major human liver cytochrome P450 isoform responsible for the primary metabolites of ziprasidone and prediction of possible drug interactions. Br J Clin Pharmacol. 2000;49(Suppl. 1):35S–42S. doi: 10.1046/j.1365-2125.2000.00151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meyer MC, Baldessarini RJ, Goff DC, et al. Clinically significant interactions of psychotropic agents with antipsychotic drugs. Drug Safety. 1996;15:333–436. doi: 10.2165/00002018-199615050-00004. [DOI] [PubMed] [Google Scholar]
- 3.Lane RM. Pharmacokinetic drug interaction potential of selective serotonin reuptake inhibitors. Int Clin Psychopharmacol. 1996;11(Suppl 5):31–61. doi: 10.1097/00004850-199612005-00005. [DOI] [PubMed] [Google Scholar]
- 4.Spatzenegger M, Jaegger W. Clinical importance of hepatic cytochrome P450 in drug metabolism. Drug Metab Rev. 1995;27:397–417. doi: 10.3109/03602539508998329. [DOI] [PubMed] [Google Scholar]
- 5.Ereshefsky LE, Riesenman C, Lam YWF. Antidepressant drug interactions and the cytochrome P450 system; the role of CYP 2D6. Clin Pharmacokinet. 1995;29(Suppl):10–19. doi: 10.2165/00003088-199500291-00004. [DOI] [PubMed] [Google Scholar]
- 6.Jacqz-aigran E, Funck-bretano C, Cresteil T. CYP2D6 and CYP3A dependent metabolism of dextromethorphan in humans. Pharmacogenetics. 1993;3:197–204. doi: 10.1097/00008571-199308000-00004. [DOI] [PubMed] [Google Scholar]
- 7.Riesenman CL, Lam YWF, Ereshefsky L. Intraindividual variability of the dextromethorphan-dextrorphan ratio in extensive metabolizers. Pharmacol Ther. 1996;59:137. [Google Scholar]
- 8.Centorrhino F, Baldessarini RJ, Frankenberg FR, et al. Serotonin reuptake inhibitors alter metabolism of clozapine. Am J Psychiatry. 1996;153:820–822. doi: 10.1176/ajp.153.6.820. [DOI] [PubMed] [Google Scholar]
- 9.Centorrhino F, Baldessarini RJ, Kando J, et al. Clozapine and metabolites: serum concentrations and clinical findings during treatment of chronically psychotic patients. J Clin Psychopharmacol. 1994;4:119–125. [PubMed] [Google Scholar]
- 10.Metropolitan height and weight tables Stat Bull Metrop Life Found. 1983;64:3–9. [PubMed] [Google Scholar]
- 11.Janiszewski JS, Fouda HG, Cole RO. Development and validation of a high-sensitivity assay for an antipsychotic agent, CP-88,059, with solid-phase extraction and narrow-bore high-performance liquid chromatography. J Chromatogr. 1995;668:133–139. doi: 10.1016/0378-4347(95)00071-p. [DOI] [PubMed] [Google Scholar]
- 12.Brett MA, Diendorf H-D. Determination of paroxetine in human plasma using high performance liquid chromatography with fluorescence detection. J Chromatogr. 1987;419:438–444. doi: 10.1016/0378-4347(87)80313-4. [DOI] [PubMed] [Google Scholar]
- 13.Wenk M, Todesco L, Keller B, et al. Determination of dextromethorphan and dextrorphan in urine by high performance liquid chromatography after solid-phase extraction. J Pharmaceut Biomed Anal. 1991;9:341–344. doi: 10.1016/0731-7085(91)80203-l. [DOI] [PubMed] [Google Scholar]
- 14.Miceli JJ, Wilner KD, Hansen RA. Single- and multiple-dose pharmacokinetics of ziprasidone under non-fasting conditions in healthy male volunteers. Br J Clin Pharmacol. 2000;49(Suppl. 1):5S–13S. doi: 10.1046/j.1365-2125.2000.00147.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilner KD, Tensfeldt TG, Baris B, et al. Single- and multiple-dose pharmacokinetics of ziprasidone in healthy young and elderly volunteers. Br J Clin Pharmacol. 2000;49(Suppl. 1):15S–20S. doi: 10.1046/j.1365-2125.2000.00148.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaye CW, Haddock RE, Langley PF, et al. A review of the metabolism and pharmacokinetics of paroxetine in man. Acta Psychiatr Scand. 1989;80(Suppl 350):60–75. doi: 10.1111/j.1600-0447.1989.tb07176.x. [DOI] [PubMed] [Google Scholar]
- 17.Howard HR, Prakash C, Seeger TF. Ziprasidone hydrochloride. Drugs Future. 1994;19:560–563. [Google Scholar]
- 18.Tandon R, Harrigan E, Zorn SH, et al. Ziprasidone: a novel antipsychotic with unique pharmacology and therapeutic potential. J Serotonin Res. 1997;4:159–177. [Google Scholar]