

Abstract
Background:
The cardiac sodium–calcium exchanger (NCX1) is a key sarcolemmal protein for the maintenance of calcium homeostasis in the heart. Since heart failure is associated with increased expression of NCX1, heterozygous (HET) and homozygous (HOM) transgenic mice overexpressing NCX1 were developed and evaluated.
Methods and Results:
The NCX1 transgenic mice display 2.3-fold (HET) and 3.1-fold (HOM) increases in exchanger activity from wildtype (WT) mice. Functional information was obtained by echocardiography and catheterizations before and following hemodynamic stress from pregnancy, treadmill exercise or trans-aortic constriction (TAC). HET and HOM mice exhibited hypertrophy and blunted responses with β-adrenergic stimulation. Postpartum mice from all groups were hypertrophied, but only the HOM mice exhibited premature death due to heart failure. HOM mice became exercise intolerant after 6 weeks of daily treadmill running. After 21 days TAC, HET and HOM mice exhibited significant contractile dysfunction and 15-40% mortality with clinical evidence of heart failure.
Conclusions:
Hemodynamic stress results in a compensated hypertrophy in WT mice, but NCX1 transgenic mice exhibit decreased contractile function and heart failure in proportion to their level of NCX1 expression. Thus exchanger overexpression in mice leads to abnormal calcium handling and a decompensatory transition to heart failure with stress.
Keywords: Heart Failure, Hypertrophy, Na / Ca Exchanger, E-C Coupling, Transgenic Mice
Introduction
Heart failure is a complex disease process that leads to severely depressed cardiac function and high mortality. Detrimental alterations in calcium homeostasis are generally recognized as a universal factor in the development of heart failure.1-5 Sodium-calcium (Na+-Ca2+) exchange is a key mechanism in maintaining calcium (Ca2+) homeostasis during cardiac excitation-contraction coupling.6-9 Ca2+ entering cardiomyocytes via L-type Ca2+ channels is primarily extruded by the Na+-Ca2+ exchanger (forward mode exchange). However, the net direction in which the exchanger will transport Ca2+ is dependent on ionic conditions and membrane potential. Thus, “reverse mode exchange” is a possible Ca2+ influx mechanism.4,10-13 Though the magnitude of Ca2+ influx through the exchanger is probably modest under normal physiological conditions, the exchanger is upregulated during hypertrophy and heart failure in most human and animal models when reverse mode exchange may become more prominent.3,4,10,11,14-20
Predicting the effects of upregulated exchange activity and its relationship to heart failure is not straightforward. Increased levels of exchanger may compensate for defective excitation-contraction (E-C) coupling mechanisms such as the depressed function of the sarcoplasmic reticulum (SR) Ca2+ pump (SERCA) that often accompanies hypertrophy and heart failure.21-23 Alternatively, increased exchanger activity may act to decrease SR Ca2+ stores and hence diminish contractility. Understanding the role of increased Na+-Ca2+ exchanger activity during heart failure is further complicated by the many adaptive modifications of Ca2+-related and contractile proteins that simultaneously occur.5,11
To help reveal the contributions of discrete components associated with the disease processes, we have used genetic manipulations to alter the level of the cardiac Na+-Ca2+ exchanger (NCX1) in the myocardium.24-26 Numerous studies with heterozygous (HET) transgenic α-myosin heavy chain-NCX1 overexpressing mice have reported alterations in Ca2+ handling and contractile function in myocytes.12,14,27-34 No adaptations in expression levels of other calcium handling proteins (L-type Ca2+ channels, SR calcium pump, calsequestrin, or phospholamban) were reported to occur in the myocardium of these HET mice.12,28,29
Based on these HET data and the evidence of increased exchanger levels in other models of heart failure, we hypothesized that further increases in NCX1 expression in our model would alter baseline function and predispose the myocardium to heart failure following hemodynamic stress. Thus homozygous (HOM) overexpressing mice were developed and found to have a 33% increase in NCX1 activity above that of the HET mice examined in the previous publications. Recently we reported on significant defects in the excitation-contraction coupling mechanism in myocytes from these HOM overexpressing mice.9,25,26,35 We now examine the in vivo phenotype of the HOM NCX1 overexpressing mice and contrast them with the HET and wildtype (WT) mice. Since no alterations in the expression of other Ca2+ handling proteins were also found in the HOM mice, the effects of exchanger overexpression can be studied in a normal background of other excitation-contraction coupling proteins. The increased exchanger activity in the HET and HOM mice induced a compensated cardiac hypertrophy at baseline and a blunted contractile response to β-adrenergic stimulation whose magnitude was proportional to NCX1 overexpression levels. Furthermore, mice chronically stressed by pregnancy, exercise, or pressure overload manifest an accelerated hypertrophic response and transition to decompensated heart failure in proportion to their expression levels. Thus, the pathophysiology observed in HOM mice is associated with abnormalities in Ca2+ handling directly tied to the greater expression of the Na+-Ca2+ exchanger as compared to the HET and WT mice.
Methods
Generation and Characterization of Homozygous Transgenic Mice
The α-myosin heavy chain (MHC) Na+-Ca2+ exchanger (NCX1.1) transgenic mice were generated previously described.24 These heterozygous (HET) mice were mated to homozygosity (HOM). Genotypes were confirmed by Southern blot and PCR, and homozygosity was confirmed genetically. All groups of mice (HOM, HET and wildtype) are on the same C57Bl/6 × C3HF1 background. Na+-Ca2+ exchange activity was determined in a crude fraction of cardiac membrane vesicles by measuring the Na+ gradient-dependent 45Ca2+ uptake.24 ANF and SERCA2 expression were respectively determined by Northern and Western blot analyses as previously described.24 DNA microarray analysis methods were similar to those from Ritter et al.36
Unless specifically stated otherwise, mice from all groups and under all conditions were evaluated at approximately 4 months of age. This investigation conforms to the Guide for the Care and Use of Laboratory Animals published the US National Institutes for Health (NIH publication # 85-23, revised 1996) and was approved by the UCLA Office for Protection of Research Subjects.
Echocardiography
Mice were sedated with Avertin (2,2,2, tribromo-ethanol, 2.5% solution, 0.016 ml/gm body mass, Aldrich Chemical Corp.) and ultrasonically imaged with either an ATL Interspec Apogee CX200 instrument37,38 or with a Siemens Acuson Sequoia C256 instrument (Siemens Medical Solutions, Mountain View CA).39 Digitized 2-D guided M-mode images were analyzed with SigmaScan (Systat Software Inc., Richmond CA) or AccessPoint (Freeland Systems LLC, Santa Fe NM) software for dimensions of the left ventricular cavity (end diastolic dimension - EDD and end systolic dimension - ESD) and wall thickness (posterior wall thickness - PWT and ventricular septal thickness - VST) during systole and diastole. Ejection times and heart rates were determined from Doppler images. Left ventricular mass was calculated from the EDD, PWT and VST values according to Tanaka et al.38 Left ventricular fractional shortening (%LVFS) and velocity of circumferential fiber shortening (Vcf) were calculated as indices of contractility.38 Mouse electrocardiograms (ECG) were obtained in the standard lead II arrangement with subcutaneous Pt electrodes (Grass Instruments, Warwick, RI).
Exercise Testing
WT and HOM mice (n = 6 each) were run daily on a Rotarod treadmill (Ugo Basile, Italy) for 8 weeks. The mice were conditioned to the device during the first two weeks at increasing speeds. From the third through the eighth weeks, the mice were run twice daily (30 minutes each) at a more strenuous pace about 10% less than their individual maximum determined at the end of the first week (3-4m/min). All exercise sessions were performed during the mouse's normal active (dark) circadian period in a room lit by only two 15w red bulbs. Exercise tolerance was scored by the maximum speed and the number falls per hour for each mouse.40
Trans-aortic Constriction (TAC)
A fixed pressure overload was obtained by surgically constricting (banding) the aorta between the right and left carotid arteries to the diameter of a 27 gauge needle in an anesthetized, ventilated mouse.41,42 At the end of the study period (7 or 21 days), the TAC mice were evaluated a second time via echocardiography prior to the terminal physiological and morphometric assessment. For confirmation of the TAC pressure gradient, the right carotid and femoral arteries were catheterized with flame-stretched PE-50 tubing and pressures were simultaneously measured.
Physiological Assessment
Mice were anesthetized with a mixture of Ketamine (100 mg/Kg body mass), Xylazine (2 mg/Kg) and Buprenorphine (0.1 mg/Kg), intubated and hemodynamic data obtained. For assessment of contractility, the right carotid artery was catheterized with a 1.4Fr catheter (Millar Instruments, Houston, TX) advanced into the left ventricle. After stabilization of baseline LV pressure and ±dP/dT recordings, sequential 10-150 μg/Kg dose (0.01 ml bolus, IV) infusions of Dobutamine were administered at 10 minute intervals. Hemodynamic measurements were acquired, digitized, displayed and analyzed with HEM V3.3 software (Notocord Systems, Croissy sur Seine, France). All hemodynamic data were recorded continuously for at least 30 minutes to ensure physiological levels of pressures and heart rates.
Morphometric and Histological Assessment
At the end of the study, standard morphometric measures were obtained including body, heart, and lung weights as well as tibia length. In some cases, hearts were immediately fixed in formalin for histological assessment. Routinely processed paraffin-embedded tissue sections were stained with hematoxylin and eosin (H & E) and Masson's Trichrome stains. For morphometric analysis, sections were imaged and evaluated using an automated image analysis system (Image Pro software, Media Cybernetics Inc., Silver Springs MD).
Statistical Methods
Data were compiled and are shown as means and standard errors. Data were evaluated using a student's two-tailed t-test or ANOVA with Instat V3.05 statistical software (GraphPad Inc., San Diego, CA). In all analyses, P < 0.05 was taken to be statistically significant.
Results
Characterization of Baseline NCX1 Transgenic Mice
After producing homozygous offspring of our transgenic mice, we assessed the level of NCX1 activity in the HOM, HET and WT mice. The NCX1 activity in membrane vesicles was increased 2.34-fold in HET and 3.12 fold in HOM vs. wildtype (WT) controls. HET activity was significantly greater (P<0.001) than WT controls and HOM activity was significantly (P<0.05) greater than both WT and HET values. Since NCX1 activity has been suggested to decline with age,43 it was evaluated in both young mice (2-4 months of age) as well as elderly mice (16-19 month of age). No significant differences were noted with age or gender.
Previously published studies have clearly established that there were no adaptations to expression levels of other calcium handling proteins in young adult HET mice.12,28,29 New analyses were performed to reveal whether the additional NCX1 activity in HOM mice elicited any changes. Western blot analysis for SERCA2a revealed no differences in expression in ventricles from HOM and WT mice from both genders (Figure 1A; WT 0.96±0.13; HOM, 1.00±0.11; mean±sem in arbitrary units; n=3 each). RNA expression levels determined by microarray analysis for many other Ca2+ handling proteins (calsequestrin, ryanodine receptor, L-type Ca2+ channel, the Na+/K+-ATPase and SERCA2a) as well as markers of hypertrophy (ANF and α-skeletal actin) were not significantly different between any of the groups examined (WT, HET & HOM). Since the 4 month old transgenic mice (HET & HOM) exhibited baseline hypertrophy (documented below), younger, 2 month old mice were specifically chosen for this gene chip analysis to remove any confounding variables due to the hypertrophic responses associated with aging.
Figure 1.

Baseline SERCA2a and ANF expression. Panel A illustrates a Western blot analysis for SERCA2a protein expression in WT and HOM mice. Panel B illustrates a representative Northern blot analysis for baseline ANF in WT and HOM mice at 4 months of age. See text for details.
Evaluation of Baseline Phenotypes
Baseline phenotypes were evaluated in WT, HET and HOM mice at 4 months of age. Each group of mice consisted of approximately equal numbers of males and nulliparous (never pregnant) females. Unlike at 2 months of age, ANF expression from Northern analysis is 20.7 times greater in HOM mice than in WT mice at 4 months of age (Figure 1B). Postmortem morphometry confirms the suggested hypertrophic response from the ANF data. Heart weights significantly increase from 119±4 mg in WT mice to 133±4 mg in HET and 146±4 mg in HOM mice (mean±SEM; P<0.001; n = 30, 23, 30). When normalized to both tibia length (HW/TL) or body weights (HW/BW), the hypertrophic response was greatest in the HOM mice and at intermediate values in the HET in parallel to the increased expression level of the NCX1 transgene (Table 1-baseline and Figure 2A). Though both measures revealed significant hypertrophy in HOM mice, we feel that the HW/TL index is a better index of hypertrophy than HW/BW in these mice since the HET and HOM mice were respectively 10% & 13% heavier than their WT counterparts whereas there were no differences in tibia length. Morphometric data obtained from older (12-15 months) non-failing baseline mice are consistent with the data from younger mice in all groups (data not shown). There were no significant gender differences in these data.
Table 1.
Morphometric Measurements
| WT | HET | HOM | |
|---|---|---|---|
| Heart Weight/Body Weight (mg/g) | |||
| Baseline | 4.11±0.10 | 4.15±0.12 | 4.48±0.13† |
| Postpartum | 3.52±0.19 | 5.66±0.38*† | 6.19±0.46*† |
| 21 days post TAC | 7.40±0.38*# | 8.77±0.41*†# | 9.59±0.59*†# |
| Heart Weight/Tibia Length (mg/mm) | |||
| Baseline | 6.54±0.19 | 7.32±0.16† | 8.15±0.22†‡ |
| Postpartum | 7.38±0.20* | 8.76±0.38*† | 10.18±0.62*† |
| 21 days post TAC | 10.79±0.43*# | 12.05±0.41*†# | 13.04±0.59*†# |
| Lung Weight/Tibia Length (mg/mm) | |||
| Baseline | 8.28±0.26 | 9.62±0.20† | 9.54±0.18† |
| Postpartum | 9.55±0.32* | 9.80±0.41 | 10.71±0.31*† |
| 21 days post TAC | 13.14±1.60*# | 21.85±1.46*†# | 20.90 ±1.54*†# |
= P < 0.05 to baseline mice
= P < 0.05 to WT mice of same group
= P < 0.05 to HET mice of same group
= P < 0.05 to postpartum mice
n=30, 23 & 30 for WT, HET & HOM baseline mice
n=19, 8 & 9 for WT, HET & HOM postpartum mice
n=13, 17 & 16 for WT, HET & HOM TAC mice
Figure 2.

Baseline Morphometry and Echocardiographic Analysis. Panel A plots the heart weight to tibia length (HW/TL) ratios from WT, HET, HOM and PP HOM groups [n = 9 to 30]. Panels B and C respectively plot the left ventricular mass (LVM) and end-diastolic dimension (EDD) from the WT, HET, HOM and PP HOM mice [n=15 to 23]. For all three panels, †= P<0.05 to WT; ‡= P<0.05 to HET mice; and § =P<0.05 to all baseline mice. See the text and tables for more details.
Ventricular remodeling and changes in function associated with hypertrophy were assessed by echocardiography at physiological heart rates (513±19 bpm; Table 2-baseline). Similar to the mice evaluated for Table 1, no animals in these groups showed overt clinical evidence of heart failure. Figure 2B shows the significant LV mass increase in the HOM mice consistent with the post-mortem data. This appears to be an eccentric type of hypertrophy as left ventricular chamber enlargement occurred (increased EDD - Figure 2C) without any significant changes in wall thickness (PWT & VST – Table 2-baseline). Echo-based indices of ventricular function (%LVFS & Vcf) were not significantly different between WT and HET mice. However, one index of function, %LVFS was significantly reduced in HOM mice suggesting some decompensation prior to failure (Table 2-baseline).
Table 2.
Left Ventricular Chamber Dimensions and Function from Echocardiography
| WT | HET | HOM | |
|---|---|---|---|
| Posterior Wall Thickness (mm) | |||
| Baseline | 0.68±0.02 | 0.67±0.03 | 0.67±0.02 |
| Postpartum | 0.67±0.01 | --- | 0.68±0.02 |
| 21 days post TAC | 0.83±0.03*# | 0.78±0.03* | 0.88±0.04*# |
| Ventricular Septal Thickness (mm) | |||
| Baseline | 0.61±0.01 | 0.67±0.03 | 0.65±0.02 |
| Postpartum | 0.62±0.01 | --- | 0.66±0.02 |
| 21 days post TAC | 0.76±0.03*# | 0.76±0.02* | 0.83±0.04*# |
| End Diastolic Dimension (mm) | |||
| Baseline | 3.57±0.09 | 3.64±0.08 | 3.95±0.10†‡ |
| Postpartum | 4.14±0.11* | --- | 4.52±0.09*† |
| 21 days post TAC | 3.98±0.11* | 4.64±0.12*† | 4.76±0.17*† |
| End Systolic Dimension (mm) | |||
| Baseline | 2.30±0.13 | 2.46±0.09 | 2.83±0.09†‡ |
| Postpartum | 2.95±0.10* | --- | 3.39±0.08*† |
| 21 days post TAC | 2.81±0.16* | 3.74±0.15*† | 4.10±0.13*†# |
| Left Ventricular Mass (mg) | |||
| Baseline | 74±4 | 80±6 | 91± 3† |
| Postpartum | 95±6* | --- | 117± 6*† |
| 21 days post TAC | 118±7*# | 145±8*† | 178±17*†# |
| Left Ventricular Fractional Shortening (%) | |||
| Baseline | 36.0±2.9 | 32.5±1.1 | 28.3±1.3†‡ |
| Postpartum | 28.9±1.5 | --- | 24.2±1.2*† |
| 21 days post TAC | 30.1±2.5 | 19.9±1.8*† | 12.7±1.0*†‡# |
| Velocity of Circumferential Fiber Shortening (mm/ms) | |||
| Baseline | 6.28±0.43 | 5.73±0.27 | 5.30±0.28 |
| Postpartum | 5.79±0.47 | --- | 4.76±0.24† |
| 21 days post TAC | 5.80±0.49 | 3.80±0.35*† | 2.97±0.30*†# |
= P < 0.05 to baseline mice
= P < 0.05 to WT mice of the same group
= P < 0.05 to HET mice of the same group
= P < 0.05 to Postpartum mice
= no data available for PP HET.
n=15, 16 & 17 for WT, HET & HOM baseline mice
n=12 & 23 for WT & HOM postpartum mice
n=15, 16 & 17 for WT, HET & HOM TAC mice
To further evaluate the ventricular function in vivo, we used LV catheterization. The LV pressures and heart rates were in the physiological range with no statistical differences between any of the groups. Contractility (+dP/dT) and relaxation (−dP/dT) were not different between the three groups (Figure 3-baseline). Subsequently, sequential bolus infusions of Dobutamine were given to evaluate the response of NCX1 overexpressing mice to β-adrenergic stimuli. HOM mice exhibited significantly blunted responses (Figure 3). Interestingly, HET mice generally exhibited an intermediate response at higher doses.
Figure 3.

Baseline Hemodynamics. Left ventricular contractility (Panel A: +dP/dT) and relaxation (Panel B: −dP/dT) were measured in WT, HET and HOM at baseline and after bolus infusions of Dobutamine at 10, 20, 30, 60 and 150 μg/Kg [n = 8 (WT), 5 (HET), & 9 (HOM); † = P<0.05 HOM to WT].
Heart Failure in Postpartum HOM Mice
Our initial observation suggesting that increased levels of NCX1 expression provoked cardiac dysfunction and heart failure was noted as premature mortality in female HOM breeders. Figure 4 shows a Kaplan-Meier survival curve gathered over a 12-month period for male, nulliparous (NP) female and postpartum (PP) female HOM and HET mice. Only 33% of the PP HOM group survived past 12 months of age whereas 89% of the NP HOM females and 96% of the HOM males survived. In the HET groups, 80% of the PP females, 97% of the NP females and 99% of the males survived to one year of age. All the PP HET deaths occurred between 3-4 months of age and none thereafter. Only one mouse died from all WT groups during this period (data not shown).
Figure 4.

A Kaplan-Meier survival plot showing that the postpartum (PP) homozygous (HOM) NCX1 transgenic mice have greatly accelerated morbidity compared to all other groups (PP HET mice, nulliparous (NP) HOM and HET mice or male HET and HOM mice). N = 119 for HET males, 116 for NP HET females, 15 for PP HET females, 101 for HOM males, 61 for NP HOM females, and 58 for PP HOM females.
Postmortem examinations of PP HOM mice were indicative of congestive heart failure revealing severe edema, pleural effusions and ascites as compared to the PP and baseline WT and HET mice. PP HOM mice sacrificed prior to the onset of heart failure (19±1 weeks of age) also exhibited significantly increased lung weight to tibia length ratios consistent with heart failure (Table 1-postpartum). Additionally both the PP HET and PP HOM mice exhibited a significant hypertrophic response (heart weight to body weight or tibia length) compared to baseline and PP WT mice at the same age (Table 1 and Figure 2A). Interestingly, the magnitude of the hypertrophic response in the PP HET mice was at an intermediate level roughly in proportion to the level of NCX1 overexpression.
Echocardiographic evaluation in PP WT and PP HOM mice confirmed the hypertrophy measured from morphometry and revealed significantly reduced ventricular function (Table 2-postpartum and Figures 2B & 2C). Specifically, the chamber dimensions (EDD & ESD) and left ventricular mass were significantly increased over both the PP WT and baseline HOM mice. Ventricular function in PP HOM mice was significantly reduced from PP WT mice, and in the case of the %LVFS index, also to the baseline HOM mice. Two failing PP HOM mice were successfully catheterized, and had an average +dP/dT of 3180±552 mmHg/min and a −dP/dT of 3054±595 mmHg/min. These values are nearly half of that of the non-failing mice in Figure 2.
At the molecular level, Northern blot analysis (Figure 5) revealed high-level expression of ANF in ventricular tissue of the PP HOM mice (WT, 0.1±0.2; HET, 1.1±0.6; HOM, 12.4±3.3; mean±sem in arbitrary units; n=3 for each; P<0.05 HOM to HET & WT). Histological analysis revealed widespread interstitial fibrosis, sub-endocardial calcification and foci of replacement fibrosis in both the left and right ventricles (LV and RV) in PP HOM mice at this age (Figure 6A) as compared to the normal appearing histology from male HOM mice at the same age (Figure 6B). Histology for all other baseline groups of mice, including the NP female HOM mice, was normal (data not shown).
Figure 5.

Northern Blot Analysis. The left panel shows a Northern blot of ANF re-expression in HOM, WT and HET PP mice (3 lanes each). The right panel plots the normalized intensities of the ANF re-expression from the blot [* = P<0.001 to WT & HET].
Figure 6.
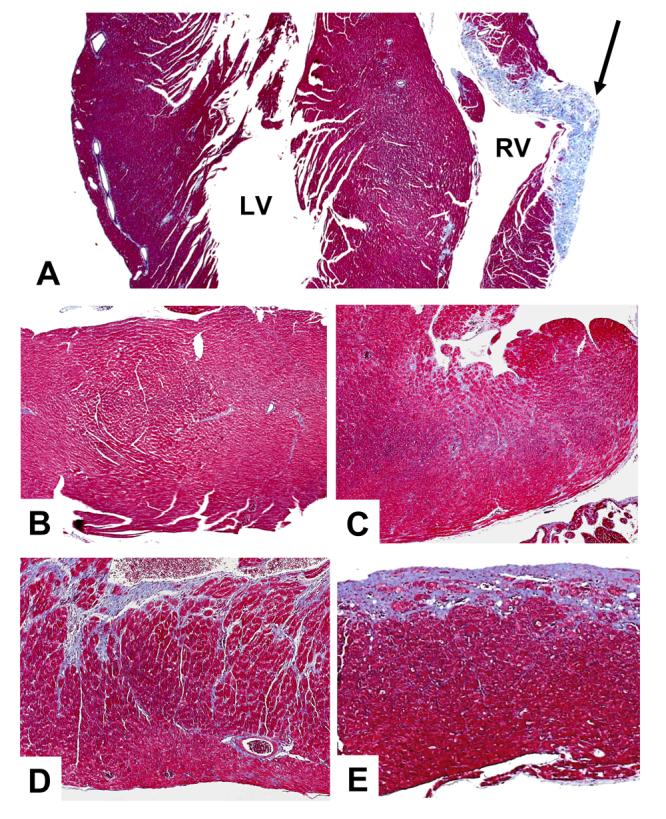
Histological Assessment of Baseline and Post-TAC Mice. Panel A is a trichrome stained section of PP HOM mouse myocardium exhibiting interstitial fibrosis and calcification. There is also a large region of replacement fibrosis in the right ventricle (arrow). Panel B is a normal appearing section from a male HOM mouse. Panels C, D & E respectively show increasing levels of fibrosis and pathology from sections from WT, HET and HOM post-TAC mice. See the text for further details.
Responses to Exercise and Pressure Overload
To evaluate the propensity toward heart failure in the HOM mice without the confounding variable of pregnancy, hemodynamic stress was induced by two methods: eight weeks of strenuous treadmill (RotaRod) exercise and by surgically induced pressure overload by trans-aortic constriction (TAC). The exercise regimen induced a modest physiological hypertrophy in both WT and HOM mice by the end of the study at 20±2 weeks of age (Figure 7A). It took 2 weeks for both groups of mice to fully condition to the exercise regimen. After the conditioning period, the WT mice ran well at full speed (4m/min) for the rest of the study. Unlike the WT mice, the HOM mice would generally not run as fast (3m/min) and fell off more often. Figure 7B plots the number of exercising mice that fell off 12 times or less per hour (averaged over the entire week). The HOM mice started to become intolerant to exercise by falling off more often by the 6th week even at a reduced speed. By the 8th week, only two mice were running well, 2 were running poorly (> 12 falls/hr) and 2 would not run at all. Though hypertrophied and exercise intolerant, none of these HOM mice showed clinical evidence of heart failure upon sacrifice.
Figure 7.

Response to Exercise. Panel A plots the heart weight to tibia length ration (HW/TL) from baseline and post-exercise WT and HOM mice [† = P < 0.05; n= 20 & 6]. Panel B plots the number of WT and HOM mice that could successfully run 60 minutes a day with 12 or less falls from the treadmill over the 8 week duration of the study [n=6 & 6].
Pressure overload was induced by trans-aortic constriction (TAC) in 14±1 week old WT, HET and HOM mice. After baseline echocardiography (Table 2), TAC was performed and the mice followed for up to 21 days post-operatively. All mice underwent repeat echo evaluation at 7 days post TAC. At 7 days post TAC, hemodynamic gradients were confirmed in half of this group, which were then sacrificed for morphometric analysis. The remaining mice from each group were observed for 21 days post TAC followed by echo, hemodynamic and morphometric analyses. All mice with trans-aortic gradients less than 40 mmHg were excluded from the analyses. All WT mice survived the entire 21 day period. Three additional WT mice were allowed to go out to 6 weeks (20 weeks of age); these all survived, exhibited a compensated hypertrophic response (HW/TL = 11.05±1.11mg/mm) and had no evidence of heart failure. All HET and HOM mice survived for at least 8 days with 80-85% of the HET and male HOM mice surviving the full period. However, only 60% of the female NP HOM mice survived for a full 21 days after TAC surgery. The mortality in both genders was associated with clinical signs of heart failure. In an attempt to evaluate the effect of age on this stress response, TAC was performed on 8 NP (female) HOM mice older than 5 months of age. Of these, 6 died within 2 days and the other 2 survived only 9 and 11 days with clinical manifestations of heart failure. No PP mice underwent the TAC procedure.
Morphometric and echo results for the mice surviving the full 21 day TAC intervention are detailed in Tables 1 and 2, respectively. While echocardiographic evidence of hypertrophy and chamber dilation occurred in all groups following 21 days of TAC, the HET and HOM mice had more significant responses than WT mice. There was an intermediate hypertrophic response for all these parameters in the 7 day TAC mice (n=15-17; data not shown).
Ventricular function (%LVFS and Vcf) decreased significantly in HET and HOM mice following 21 days TAC from their baseline and PP counterparts (Table 2, Figure 8). The dysfunction was more pronounced in the HOM mice, with the HET mice exhibiting intermediate, but significantly reduced values. A few HET and HOM mice were catheterized following 21 days TAC. Though limited, these data also suggest a graded reduction in function with baseline ±dP/dT in the 3500 to 4000 mmHg/min range. The significant decrease in ventricular function in HET and HOM mice paralleled the increase in lung to body weight ratios in 21 day post TAC mice (Table 1). Histological analyses (Figure 4 C-E) show a graded elevation of interstitial fibrosis and pathology after 21 day TAC in WT, HET and HOM mice. Though hypertrophied with some modest fibrosis, there was no evidence of heart failure in the WT mice following TAC.
Figure 8.

Ventricular Dysfunction Following TAC. Panels A and B respectively plot the left ventricular fractional shortening (LV%FS) and the velocity of circumferential fiber shortening (Vcf) from echo from the WT, HET and HOM mice at baseline and 21 days post TAC [ n=15-23; * = P<0.05 to pre-TAC; †= P<0.05 to WT; ‡= P<0.05 to HET]. See Table 2 for further details.
Discussion
Alterations in Ca2+ homeostasis are a hallmark of myocardial pathophysiology such as hypertrophy and heart failure.2-4,11,13,22 Such alterations are often due to functional changes in Ca2+ handling proteins including the L-type Ca2+ channel, calsequestrin, SERCA, phospholamban, troponin C, the ryanodine receptor (RYR) and the Na+-Ca2+ exchanger. The HET and HOM Na+-Ca2+ exchanger mouse models evaluated here provide a means to study the effects of exchanger overexpression without confounding alterations in expression of other Ca2+ handling proteins as confirmed by gene chip and Western analyses (Figure 1A). Recent work from our group, indicate that HOM myocytes have significant defects in excitation-contraction coupling including enhanced reverse-mode NCX currents, smaller Ca2+ transients, increased ICa and reduced E-C coupling gain.26,35 They also reported a high incidence of oscillatory calcium release in HOM, but not WT myocytes under their test conditions. These factors are consistent with intracellular calcium overload which is major contributing factor to the onset of hypertrophy and heart failure by multiple pathways.2,5,11,44
The data in this study extend the electrophysiology studies in cells to the whole animal. We found that increased expression of the Na+-Ca2+ exchanger directly led to a hypertrophic response and that chronic hemodynamic stresses led to decompensatory transition to heart failure in the HOM overexpressing mice. Furthermore, the magnitude of the hypertrophy and pathology increased with increasing exchanger expression. Elevated expression of the exchanger has been documented in human and other animal models of hypertrophy and heart failure.3,16-18 Though heart failure is a complex disease with numerous pathological factors, these data clearly suggest that overexpression of the Na+-Ca2+ exchanger can significantly contribute to the process. Based on our previous electrophysiology data, we believe that the pathophysiology observed in the HET and HOM mice result from perturbed calcium homeostasis, specifically intracellular calcium overload.
The baseline data (Figures 1B & 2 and Tables 1-2) indicate a hypertrophic phenotype in both HET and HOM mice by 4 months of age. Linck et al.29 reported a three fold increase in ANF expression HET mice; this value is intermediate between WT and the HOM expression reported here (Fig. 1B). Interestingly, despite numerous published reports using HET mice from this line, only a few evaluated the morphometric phenotype and none reported a hypertrophic response.12,24,27 This difference in hypertrophic phenotype is likely due to the relatively younger age of the animals in the previous studies and their normalization of heart weight to body weight rather than to tibia length as is our preference. Adachi-Akahane et al.24 and Terracciano et al.12 reported no significant differences between HET and WT cell capacitance. Unlike HET mice, previous work from our group found significantly increased capacitance in HOM myocytes indicative of cellular hypertrophy in agreement with our morphometric data.35
Direct LV catheterization revealed normal levels of baseline left ventricular contractility (+dP/dT) and relaxation (−dP/dT) in all groups (Figure 3). Despite these normal baseline ±dP/dT values, the different groups of transgenic mice did not respond uniformly to β-adrenergic stimulation. Both contractility and relaxation were significantly blunted in HOM mice at elevated doses of Dobutamine with intermediate HET values. This graded blunting is proportional to the increasing levels of NCX1 expression. Blunted adrenergic responsiveness is associated with hypertrophic and failing hearts in both human and animal models,44,45 and in cells.46
Similarly, the baseline contractile function determined from echo (%LVFS & Vcf) is in the normal range except for HOM %LVFS (Table 2-baseline). Thus the baseline contractility and relaxation are generally normal despite the reduced EC coupling gain with normal SR Ca2+ loads in HOM myocytes.35 The normal LV function in the whole animal, as opposed to the depressed function in isolated cells, is not surprising since the mouse can compensate for the reduced EC coupling gain by increasing its adrenergic stimulation. Such chronic stimulation might also contribute to increased calcium overload via enhanced L-type calcium channel function and eventually lead to heart failure via calcium-triggered decompensatory pathways. An enhanced relaxation rate might also be expected from the higher exchange activity. However, the extra exchange activity could simply compensate for an increased Ca2+ influx or SR Ca2+ release.
Though the Na+-Ca2+ exchanger overexpressing mice did not exhibit heart failure under normal conditions, various forms of chronic hemodynamic stress did lead to decompensation and heart failure. For these studies we evaluated mice following pregnancy, treadmill exercise and arterial pressure overload from aortic constriction. Pregnancy increases the hemodynamic load (volume overload) upon the heart and can be associated with chamber dilation, hypertension, hypertrophy and heart failure.47,48 Treadmill exercise intermittently increases the hemodynamic load which leads to a “physiological hypertrophy” in normal subjects, but can also reveal reduced cardiac function in subjects with compromised hearts.49 Finally, aortic constriction provides a chronic pressure overload which the heart must overcome to sustain normal cardiac output.42 Though different, all three of these hemodynamic stress modalities precipitated cardiac dysfunction in the Na+-Ca2+ exchanger overexpressing mice.
To evaluate the propensity toward heart failure in the NCX1 overexpressors, mice were stressed by either exercise or pressure overload via TAC. A daily exercise program induced a moderate physiological hypertrophy in both WT and HOM mice, but only the WT mice sustained their running ability (Figure 7). The exercise intolerance exhibited in four of the six HOM mice at the 6th week of training suggests that the additional demand from exercise in HOM mice starts a stress-induced decompensation toward heart failure. With exercise, increased adrenergic stimulation leads to higher heart rates and contractility. These would likely trigger higher intracellular calcium levels in an already calcium overloaded heart.
Unlike exercise, a substantial hypertrophic response was evident in TAC-induced pressure overload in all groups by 7 days and continued to increase until the end of the 21-day study (Tables 1 & 2). As with exercise, WT mice were able to sustain their function and had no post-surgical mortality. But the HET and HOM mice had a graded reduction in function, increased fibrosis, and increased mortality prior to 21 days post-TAC (Table 2 and Figures 6C-E & 8). Interestingly, the NP (female) HOM mice appeared to be more severely effected by TAC with a somewhat greater mortality than the males. The source or this gender difference in mortality is unclear and is in contrast to increased male mortality following ischemia/reperfusion injury in HET mice.28 Older HOM mice were completely intolerant of the TAC procedure. Taken as a whole, these data suggest that the WT, HET and HOM mice have increasing levels of dysfunction and pathology following pressure overload in proportion to their increased expression of NCX1. Thus the level of NCX1 expression likely modulates the intracellular calcium levels which, in combination with pressure overload, accelerate the hypertrophic response sufficiently to decompensate into heart failure.
The high rate of premature death due to heart failure in PP HOM mice was unexpected (Figures 4 & 6A and Tables 1 & 2). The improved survival, graded reduction in hypertrophic response (PP ANF – Figure 5, morphometry and echo), the lack of heart failure in the PP WT and PP HET mice supports the concept of a direct relationship between NCX1 activity and pathophysiological consequences. The hemodynamic stress associated with pregnancy appears sufficient to induce a decompensatory transition to heart failure accompanied by tissue calcification and fibrosis in at least two thirds of the PP HOM mice by 9 months of age. As with TAC, the added stress of the volume overload commonly associated with pregnancy triggers this accelerated decompensation.
Why does overexpression of NCX1 lead to a hypertrophic phenotype? Hypertrophy, with or without fibrosis and heart failure, is a complex process resulting from a combination of multiple factors: genetic, structural (remodeling), autocrine and paracrine.2,4,44 Indeed, a large number of changes in gene product expression have been elucidated in several experimental models. It is clear that multiple biochemical signaling events are involved in the pathogenesis and progression of hypertrophy and eventual failure. As hypertrophy progresses it is almost always accompanied by fibrous tissue proliferation and loss of myocytes (apoptosis and/or necrosis), that play a role in the ultimate decompensation of the hypertrophied heart. Additionally, an alteration in calcium homeostasis is a major contributing factor to the cascade of pathways leading to hypertrophy and failure.3,5,11,22
In our NCX overexpressing mouse model, one might expect intracellular Ca2+ levels to be lower with more exchanger available for extrusion, but this is not necessarily the case. Rodent cardiomyocytes have elevated internal Na+ levels compared to cardiomyocytes of rabbit and other larger mammals.34,50,51 Increased Na+ levels in mice favor increased Ca2+ influx or decreased Ca2+ efflux during diastole via the exchanger leading to chronic calcium overload.12,13,52 Similar to previous studies on HET myocytes,12,24,31-34 the HOM myocytes appear to have an enhanced reverse-mode exchange and also a reduced gain of excitation-contraction coupling35 which may make them more susceptible to the stresses of pregnancy, exercise or TAC.
In summary, overexpression of the cardiac Na+-Ca2+ exchanger in HET and HOM transgenic mice leads to 2 to 3 fold increases in NCX1 activity without alterations in other Ca2+handling proteins. Increased NCX1 activity provokes a dilated ventricular phenotype, induces exercise intolerance and leads to premature mortality and heart failure in mice following pregnancy or pressure overload. Increasing levels of exchanger expression lead to a graded development of hypertrophy and susceptibility to a decompensatory transition to heart failure. The NCX overexpressing model developed and evaluated in our study should also be useful in future studies designed to elucidate the numerous steps in the progression from hypertrophy to heart failure.
Acknowledgements
We wish to thank Dr. Scott Henderson for his helpful discussions. We also thank Jeanne Kim, Liyan Lu, and Yujuan Lu for their technical assistance.
Footnotes
These studies were supported by an American Heart Association Grant-in-Aid (KPR), American Heart Association (Western States) Undergraduate Fellowship (HCC & PR), Laubisch Endowment Funds (KPR & RSR), the Piansky Family Trust (MCF) and NIH HL-48509 (KDP).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Houser SR. When does spontaneous sarcoplasmic reticulum Ca2+ release cause a triggered arrhythmia? Cellular versus tissue requirements. Circ Res. 2000;87:725–7. doi: 10.1161/01.res.87.9.725. [DOI] [PubMed] [Google Scholar]
- 2.Mann DL, Bristow MR. Mechanisms and models in heart failure: The biomechanical model and beyond. Circulation. 2005;111:2837–49. doi: 10.1161/CIRCULATIONAHA.104.500546. [DOI] [PubMed] [Google Scholar]
- 3.Pogwizd SM, Schlotthauer K, Li L, Yuan WL, Bers DM. Arrhythmogenesis and contractile dysfunction in heart failure - Roles of sodium-calcium exchange, inward rectifier potassium current, and residual beta-adrenergic responsiveness. Circ Res. 2001;88:1159–67. doi: 10.1161/hh1101.091193. [DOI] [PubMed] [Google Scholar]
- 4.Sipido KR, Volders PGA, Vos MA, Verdonck F. Altered Na/Ca exchange activity in cardiac hypertrophy and heart failure: a new target for therapy? Cardiovasc Res. 2002;53:782–805. doi: 10.1016/s0008-6363(01)00470-9. [DOI] [PubMed] [Google Scholar]
- 5.Yano M, Ikeda Y, Matsuzaki M. Altered intracellular Ca2+ handling in heart failure. J Clin Invest. 2005;115:556–64. doi: 10.1172/JCI24159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 7.Hilgemann DW. New insights into the molecular and cellular workings of the cardiac Na+/Ca2+ exchanger. Am J Physiol. 2004;287:C1167–C1172. doi: 10.1152/ajpcell.00288.2004. [DOI] [PubMed] [Google Scholar]
- 8.Philipson KD, Nicoll DA. Sodium-calcium exchange: A molecular perspective. Annu Rev Physiol. 2000;62:111–33. doi: 10.1146/annurev.physiol.62.1.111. [DOI] [PubMed] [Google Scholar]
- 9.Reuter H, Pott C, Goldhaber JI, Henderson SA, Philipson KD, Schwinger RHG. Na+-Ca2+exchange in the regulation of cardiac excitation-contraction coupling. Cardiovasc Res. 2005;67:198–207. doi: 10.1016/j.cardiores.2005.04.031. [DOI] [PubMed] [Google Scholar]
- 10.Gaughan JP, Furukawa S, Jeevanandam V, Hefner CA, Kubo H, Margulies KB, et al. Sodium/calcium exchange contributes to contraction and relaxation in failed human ventricular myocytes. Amer J Physiol. 1999;277:H714–H724. doi: 10.1152/ajpheart.1999.277.2.H714. [DOI] [PubMed] [Google Scholar]
- 11.Houser SR, Piacentino V, Weisser J. Abnormalities of calcium cycling in the hypertrophied and failing heart. J Mol Cell Cardiol. 2000;32:1595–607. doi: 10.1006/jmcc.2000.1206. [DOI] [PubMed] [Google Scholar]
- 12.Terracciano CMN, DeSouza AI, Philipson KD, MacLeod KT. Na+ - Ca2+ exchange and sarcoplasmic reticular Ca2+ regulation in ventricular myocytes overexpressing the Na+ - Ca2+ exchanger. J Physiol London. 1998;512:651–67. doi: 10.1111/j.1469-7793.1998.651bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weisser-Thomas J, Piacentino V, Gaughan JP, Margulies K, Houser SR. Calcium entry via Na/Ca exchange during the action potential directly contributes to contraction of failing human ventricular myocytes. Cardiovasc Res. 2003;57:974–85. doi: 10.1016/s0008-6363(02)00732-0. [DOI] [PubMed] [Google Scholar]
- 14.Hampton TG, Wang JF, DeAngelis J, Amende I, Philipson KD, Morgan JP. Enhanced gene expression of Na+/Ca2+ exchanger attenuates ischemic and hypoxic contractile dysfunction. Amer J Physiol. 2000;279:H2846–H2854. doi: 10.1152/ajpheart.2000.279.6.H2846. [DOI] [PubMed] [Google Scholar]
- 15.Hobai IA, ORourke B. Enhanced Ca2+-activated Na+-Ca2+ exchange activity in canine pacing-induced heart failure. Circ Res. 2000;87:690–8. doi: 10.1161/01.res.87.8.690. [DOI] [PubMed] [Google Scholar]
- 16.Kent RL, Rozich JD, McCollam PL, McDermott DE, Thacker UF, Menick DR, et al. Rapid expression of the Na+-Ca2+ exchanger in response to cardiac pressure overload - rapid communication. Am J Physiol. 1993;265:H1024–H1029. doi: 10.1152/ajpheart.1993.265.3.H1024. [DOI] [PubMed] [Google Scholar]
- 17.Schillinger W, Fiolet JW, Schlotthauer K, Hasenfuss G. Relevance of Na+-Ca2+ exchange in heart failure. Cardiovasc Res. 2003;57:921–33. doi: 10.1016/s0008-6363(02)00826-x. [DOI] [PubMed] [Google Scholar]
- 18.Studer R, Reinecke H, Bilger J, Eschenhagen T, Bohm M, Hasenfuss G, et al. Gene expression of the cardiac Na+ - Ca2+ exchanger in end-stage human heart failure. Circ Res. 1994;75:443–53. doi: 10.1161/01.res.75.3.443. [DOI] [PubMed] [Google Scholar]
- 19.Wang ZY, Nolan B, Kutschke W, Hill JA. Na+-Ca2+ exchanger remodeling in pressure overload cardiac hypertrophy. J Biol Chem. 2001;276:17706–11. doi: 10.1074/jbc.M100544200. [DOI] [PubMed] [Google Scholar]
- 20.Zhang XQ, Song JL, Rothblum LI, Lun MY, Wang XJ, Ding F, et al. Overexpression of Na+/Ca2+ exchanger alters contractility and SR Ca2+ content in adult rat myocytes. Amer J Physiol. 2001;281:H2079–H2088. doi: 10.1152/ajpheart.2001.281.5.H2079. [DOI] [PubMed] [Google Scholar]
- 21.Gomez AM, Valdivia HH, Cheng H, Lederer MR, Santana LF, Cannell MB, et al. Defective excitation-contraction coupling in experimental cardiac hypertrophy and heart failure. Science. 1997;276:800–6. doi: 10.1126/science.276.5313.800. [DOI] [PubMed] [Google Scholar]
- 22.Schultz JEJ, Glascock BJ, Witt SA, Nieman ML, Nattamai KJ, Liu LH, Lorenz JN, et al. Accelerated onset of heart failure in mice during pressure overload with chronically decreased SERCA2 calcium pump activity. Am J Physiol. 2004;286:H1146–H1153. doi: 10.1152/ajpheart.00720.2003. [DOI] [PubMed] [Google Scholar]
- 23.Weisser-Thomas J, Kubo H, Hefner CA, Gaughan JP, McGowan BS, Ross R, et al. The Na+/Ca2+ exchanger/SR Ca2+ ATPase transport capacity regulates the contractility of normal and hypertrophied feline ventricular myocytes. J Card Failure. 2005;11:380–7. doi: 10.1016/j.cardfail.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 24.Adachi-Akahane S, Lu LY, Li ZP, Frank JS, Philipson KD, Morad M. Calcium signaling in transgenic mice overexpressing cardiac Na+ - Ca2+ exchanger. J Gen Physiol. 1997;109:717–29. doi: 10.1085/jgp.109.6.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goldhaber JI, Henderson SA, Reuter H, Pott C, Philipson KD. Effects of Na+-Ca2+ exchange expression on excitation-contraction coupling in genetically modified mice. Annals N Y Acad Sci. 2005;1047:122–6. doi: 10.1196/annals.1341.011. [DOI] [PubMed] [Google Scholar]
- 26.Pott C, Goldhaber JI, Philipson KD. Genetic manipulation of cardiac Na+/Ca2+ exchange expression. Biochem Biophys Res Comm. 2004;322:1336–40. doi: 10.1016/j.bbrc.2004.08.038. [DOI] [PubMed] [Google Scholar]
- 27.Baumer AT, Flesch M, Kilter H, Philipson KD, Bohm M. Overexpression of the Na+ - Ca2+ exchanger leads to enhanced inotropic responsiveness to Na+ - channel agonist without sarcoplasmic reticulum protein changes in transgenic mice. Biochem Biophys Res Comm. 1998;249:786–90. doi: 10.1006/bbrc.1998.9248. [DOI] [PubMed] [Google Scholar]
- 28.Cross HR, Lu LY, Steenbergen C, Philipson KD, Murphy E. Overexpression of the cardiac Na+ / Ca2+ exchanger increases susceptibility to ischemia/reperfusion injury in male, but not female, transgenic mice. Circ Res. 1998;83:1215–23. doi: 10.1161/01.res.83.12.1215. [DOI] [PubMed] [Google Scholar]
- 29.Linck B, Boknik P, Huke S, Kirchhefer U, Knapp J, Luss H, et al. Functional properties of transgenic mouse hearts overexpressing both calsequestrin and the Na+-Ca2+ exchanger. J Pharmacol Exp Ther. 2000;294:648–57. [PubMed] [Google Scholar]
- 30.Stagg MA, Malik AH, MacLeod KT, Terracciano CMN. The effects of overexpression of the Na+/Ca2+ exchanger on calcium regulation in hypertrophied mouse cardiac myocytes. Cell Calcium. 2004;36:111–8. doi: 10.1016/j.ceca.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 31.Su Z, Bridge JHB, Philipson KD, Spitzer KW, Barry WH. Quantitation of Na Ca exchanger function in single ventricular myocytes. J Mol Cell Cardiol. 1999;31:1125–35. doi: 10.1006/jmcc.1999.0949. [DOI] [PubMed] [Google Scholar]
- 32.Sugishita K, Su Z, Li FH, Philipson KD, Barry WH. Gender influences [Ca2+](I) during metabolic inhibition in myocytes overexpressing the Na+-Ca2+ exchanger. Circulation. 2001;104:2101–6. doi: 10.1161/hc4001.097038. [DOI] [PubMed] [Google Scholar]
- 33.Terracciano CMN, Philipson KD, MacLeod KT. Overexpression of the Na+/Ca2+ exchanger and inhibition of the sarcoplasmic reticulum Ca2+-ATPase in ventricular myocytes from transgenic mice. Cardiovasc Res. 2001;49:38–47. doi: 10.1016/s0008-6363(00)00205-4. [DOI] [PubMed] [Google Scholar]
- 34.Yao AS, Su Z, Nonaka A, Zubair I, Lu LY, Philipson KD, et al. Effects of overexpression of the Na+ - Ca2+ exchanger on [Ca2+]i transients in murine ventricular myocytes. Circ Res. 1998;82:657–65. doi: 10.1161/01.res.82.6.657. [DOI] [PubMed] [Google Scholar]
- 35.Reuter H, Han T, Motter C, Philipson KD, Goldhaber JI. Mice overexpressing the cardiac sodium-calcium exchanger: Defects in excitation-contraction coupling. J Physiol London. 2004;544:779–89. doi: 10.1113/jphysiol.2003.055046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ritter MR, Dorrell MI, Edmonds J, Friedlander SF, Friedlander M. Insulin-like growth factor 2 and potential regulators of hemangioma growth and involution identified by large-scale expression analysis. Proc Nat Acad Sci (USA) 2002;99:7455–60. doi: 10.1073/pnas.102185799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shai SY, Harpf AE, Babbitt CJ, Jordan MC, Fishbein MC, Chen J, et al. Cardiac myocyte-specific excision of the beta 1 integrin gene results in myocardial fibrosis and cardiac failure. Circ Res. 2002;90:458–64. doi: 10.1161/hh0402.105790. [DOI] [PubMed] [Google Scholar]
- 38.Tanaka N, Dalton N, Mao L, Rockman HA, Peterson KL, Gottshall KR, et al. Transthoracic echocardiography in models of cardiac disease in the mouse. Circulation. 1996;94:1109–17. doi: 10.1161/01.cir.94.5.1109. [DOI] [PubMed] [Google Scholar]
- 39.Henderson SA, Goldhaber JI, So JM, Han T, Motter C, Ngo A, et al. Functional adult myocardium in the absence of Na+ - Ca2+ exchange. Cardiac-specific knockout of NCX1. Circ Res. 2004;95:604–11. doi: 10.1161/01.RES.0000142316.08250.68. [DOI] [PubMed] [Google Scholar]
- 40.Huynh L, Scremin OU, Jordan MC, Roos KP. Rota-rod treadmill exercise enhances murine ventricular mass. FASEB J. 2001;15(4):A793. Abstract. [Google Scholar]
- 41.Keller RS, Shai SY, Babbitt CJ, Pham CG, Solaro RJ, Valencik ML, et al. Disruption of integrin function in the murine myocardium leads to perinatal lethality, fibrosis, and abnormal cardiac performance. Am J Pathol. 2001;158:1079–90. doi: 10.1016/S0002-9440(10)64055-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rockman HA, Ross RS, Harris AN, Knowlton KU, Steinhelper ME, Field LJ, et al. Segregation of atrial-specific and inducible expression of an atrial natriutetic factor transgene and in vivo murine model of cardiac hypertrophy. Proc Natl Acad Sci USA. 1991;88:8277–81. doi: 10.1073/pnas.88.18.8277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lim CC, Liao RL, Varma N, Apstein CS. Impaired lusitropy-frequency in the aging mouse: role of Ca2+- handling proteins and effects of isoproterenol. Amer J Physiol. 1999;277:H2083–H2090. doi: 10.1152/ajpheart.1999.277.5.H2083. [DOI] [PubMed] [Google Scholar]
- 44.Brodde OE, Michel MC, Zerkowski HR. Signal transduction mechanisms controlling cardiac contractility and their alterations in chronic heart failure. Cardiovasc Res. 1995;30:570–84. [PubMed] [Google Scholar]
- 45.Feldman AM, Tena RG, Kessler PD, Weisman HF, Schulman SP, Blumenthal RS, et al. Diminished β-andrenergic receptor responsiveness and cardiac dilation in hearts of myopathic Syrian hamsters (BIO 53.58) are associated with a functional abnormality of the G stimulatory protein. Circ Res. 1990;81:1341–52. doi: 10.1161/01.cir.81.4.1341. [DOI] [PubMed] [Google Scholar]
- 46.Sato M, Gong H, Terracciano CMN, Ranu H, Harding SE. Loss of β-adrenoceptor response in myocytes overexpressing the Na+/Ca2+-exchanger. J Molec Cell Cardiol. 2004;36:43–8. doi: 10.1016/j.yjmcc.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 47.Eghbali M, Deva R, Alioua A, Minosyan TY, Ruan H, Wang Y, et al. Molecular and functional signature of heart hypertrophy during pregnancy. Circ Res. 2005;96:1208–16. doi: 10.1161/01.RES.0000170652.71414.16. [DOI] [PubMed] [Google Scholar]
- 48.Wong AYH, Kulandavelu S, Whiteley KJ, Qu D, Langille BL, Adamson SL. Maternal cardiovascular changes during pregnancy and postpartum in mice. Am J Physiol. 2002;282:H918–H925. doi: 10.1152/ajpheart.00641.2001. [DOI] [PubMed] [Google Scholar]
- 49.Haubold KW, Allen DL, Capetanaki Y, Leinwand LA. Loss of desmin leads to impaired voluntary wheel running and treadmill exercise performance. J Appl Physiol. 2003;95:1617–22. doi: 10.1152/japplphysiol.00408.2003. [DOI] [PubMed] [Google Scholar]
- 50.Despa S, Islam MA, Pogwizd SM, Bers DM. Intracellular [Na+] and Na+ pump rate in rat and rabbit ventricular myocytes. J Physiol (Lond) 2002;539:133–43. doi: 10.1113/jphysiol.2001.012940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pogwizd SM, Sipido KR, Verdonck F, Bers DM. Intracellular Na in animal models of hypertrophy and heart failure: contractile function and arrhythmogenesis. Cardiovasc Res. 2003;57:887–96. doi: 10.1016/s0008-6363(02)00735-6. [DOI] [PubMed] [Google Scholar]
- 52.Baartscheer A, Schumacher CA, Belterman CNW, Coronel R, Fiolet JWT. [Na+](I) and the driving force of the Na+/Ca2+-exchanger in heart failure. Cardiovasc Res. 2003;57:986–95. doi: 10.1016/s0008-6363(02)00848-9. [DOI] [PubMed] [Google Scholar]