

Abstract
We sought to define the relationship between cytokine stimulated release of matrix metalloproteinases (MMPs) and cell migration using adult rat cardiac fibroblasts. Interleukin-1β (IL-1β) increased release of MMP-2, 3, and 9, and TIMP-1, by 3–6-fold, measured by immunoblotting and gel zymography. Tumor necrosis factor-α (TNFα) augmented IL-1 stimulated release of MMP-9, but not MMP-2 or -3. Transforming growth factor-β1 (TGFβ1) attenuated all the responses to IL-1β. IL-1β was also the most robust stimulus of adult rat cardiac fibroblast migration, measured in Boyden chamber assays. The combination of IL-1β plus TNFα substantially enhanced migration, whereas TGFβ1 strongly inhibited the migratory response to IL-1β. The pan-selective MMP inhibitor GM 6001 effectively blocked IL-1β stimulated migration. Pharmacologic inhibitors selective for ERK, JNK, and p38 MAP kinase pathways inhibited the IL-1β regulation of individual MMPs. Increased MMP activity associated with migration of cardiac fibroblasts may be important determinants of cytokine-directed remodeling of injured myocardium.
Keywords: Cytokines, fibroblasts, MAP kinases, matrix metalloproteinases, migration
INTRODUCTION
In response to myocardial injury or infarction, activated cardiac fibroblasts migrate into the infarct zone and release matrix metalloproteinases (MMPs), counterbalanced by tissue inhibitors of matrix metalloproteinases (TIMPs), leading to net degradation of damaged ECM.[1] These fibroblast responses are regulated by pro-inflammatory cytokines including IL-1β and TNFα produced by resident cells at sites of injury, including fibroblasts themselves, and by infiltrating immune/inflammatory cells.[2] The cytokines exert their effects through intracellular signaling pathways converging on regulated gene transcription. Collagen re-synthesis by cardiac fibroblasts, driven by the pro-fibrotic cytokine TGFβ1, ultimately results in remodeling of the infarct scar and wound healing.[3; 4] Cardiac fibroblast phenotype is therefore likely to reflect complex interactions among the combinations of cytokines present through the temporal stages of post-infarct remodeling.[5] Maladaptive alterations in this scheme are hypothesized to contribute to the progression to heart failure, where chronic elevations of intracardiac cytokines, MMPs, and aberrant collagen synthesis have been observed.[6; 7]
Cytokine-stimulated fibroblast migration in other systems is important to recruit activated fibroblasts into wound sites as part of the inflammatory and healing process (reviewed in [8; 9]). In cultured neonatal rat cardiac fibroblasts, we have shown that IL-1β stimulates robust cell migration (20-fold over control), TNFα elicits a modest increase (2–4 fold), while IL-6 had no effect. We further demonstrated that fibroblast migration is mediated by the MAP kinase signaling pathway.[10]
In addition to contributing to normal and pathological tissue remodeling, MMP production likely facilitates cell migration through the ECM. Transgenic mice deficient in MMP-9 were protected from acute cardiac rupture following surgical MI, but subsequently exhibited reduced inflammatory infiltrates and defective healing.[11] MMP inhibition has been shown to prevent vascular smooth muscle cell migration into neointima following balloon injury in vivo.[12] Extensive studies in cancer chemotherapy have shown that MMP inhibitors block tumor cell invasiveness both in vivo and in vitro.[13]
Taken together, these results point to the hypothesis that cytokine regulation of cardiac fibroblast MMP production and cell migration are functionally related. However, this question has not been systematically evaluated in this cell type. Furthermore, the interactions between physiologically relevant combinations of pro-inflammatory and pro-fibrotic cytokines that co-exist in the wound environment have not been explored. In the present study, we addressed these issues using cultured cardiac fibroblasts from adult rats. In addition we examined the role of MAP kinase activation in cytokine stimulated MMP production extending our previous findings on the role of MAP kinases in cytokine stimulated migration.
MATERIALS AND METHODS
Cell Isolation
The investigation conforms with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1996). All experiments were conducted under authorization of the Institutional Animal Care and Use Committee of University of Colorado Health Sciences Center.
Cardiac fibroblasts were isolated from hearts of adult Sprague-Dawley rats (250–325 gm, Charles River) by retrograde Langendorff perfusion with trypsin and collagenase and differential centrifugation to remove cardiac myocytes.[14] Cells were plated in complete medium composed of DMEM containing 10% fetal bovine serum, and placed in a tissue culture incubator at 37°C with a 10% CO2 atmosphere. Antibiotics (penicillin, 100 U/ml; streptomycin, 50 μg/ml) were included in all culture media. After two hours, dishes were washed 3X with DMEM to remove unattached cells and debris. Adherent fibroblasts remaining on the plates were incubated with 10 ml complete medium. Medium was changed after 24 hr and at regular intervals until cultures were confluent, at which point they were washed with PBS and passaged into experimental cultures using 0.2% trypsin-EDTA.
Experimental Treatments for MMP analysis
At cell confluence, experimental cultures were rinsed 3X with DMEM and changed to serum free medium composed of DMEM with bovine serum albumin, 1 mg/ml, for 48 hours. Following this interval, the medium was replaced with fresh DMEM containing the indicated experimental agents or corresponding vehicle for an additional 48 hrs. All cytokines were used at final concentrations of 10 ng/ml. These concentrations were shown in our previous reports to produce maximum biological effects.[10; 15] Pharmacological MAP kinase inhibitors or vehicle (0.1% DMSO, v/v) were added 20 min prior to IL-1β. At the end of the treatment interval, supernatants from duplicate dishes were pooled and concentrated by centrifugation using Centricon Plus-20 spin filters (10,000 MW cut-off, Amicon-Millipore). Samples were stored at 4° C. Protein concentration was determined using the Bradford method. In control experiments (not shown), exposure of cultures to cytokines, pharmacological agents, or DMSO vehicle under these conditions did not affect cell viability. The pharmacological agents and DMSO vehicle did not affect basal cell functions of MMP production, migration, or MAP kinase phosphorylation.
In-gel zymography
Supernatant samples containing 500 ng total protein were mixed with equal volumes of 2X zymography sample buffer (125 mM Tris-HCl, pH 6.8, 50% glycerol, 8% SDS, 0.02% bromophenol blue), loaded onto pre-cast 10% polyacrylamide zymogram gels containing gelatin or casein (BioRad), and electrophoresed with 2.5 mM Tris-HCl, 19.2 mM glycine, 0.01% SDS, pH 8.3, at 100 V until the tracking dye reached the bottom of the gel. After electrophoresis, gels were equilibrated for 30 min at room temperature with renaturing buffer (2.5% Triton) with gentle agitation. Zymograms were developed overnight at 37° C in developing buffer, 50 mM Tris-HCl, pH 7.5, 200 mM NaCl, 5 mM CaCl2, 0. 02% Brij-35. Gels were stained with 0.5% Coomassie Blue at RT for 1 hr, destained with methanol:glacial acetic acid:water (50:10:40), rehydrated in methanol:glacial acetic acid:water (5:7:88), and dried. Areas of MMP activity appeared as clear bands. Zymograms were scanned using an HP 600 flatbed scanner.
Western Blots
Supernatant samples (15 μg total protein per lane) were denatured in 2X Laemmli sample buffer. SDS-PAGE and immunoblotting were performed as described previously.[10] Immunoreactive species corresponding to the active MMP enzymes were verified by their predicted Mr. Blots were visualized with enhanced chemiluminescence (Pierce, Rockford, IL). Densitometry was performed using the UMAX Power Look II scanner with BioImage software (UMAX Tech., Inc, Dallas, TX) or with a FluorChem SP imaging system (Alpha Innotech, San Leandro, CA).
Cell migration assay
Migration of adult cardiac fibroblasts was assayed with minor modification of procedures previously established for neonatal fibroblasts.[10] Duplicate or triplicate determinations were performed for each experimental condition.
Reagents
Cell culture reagents were from Gibco-Life Technologies or Sigma. Fetal bovine serum was from Gemini Bio-Products. Cytokines (recombinant rat IL-1β, TNFα, and recombinant human TGFβ1) were from R&D Systems. Pre-formulated zymography buffers and pre-cast gels were from Bio-Rad. The MEK1/2 MAP kinase inhibitor U0126 was from Promega. The p-38 and JNK MAP kinase inhibitors SB 202190, and SP600125, respectively, and MMP inhibitor GM 6001 and its biologically inactive congener (cat. no. 364210), were from Calbiochem. The following rat-reactive MMP and TIMP antibodies were obtained from Chemicon: MMP-2, AB809; MMP-3, AB810; MMP-9, AB805; MMP-13, AB8120; TIMP-2, AB-801; TIMP-3, AB802. Antibody for TIMP-1 was from R&D Systems (cat. no. AF580). Additional reagents and their sources are described above. All other chemicals were of the highest purity available from standard commercial sources.
Statistics
All values represent mean ± SEM from three or more separate experiments performed on different cell preparations. Statistical comparisons among groups were performed by one-way ANOVA with post hoc test using Graph-PAD Prism software (San Diego, CA). A p-value <0.05 was used to determine significance.
RESULTS
Cytokine regulation of MMP activity and abundance
Cytokine induced expression and release of active MMP species into the culture supernatant by cardiac fibroblasts was evaluated by gel zymography to assess MMP activity, and by immunoblotting to determine relative protein abundance. Representative zymography experiments shown in Figure 1 indicated that activities corresponding to MMP-2, -3, and -9 were stimulated by IL-1β. The IL-1β induced increases in zymographic activities of MMP-2, 3, and 9 reflects increased abundances of the corresponding proteins detected on immunoblots. The elevation of MMP activities by IL-1β contrasts with the pro-fibrotic cytokine TGFβ1, which did not increase any of these MMP activities. The combination of TGFβ1 with IL-1β reduced the IL-1β stimulated increase in MMP-2, 3, and 9 enzyme activities and protein abundances in concert.
Figure 1.
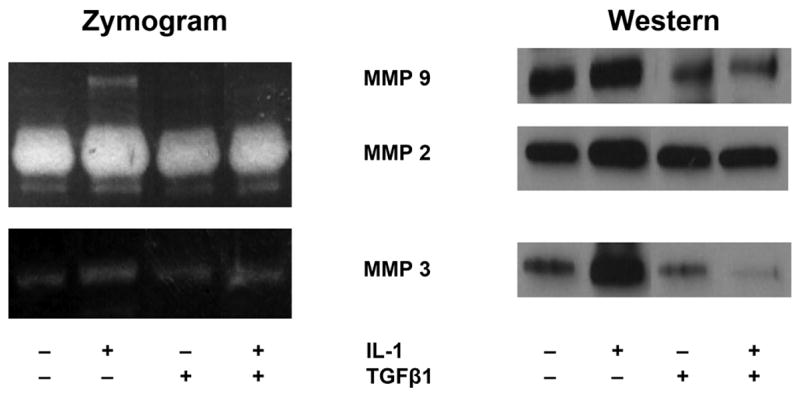
IL-1β and TGFβ1 regulation of MMP activity and abundance. Cardiac fibroblast cultures were grown to confluence, stimulated with the indicated concentrations of cytokines, and supernatants analyzed for MMP activity by gel zymography, or enzyme abundance by immunoblotting with antibodies against specified MMPs, as described in Materials and Methods. Representative experiments are shown.
In order to assess quantitatively the regulation of MMP abundance, we used immunoblotting with antibodies recognizing individual MMPs combined with imaging densitometry, as shown in Figure 2. IL-1β stimulated approximately three-fold increases in abundance of MMPs -2, -3, and -9. TGFβ1 did not increase abundance of MMPs-2, 3, or 9. Co-treatment with TGFβ1 strongly and consistently reversed the IL-1β induction of each MMP back to control levels. These results are in agreement with the effects of TGFβ1 on MMP enzymatic activities shown in Figure 1.
Figure 2.

Interactions of IL-1β and TGFβ1 on regulation of MMP abundance. Cardiac fibroblasts were treated with indicated combinations of IL-1β and TGFβ1 and the culture supernatants were analyzed for MMP expression by immunoblotting with quantitative densitometry as described in Materials and Methods. Data represent mean ± SEM from 4–7 separate experiments for each treatment and are normalized relative to control (CON). *, p < 0.05 versus control; †, p< 0.05 versus IL-1β.
Densitometric immunoblot analysis was extended to examine the actions of the pro-inflammatory cytokine TNFα, and its interactions with IL-1β, as shown in Supplemental Figure 1. TNFα alone had nominal effects on MMP production, with a trend toward increased MMP-9. The combination of IL-1β plus TNFα significantly enhanced MMP-9 production relative to IL-1β or TNFα alone but did not increase MMP-2 or 3 compared to IL-1β. We observed enhancements of MMP zymographic activity by TNFα that were consistent with the immunoblot findings on MMP abundance (data not shown).
In addition to its effects on MMP production, IL-1β stimulated a robust increase (six-fold) in the release of TIMP-1 (Supplemental Figure 1). The addition of TNFα together with IL-1β did not further modulate TIMP-1 induction by IL-1β. We also detected constitutive TIMP-2 expression in cardiac fibroblast culture supernatants, but its abundance was not affected by cytokines. Release of TIMP-3 and -4 were not detected (data not shown).
Cytokine regulation of adult cardiac fibroblast migration
The influence of pro-inflammatory and pro-fibrotic cytokines to regulate cardiac fibroblast migration was evaluated in comparison with the foregoing studies on regulation of MMP expression. Figure 3 demonstrates that IL-1β increased migration of adult rat cardiac fibroblasts approximately 12-fold compared to control cells. TNFα also stimulated migration but elicited a smaller and more variable response. The combination of IL-1β plus TNFα strongly augmented cardiac fibroblast migration greater than IL-1β alone, up to 25-fold over control cells. By contrast, TGFβ1 did not stimulate migration by itself, and strikingly suppressed IL-1β stimulated fibroblast migration.
Figure 3.

Cytokine regulation of cardiac fibroblast migration. Cardiac fibroblasts were treated with the indicated cytokines and assayed for cell migration as described in Materials and Methods. Data represent mean ± SEM from 3 separate experiments for each treatment and are normalized relative to the response to IL-1β. *, p < 0.05 versus control; †, p< 0.05 versus IL-1β.
In order to directly demonstrate the requirement for MMP activation in cell migration, the migratory response to IL-1β was measured in the presence of the pan-selective MMP inhibitor GM 6001. As shown in Supplemental Figure 2, GM 6001 (10 nM) effectively blocked IL-1β stimulated migration, whereas a chemically related but inactive congener had no effect at equal concentration.
Role of MAP Kinase signaling in IL-1β induced MMP expression
We previously demonstrated involvement of MAP kinase signaling in IL-1β stimulated migration of neonatal rat cardiac fibroblasts.[10] In this context, it was of interest to determine the role of MAP kinase signaling in IL-1β regulated MMP expression in adult rat cardiac fibroblasts. As shown in Supplemental Figure 3, stimulation with IL-1β elicits rapid increases in the active phosphorylated species of ERK, JNK, and p38 MAP kinases detected by immunoblotting with phosphoprotein-specific antibodies.
To determine the effects of individual MAP kinases on MMP expression, cells were treated with IL-1β in the absence or presence of an inhibitor of MEK1/2, U0126 (10 μM), to block ERK-MAP kinase activation, SP600125 (10 μM) to block JNK MAP kinase activation, or SB 202190 (1 μM) to block p38 MAP kinase. These inhibitor concentrations were validated in our previous study.[10] MMP expression was quantitated by immunoblotting and densitometry as before. Results are shown in Figure 4. IL-1β stimulated MMP-2 production was not affected by any of the MAP kinase inhibitors. IL-1β induction of MMP-3 was significantly attenuated by MEK and JNK inhibition but not by p38 inhibition. Conversely, MMP-9 induction was selectively reduced by the p38 inhibitor. These results suggest that IL-1β differentially regulates individual MMP species through distinct branches of the MAP kinase signaling pathways.
Figure 4.
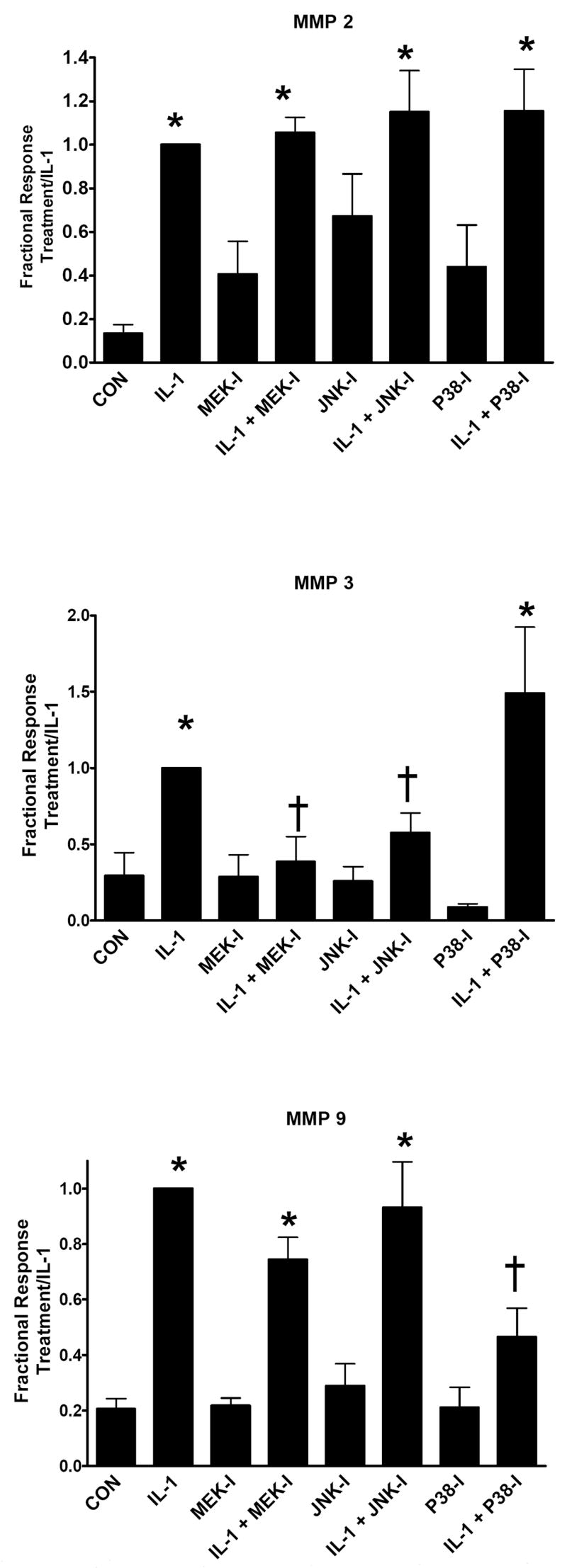
Dependence of IL-1β stimulated MMP production on MAP kinase pathways. Cardiac fibroblasts were treated with IL-1β or no cytokine addition (CON) in the absence and presence of specified MAP kinase inhibitors. Production of MMP-2, 3, and 9 were quantitated by immunoblotting and densitometry as described in Materials and Methods. Data represent mean ± SEM from 4 separate experiments for each treatment and are normalized relative to control (CON). MEK-I, U01216, 10 μM; JNK-I, SP 600125, 10 μM; p38-I, SB 202190, 1 μM. *, p < 0.05 versus control; †, p < 0.05 versus IL-1β.
DISCUSSION
This study provides novel insights into the relation between MMP expression and migration of cardiac fibroblasts, and regulation of these processes by combinations of cytokines present in the post-injury myocardium. Thus, IL-1β stimulates a spectrum of MMPs including MMP-2, 3, and 9. Compared to IL-1β, the pro-inflammatory cytokine TNFα only modestly increases MMP activity, interacting with IL-1β to selectively enhance MMP-9 release. Activation of MMP-2 and 9 by IL-1β is consistent with previous reports in cardiac fibroblasts.[16–18] Additionally, our data directly demonstrate increased MMP-3 expression and activity, extending a previous observation of IL-1β induced increase in MMP-3 mRNA.[17] In contrast to that study, we observe no evidence for IL-1β activation of MMP-13. A study of acute post-MI induction of MMPs in rabbit showed a similar pattern of elevated MMP-2, 3, and -9, without evidence of increased MMP-13.[19]
This complement of MMPs induced by IL-1β, combined with the absence of MMP-13 (rodent native fibrillar collagenase), suggests that IL-1β directs metabolism of degraded fibrillar collagens, native collagen III, or non-collagenous ECM such as basement membrane, rather than indiscriminate degradation of structurally sound fibrillar collagen I, the majority ECM constituent.[20] Further, IL-1β strongly increased TIMP-1, supporting the interpretation of selective and tightly controlled ECM metabolism.
In contrast, TGFβ1 consistently attenuated the induction of cardiac fibroblast MMPs and cell migration elicited by IL-1β. These results point to important physiological antagonism of the pro-inflammatory actions of IL-1β by the pro-fibrotic cytokine TGFβ1. In this regard the MMP promoters contain TGFβ1 inhibitory elements that are the targets for MMP down-regulation.[21] Previous studies from our group showed that Angiotensin II stimulation of cardiac fibroblasts upregulates TGFβ1, suggesting that multiple fibrotic stimuli exert actions through TGFβ1.[22] We have also described elevation of TGFβ1 mRNA upon stimulation with IL-1β, emphasizing the importance of feedback interactions between these two cytokines to regulate cardiac fibroblast phenotype.[8]
We further addressed the dependence of IL-1β stimulated MMP production on signaling through MAP kinase pathways. We observed differential regulation of MMP-2, 3, and 9 with respect to the ERK, JNK, and p38 MAP kinase pathways. The insensitivity of MMP-2 expression to MAP kinase inhibition is consistent with the absence of binding sites for AP-1 transcription factors in the MMP-2 promoter for this constitutively expressed enzyme.[21] In agreement with our data, previous reports by Xie et al., showed no effect of MAP kinase inhibitors on IL-1β stimulated MMP-2 expression in cardiac fibroblasts.[18] However, these authors reported that MMP-9 was sensitive to inhibition of ERK and JNK, whereas we see dependence on p38 MAP kinase but not ERK or JNK. MAP kinase regulation of MMP-3 has not been studied previously in cardiac fibroblasts. Similar to our results in cardiac fibroblasts, MMP-3 was attenuated by inhibition of ERK and JNK MAP kinases in human skin fibroblasts.[23] These fingerprints of MMP regulation by pro-inflammatory cytokines in specific cell types and experimental conditions may reflect interactions in upstream intracellular signaling pathways converging on elements of the MMP gene promoters.
Taken together, these data provide novel insights into the association of MMP expression with cell migration: IL-1β and TNFα stimulate migration and MMP production with similar relative potencies, whereas TGFβ1 opposes both MMP production and migration. Inhibition of MMP activity blocks migration. Further, IL-1β stimulates both migration and MMP production via MAP kinase signaling pathways. We suggest that activation of MMP species plays a permissive role to release ECM constraints on cell motility in cytokine directed fibroblast migration. Cardiac fibroblasts do constitutively synthesize collagen in vitro,[8; 17] consistent with a requirement for MMP activity to facilitate motility. Other aspects of MMP involvement to facilitate migration may also occur. For example, release of extracellular pro-migratory cytokines including heparin-bound EGF [24] and TNFα [25] by metalloproteinase activation has been reported in some systems.
In conclusion, this study shows that cytokines regulate MMP expression and activity coordinately with cell migration in cardiac fibroblasts. The data further emphasize the physiological importance of interactions among the multiple pro- and anti-inflammatory cytokines present in the post-injury environment to determine cardiac fibroblast phenotype. These considerations have important implications for efforts to develop therapeutic agents targeted at MMPs to limit adverse consequences of ECM remodeling in the injured and failing myocardium.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge Drs. M. Darren Mitchell and S. Kelly Ambler for numerous helpful discussions. Dr. Ambler also critically reviewed the manuscript. This work was supported by NIH (GM 59428 and HL79160).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wilson EM, Spinale FG. Myocardial remodelling and matrix metalloproteinases in heart failure: turmoil within the interstitium. Ann Med. 2001;33:623–634. doi: 10.3109/07853890109002108. [DOI] [PubMed] [Google Scholar]
- 2.Frangogiannis NG, Smith CW, Entman ML. The inflammatory response in myocardial infarction. Cardiovasc Res. 2002;53:31–47. doi: 10.1016/s0008-6363(01)00434-5. [DOI] [PubMed] [Google Scholar]
- 3.Leask A, Abraham DJ. TGF-beta signaling and the fibrotic response. FASEB J. 2004;18:816–827. doi: 10.1096/fj.03-1273rev. [DOI] [PubMed] [Google Scholar]
- 4.Weber KT, Sun Y, Katwa LC. Wound healing following myocardial infarction. Clin Cardiol. 1996;19:447–455. doi: 10.1002/clc.4960190602. [DOI] [PubMed] [Google Scholar]
- 5.Eghbali M. Cardiac fibroblasts: function, regulation of gene expression, and phenotypic modulation. Basic Res Cardiol. 1992;87(Suppl 2):183–189. doi: 10.1007/978-3-642-72477-0_16. [DOI] [PubMed] [Google Scholar]
- 6.Brown RD, Ambler SK, Mitchell MD, Long CS. The cardiac fibroblast: Therapeutic target in myocardial remodeling and failure. Annu Rev Pharmacol Toxicol. 2005;45:657–687. doi: 10.1146/annurev.pharmtox.45.120403.095802. [DOI] [PubMed] [Google Scholar]
- 7.Seta Y, Shan K, Bozkurt B, Oral H, Mann DL. Basic mechanisms in heart failure: the cytokine hypothesis. J Card Fail. 1996;2:243–249. doi: 10.1016/s1071-9164(96)80047-9. [DOI] [PubMed] [Google Scholar]
- 8.Brown RD, Mitchell MD, Long CS. Proinflammatory cytokines and cardiac extracellular matrix: Regulation of fibroblast phenotype. In: Villarreal FJ, editor. Interstitial Fibrosis in Heart Disease. Springer; New York: 2005. pp. 57–82. [Google Scholar]
- 9.Postlethwaite AE, Kang AH. Fibroblasts and Matrix Proteins. In: Gallin JI, Snyderman R, editors. Inflammation. Basic Principles and Clinical Correlates. Lippincott Williams & Wilkins; Philadelphia: 1999. pp. 227–257. [Google Scholar]
- 10.Mitchell MD, Laird RE, Brown RD, Long CS. IL-1beta stimulates rat cardiac fibroblast migration via MAP kinase pathways. Am J Physiol Heart Circ Physiol. 2007;292:H1139–H1147. doi: 10.1152/ajpheart.00881.2005. [DOI] [PubMed] [Google Scholar]
- 11.Heymans S, Luttun A, Nuyens D, Theilmeier G, Creemers E, Moons L, Dyspersin GD, Cleutjens JP, Shipley M, Angellilo A, Levi M, Nube O, Baker A, Keshet E, Lupu F, Herbert JM, Smits JF, Shapiro SD, Baes M, Borgers M, Collen D, Daemen MJ, Carmeliet P. Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure. Nat Med. 1999;5:1135–1142. doi: 10.1038/13459. [DOI] [PubMed] [Google Scholar]
- 12.Dollery CM, Humphries SE, McClelland A, Latchman DS, McEwan JR. Expression of tissue inhibitor of matrix metalloproteinases 1 by use of an adenoviral vector inhibits smooth muscle cell migration and reduces neointimal hyperplasia in the rat model of vascular balloon injury. Circulation. 1999;99:3199–3205. doi: 10.1161/01.cir.99.24.3199. [DOI] [PubMed] [Google Scholar]
- 13.Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 2002;295:2387–2392. doi: 10.1126/science.1067100. [DOI] [PubMed] [Google Scholar]
- 14.Xiao L, Pimental DR, Amin JK, Singh K, Sawyer DB, Colucci WS. MEK1/2-ERK1/2 mediates α1-adrenergic receptor-stimulated hypertrophy in adult rat ventricular myocytes. J Mol Cell Cardiol. 2001;33:779–787. doi: 10.1006/jmcc.2001.1348. [DOI] [PubMed] [Google Scholar]
- 15.Long CS. Autocrine and paracrine regulation of myocardial cell growth in vitro. The TGFβ paradigm. Trends Cardiovasc Med. 1996;6:217–226. doi: 10.1016/S1050-1738(96)00090-4. [DOI] [PubMed] [Google Scholar]
- 16.Meiners S, Hocher B, Weller A, Laule M, Stangl V, Guenther C, Godes M, Mrozikiewicz A, Baumann G, Stangl K. Downregulation of matrix metalloproteinases and collagens and suppression of cardiac fibrosis by inhibition of the proteasome. Hypertension. 2004;44:471–477. doi: 10.1161/01.HYP.0000142772.71367.65. [DOI] [PubMed] [Google Scholar]
- 17.Siwik DA, Chang DL, Colucci WS. Interleukin-1β and tumor necrosis factor-α decrease collagen synthesis and increase matrix metalloproteinase activity in cardiac fibroblasts in vitro. Circ Res. 2000;86:1259–1265. doi: 10.1161/01.res.86.12.1259. [DOI] [PubMed] [Google Scholar]
- 18.Xie Z, Singh M, Singh K. Differential regulation of matrix metalloproteinase-2 and -9 expression and activity in adult rat cardiac fibroblasts in response to interleukin-1beta. J Biol Chem. 2004;279:39513–39519. doi: 10.1074/jbc.M405844200. [DOI] [PubMed] [Google Scholar]
- 19.Romanic AM, Burns-Kurtis CL, Gout B, Berrebi-Bertrand I, Ohlstein EH. Matrix metalloproteinase expression in cardiac myocytes following myocardial infarction in the rabbit. Life Sci. 2001;68:799–814. doi: 10.1016/s0024-3205(00)00982-6. [DOI] [PubMed] [Google Scholar]
- 20.Barrett AJ, Rawlins ND, Woessner JF. Handbook of Proteolytic Enzymes. Academic Press; San Diego: 1998. [Google Scholar]
- 21.Chakraborti S, Mandal M, Das S, Mandal A, Chakraborti T. Regulation of matrix metalloproteinases: an overview. Mol Cell Biochem. 2003;253:269–285. doi: 10.1023/a:1026028303196. [DOI] [PubMed] [Google Scholar]
- 22.Gray MO, Long CS, Kalinyak JE, Li HT, Karliner JS. Angiotensin II stimulates cardiac myocyte hypertrophy via paracrine release of TGF-β1 and endothelin-1 from fibroblasts. Cardiovasc Res. 1998;40:352–363. doi: 10.1016/s0008-6363(98)00121-7. [DOI] [PubMed] [Google Scholar]
- 23.Park CH, Lee MJ, Ahn J, Kim S, Kim HH, Kim KH, Eun HC, Chung JH. Heat shock-induced matrix metalloproteinase (MMP)-1 and MMP-3 are mediated through ERK and JNK activation and via an autocrine interleukin-6 loop. J Invest Dermatol. 2004;123:1012–1019. doi: 10.1111/j.0022-202X.2004.23487.x. [DOI] [PubMed] [Google Scholar]
- 24.Lucchesi PA, Sabri A, Belmadani S, Matrougui K. Involvement of metalloproteinases 2/9 in epidermal growth factor receptor transactivation in pressure-induced myogenic tone in mouse mesenteric resistance arteries. Circulation. 2004;110:3587–3593. doi: 10.1161/01.CIR.0000148780.36121.47. [DOI] [PubMed] [Google Scholar]
- 25.Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, Castner BJ, Stocking KL, Reddy P, Srinivasan S, Nelson N, Boiani N, Schooley KA, Gerhart M, Davis R, Fitzner JN, Johnson RS, Paxton RJ, March CJ, Cerretti DP. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature. 1997;385:729–733. doi: 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.