

Abstract
Culture of Desulfovibrio vulgaris in a medium supplemented with 5-aminolevulinic acid and l-methionine-methyl-d3 resulted in the formation of porphyrins (sirohydrochlorin, coproporphyrin III, and protoporphyrin IX) in which the methyl groups at the C-2 and C-7 positions were deuterated. A previously unknown hexacarboxylic acid was also isolated, and its structure was determined to be 12,18-didecarboxysirohydrochlorin by mass spectrometry and 1H NMR. These results indicate a primitive pathway of heme biosynthesis in D. vulgaris consisting of the following enzymatic steps: (i) methylation of the C-2 and C-7 positions of uroporphyrinogen III to form precorrin-2 (dihydrosirohydrochlorin); (ii) decarboxylation of acetate groups at the C-12 and C-18 positions of precorrin-2 to form 12,18-didecarboxyprecorrin-2; (iii) elimination of acetate groups of the C-2 and C-7 positions of 12,18-didecarboxyprecorrin-2 to form coproporphyrinogen III; and (iv) conversion of coproporphyrinogen III to protoporphyrin IX via protoporphyrinogen IX. We isolated the following three enzymatic activities involved in steps i–iii from the soluble fraction of the cells by anion-exchange chromatography: S-adenosyl-l-methionine:uroporphyrinogen III methyltransferase, precorrin-2 12,18-acetate decarboxylase, and 12,18-didecarboxyprecorrin-2 2,7-decarboxymethylase; all enzymic products were converted into autooxidized methyl esters and analyzed by thin-layer chromatography, UV–visible (UV-VIS) absorption, and mass spectrometry. The enzymatic reactions in D. vulgaris shed new light on porphyrin biosynthesis at an early stage in the evolution of prokaryotes.
Porphyrin biosynthesis in aerobic organisms has been extensively investigated and the pathway is well established (1). Although the source of 5-aminolevulinic acid (ALA) can be either glycine plus succinyl-CoA or glutamate, depending on the species, the biosynthetic pathway from ALA to protoporphyrin IX is common to all aerobic organisms so far examined. However, Akutsu, Park, and Sano discovered in 1993 that the methyl groups at the C-2 and C-7 positions of heme c in cytochrome c3 from the obligate anaerobe Desulfovibrio vulgaris Miyazaki F arise, not from C-2 of ALA, as in the established pathway, but from the methyl group of l-methionine (2). This finding suggested that an alternative pathway from uroporphyrinogen III to protoheme operates in this sulfate-reducing bacterium.
Sulfate-reducing bacteria belonging to the genus Desulfovibrio are included among the domain eubacteria and are considered to be closely related phylogenetically to the earliest living organisms, which appeared on earth approximately 3 billion years ago (3). Therefore, it is important to establish the biosynthetic pathway of porphyrins in D. vulgaris, not only from the biochemical point of view, but also from the viewpoint of molecular evolution. In this paper, we describe a sequence of intermediates in the conversion of uroporphyrinogen III to coproporphyrinogen III and their stepwise enzymic conversion.
MATERIALS AND METHODS
Materials.
Uroporphyrin III octamethyl ester, coproporphyrin III tetramethyl ester, and protoporphyrin IX dimethyl ester were obtained from Sigma. ALA was obtained from Wako Pure Chemical (Kyoto). Deuterated l-methionine-methyl-d3 was purchased from Cambridge Isotope Laboratories (Cambridge, MA).
Culture Conditions.
D. vulgaris Miyazaki F and Hildenborough cells were cultured anaerobically on Postgate medium C (4) containing ALA (10 mg/liter) and l-methionine (0.2 g/liter) for 18 h at 37°C.
Isolation and Characterization of Porphyrins.
The bacterial cells were collected by centrifuging the culture medium at 11,000 × g. The pellets were resuspended in 5 vol of l0 mM Tris⋅HCl, pH 7.3, containing 5 mM dithiothreitol (DTT). The mixture was sonicated with 10–15 1-min bursts at 0°C. After DNase I (Sigma) was added, the mixture was centrifuged at 45,000 × g for 1 h at 4°C. The supernatant and the pellet were regarded as the soluble and cell membrane fractions, respectively.
Porphyrins were extracted from the soluble and the membrane fractions with 10–20 vol of 0.1 M HCl/acetone and 0.5 M HCl/acetone, respectively, according to a method described previously (5, 6). The HCl/acetone solution was evaporated to dryness under reduced pressure, and the solid was subjected to methyl esterification. An efficient way to prepare methyl esters of siro- and siro-type hydrochlorin without formation of lactone is as follows. The extracted porphyrins (about 1 μmol) were kept in one arm of a Thunberg-type tube and anhydrous HCl in methanol (5–7%, 5–7 ml) containing 50 mg of powdered anhydrous FeSO4 was kept in the other arm of the tube. The system was flushed slowly with nitrogen and the contents were mixed, followed by the passage of nitrogen gas into the solution at a very fast flow for 2 min at 50°C. After the tube was closed, the mixture was kept overnight at room temperature. The mixture was then neutralized with saturated sodium acetate and the ester was extracted into ethyl acetate, washed with water, dried over sodium sulfate, and evaporated to dryness under reduced pressure.
For purification of porphyrin esters, preparative TLC was performed on silica gel plates (Kieselgel 60, Merck) with benzene/ethyl acetate/methanol (85:12:3, vol/vol) as an eluent. The methyl esters of porphyrins were also separated by HPLC using a YMC porous silica column (s-5 120A SIL, Yamamura Chemical Laboratories, Kyoto; 2 × 25 cm) or a Cosmosil 5SL column (Nacalai Tesque, Kyoto; 4.6 × 150 mm). The various porphyrin esters were eluted with n-heptane/ethyl acetate/dichloromethane/methanol (60:25:15:2.7, vol/vol) and were detected by monitoring fluorescence at 600 nm with excitation at 380 nm (siro- or siro-type porphyrins) or fluorescence at 620 nm with excitation at 400 nm (uroporphyrin, coproporphyrin, protoporphyrin, etc.) by means of a Shimadzu RF-535 fluorescence detector.
Absorption spectra were recorded by a Shimadzu UV-3100PC UV-VIS-NIR scanning spectrophotometer or a Shimadzu UV-2200 spectrophotometer. Laser desorption ionization mass spectrometry (LDIMS) was performed on a Shimadzu/Kratos Kompact MALDI III laser ionization time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm). Ions were accelerated to a kinetic energy of 5 keV and were analyzed in the positive linear mode of operation. The laser power density was 106 W⋅cm−2. High-resolution liquid secondary ion mass spectrometry (LSIMS) was performed with a Shimadzu/Kratos Concept I.H. instrument. 1H NMR spectra were obtained with a JEOL JNM-GX-270 NMR spectrometer.
Preparation of Enzymes.
Frozen D. vulgaris Miyazaki F or Hildenborough cells (2 g wet mass) were resuspended in 6 ml of 25 mM Tris⋅HCl, pH 7.7/5 mM DTT and disrupted by sonication, then treated with DNase I for 30 min. The homogenate was centrifuged at 100,000 × g for 30 min. The supernatant was diluted 10-fold with cold distilled water and applied to a DEAE-Toyopearl column (Tosoh, Tokyo; 1 × 5 cm) equilibrated with 25 mM Tris⋅HCl, pH 7.7. After washing with the same buffer, S-adenosyl-l-methionine:uroporphyrinogen III methyltransferase (SUMT) activity was eluted with the same Tris buffer containing 0.1 M NaCl. After desulfoviridin (dark green fraction) was eluted with the Tris buffer containing 0.2 M NaCl, precorrin-2 12,18-acetate decarboxylase (precorrin-2 decarboxylase) activity was eluted with the same buffer containing 0.3 M NaCl. 12,18-Didecarboxyprecorrin-2 2,7-decarboxymethylase (“coproporphyrinogen synthase”) was eluted with the same buffer containing 0.3 M NaCl closely after precorrin-2 decarboxylase was eluted.
SUMT was further purified to homogeneity as outlined below. The cells (27 g wet weight) were homogenized in 54 ml of 50 mM Tris⋅HCl, pH 7.7, containing 5 mM DTT and 1 mM EDTA by sonication. The homogenate was centrifuged at 20,800 × g for 20 min. The supernatant was applied to a DEAE-Toyopearl column (2.5 × 20 cm) equilibrated with the same buffer. The SUMT activity was eluted with the same buffer but containing 0.1 M NaCl. The active fractions were pooled, concentrated to about 2 ml with a Centriflo CF 25 membrane concentrator (Amicon), and applied to a TSKgel G3000SW column (Tosoh, Tokyo; 2 × 35 cm) equilibrated with 50 mM Tris⋅HCl, pH 7.4, containing 0.2 M KCl. The enzyme was eluted with the same buffer. The active fractions were collected, concentrated, diluted 2-fold with cold distilled water, and applied to a Mono Q HR 5/5 column (Pharmacia Biotech) equilibrated with 20 mM Tris⋅HCl, pH 7.4. SUMT was eluted with a linear gradient of NaCl, 0–0.5 M, in the Tris buffer. The active fractions were collected, concentrated, and further purified by gel filtration on a TSKgel G3000SWXL column (Tosoh, Tokyo; 7.8 × 300 mm) equilibrated with 20 mM Tris⋅HCl, pH 7.4/0.2 M KCl.
Enzyme Assays.
(i) SUMT assay. SUMT catalyzes the conversion of uroporphyrinogen III into precorrin-2. The methylase activity was measured in a final volume of 2 ml containing 0.1 M Tris⋅HCl buffer at pH 7.7, 0.1 M NaCl, 5 mM DTT, 0.5 mM S-adenosyl-l-methionine (SAM), 5 μM uroporphyrinogen III [prepared by the reduction of uroporphyrin III with sodium amalgam (7)], and the enzyme solution. The same experiment was done in the absence of SAM. The mixture was incubated anaerobically in the dark at 37°C for 1–3 h, and the reaction was stopped by the addition of 20 ml of HCl/acetone (0.5 M) at 0°C. After being centrifuged, the supernatant was completely dried under reduced pressure. The extracted porphyrins were subjected to methyl esterification and separated by TLC. SUMT activity was determined by the amount of sirohydrochlorin octamethyl ester recovered from the reaction mixtures.
(ii) Precorrin-2 decarboxylase assay. Precorrin-2 decarboxylase catalyzes the conversion of precorrin-2 to 12,18-didecarboxyprecorrin-2. Because precorrin-2 is extremely sensitive to oxidation and the reduction of sirohydrochlorin with sodium amalgam resulted in a compound different from precorrin-2, the precorrin-2 was generated from uroporphyrinogen III and SAM by using partially purified SUMT. Thus, the decarboxylase activity was anaerobically measured in a final volume of 2 ml containing 0.1 M Tris⋅HCl at pH 7.7, 0.1 M NaCl, 5 mM DTT, 0.5 mM SAM, 0.5 mM NADH, 0.5 mM NADPH, 5 μM uroporphyrinogen III, partially purified SUMT, and the enzyme solution. As a control, the same experiment was done in the absence of SAM. After incubation, the porphyrins were extracted and subjected to methyl esterification as described for SUMT assay. The decarboxylase activity was determined by the amounts of 12,18-didecarboxysirohydrochlorin hexamethyl ester and 12/18-monodecarboxysirohydrochlorin heptamethyl ester recovered from the reaction mixtures.
(iii) Coproporphyrinogen synthase assay. Acetate eliminase catalyzes the conversion of 12,18-didecarboxyprecorrin-2 to coproporphyrinogen III. The substrate, 12,18-didecarboxyprecorrin-2, was enzymically formed from uroporphyrinogen III and SAM in the reaction mixtures. The acetate eliminase activity was anaerobically measured in a final volume of 2 ml containing 0.1 M Tris⋅HCl at pH 7.7, 0.1 M NaCl, 5 mM DTT, 0.5 mM SAM, 0.5 mM NADH, 0.5 mM NADPH, 0.5 mM NAD+, 0.5 mM NADP+, 5 μM uroporphyrinogen III, partially purified SUMT and precorrin-2 decarboxylase, and the enzyme solution. As a control, the same experiment was done in the absence of SAM. After incubation, the porphyrins were extracted and subjected to methyl esterification as described for SUMT assay. The eliminase activity was determined by the amounts of coproporphyrin III tetramethyl ester and 2/7-monodecarboxymethyl-12,18-didecarboxysirohydrochlorin pentamethyl ester recovered from the reaction mixtures.
For the usual anaerobic enzyme assay, we used an aluminum-seal vial (3 ml) with a rubber septum (usually used for headspace method in gas chromatography; Maruemu, Osaka, Japan). The sealed vial containing the reaction mixture was flushed with argon for 20 min prior to the addition of porphyrinogen via the septum with a needle. We confirmed by the catechol-2,3-dioxygenase method (8) that the O2 concentration of 0.1 M Tris⋅HCl, pH 7.7, in the vial was less than 0.5 μM immediately after 20-min argon flushing. However, it was difficult to check for every experiment whether the initial anaerobic condition (<0.5 μM O2) was maintained throughout the incubation (1–3 h), because it was difficult to avoid leakage of air into the vial through the pin-hole of the rubber septum made by the needle. Absolutely anaerobic conditions (<0.3 μM O2) were obtained as follows. A specially designed glass vessel containing the reaction mixture was connected to a high-vacuum line and flushed three times with argon. The vessel was then closed by fusing its neck. To determine residual O2 in this method, a Thunberg-type tube consisting of two arms and a quartz cell was used, and the O2 concentration was determined by measuring the amount of 2-hydroxymuconate semialdehyde, the product of enzymic dioxygenation of catechol. Microaerobic conditions (2–5 μM) were made by argon bubbling followed by injection of a calculated aliquot of air-saturated water by a gas-tight microsyringe into the incubation mixture.
RESULTS
Three important techniques were developed for this study. First, the best medium composition for overproduction of porphyrin intermediates was determined. The absorbance at 377 or 405 nm of porphyrin methyl esters per mg of protein of the culture was used as a measure of the efficiency of porphyrin production. The growth of Desulfovibrio with the supplement of ALA and l-methionine under optimal conditions enhanced the formation of uro-, copro-, and protoporphyrin 4–5 times and siro- or siro-type porphyrins 2–3 times compared with normal growth without the supplement. Second, an improved method was developed to avoid lactone formation (9, 10) during methyl esterification of sirohydrochlorin. This method greatly facilitated the isolation and characterization of intermediates of porphyrin biosynthesis. Moreover, the method was useful for delactonization of (monolactone) sirohydrochlorin heptamethyl ester (Mr 959) and (dilactone) sirohydrochlorin hexamethyl ester (Mr 943) into sirohydrochlorin octamethyl ester (Mr 975). Third, LDIMS (11) was used to determine the molecular weights of unfragmented porphyrins.
Identification of Porphyrins Accumulated by Cell Culture of D. vulgaris Miyazaki F and Hildenborough.
The major component of the porphyrin extracted from whole bacterial cells was sirohydrochlorin. Small amounts of sirohydrochlorin monoamide, uroporphyrin III, coproporphyrin III, and protoporphyrin IX were present. Besides these, trace amounts of an unidentified compound, referred to as “pigment X,” were found. Protoporphyrin IX and coproporphyrin III were mainly present in the bacterial membrane fraction. The Mr value of each porphyrin ester purified by repeated TLC and/or HPLC was found to be as follows by mass spectrometry ([M + H]+): sirohydrochlorin octamethyl ester, 975 by LDIMS and 975.4245 by LSIMS (calculated value 975.4222 for C50H63N4O16); sirohydrochlorin monoamide, 960; uroporphyrin III octamethyl ester, 943; coproporphyrin III tetramethyl ester, 711; and protoporphyrin IX dimethyl ester, 591. The UV-VIS absorption spectra, HPLC, and 1H NMR data for the methyl esters were identical with those of the corresponding authentic samples.
Mass Spectra of Porphyrin Intermediates Isolated from the Bacteria Cultured with l-Methionine-methyl-d3.
Because methyl transfer from l-methionine to the C-2 and C-7 positions of heme c in cytochrome c3 occurs in D. vulgaris (2), the cells were grown in medium supplemented with ALA and l-methionine-methyl-d3, and porphyrin intermediates in those cells were analyzed. Mass spectra of each porphyrin ester isolated from the deuterated cells are presented in Fig. 1.
Figure 1.

Laser desorption ionization mass spectra of purified porphyrin intermediates of D. vulgaris cells cultured in the presence of l-methionine-methyl-d3. Peaks A, B, C, and D were assigned to uroporphyrin III octamethyl ester (943), 2,7-methyl-d3 sirohydrochlorin octamethyl ester (980), 2,7-methyl-d3 coproporphyrin III tetramethyl ester (717), and 2,7-methyl-d3 protoporphyrin IX dimethyl ester (596), respectively.
Whereas the observed Mr value of uroporphyrin III octamethyl ester did not change at all (943), those of deuterated sirohydrochlorin octamethyl ester, deuterated coproporphyrin III tetramethyl ester, and deuterated protoporphyrin IX dimethyl ester were 980, 717, and 596, respectively, and these Mr values were larger by 5 or 6 compared with those of the corresponding compounds isolated from the nondeuterated cells. This result strongly suggests that deuterated methyl groups transferred from l-methionine into precorrin-2 are carried over to protoporphyrinogen IX and eventually heme c biosynthesis, consistent with the conclusion of Akutsu et al. (2).
Isolation and Structure of Pigment X.
To clarify the structure of pigment X, a large-scale (40- to 50-liter) culture was prepared to allow isolation of the pigment. TLC revealed a new intense porphyrin band showing orange fluorescence at the Rf value corresponding to hexacarboxylate methyl ester. The quantitative ratio of the new compound to sirohydrochlorin octamethyl ester was approximately 1:10. The structure of this compound was established as 12,18-didecarboxysirohydrochlorin hexamethyl ester (Fig. 2, 3ox) by mass, UV-VIS, and 1H NMR spectroscopy.
Figure 2.

Absorption (A) and 1H NMR (B) spectra of 12,18-didecarboxysirohydrochlorin hexamethyl ester. Samples were dissolved in pyridine (A) or d-chloroform (B), respectively. The absorption spectrum showed maxima at 381.5, 510.5, 546.0, 586.5, and 635.0 nm. In the 1H NMR spectrum, assignments are given on the top of each signal. The structure of 12,18-didecarboxysirohydrochlorin hexamethyl ester (3ox) is shown along with the numbering system. Rings A, B, C, and D are named clockwise from the upper left.
High-resolution LSIMS of pigment X methyl ester showed a molecular ion of 859.4120 (calculated 859.4114 for C46H59N4O12 as [M + H]+). The absorption spectrum of the pigment (Fig. 2A) was similar to that of sirohydrochlorin octamethyl ester, indicating that A and B rings are still reduced as in sirohydrochlorin. The 1H NMR spectrum (Fig. 2B) shows four meso bridge protons (Left), and the signal at 4.28 ppm in the high-field region due to the methylene protons of the acetate groups at the C-12 and C-18 positions of sirohydrochlorin octamethyl ester (12) was completely lost (Right). The chemical shifts and intensities of other signals were similar to those of sirohydrochlorin octamethyl ester, observed under the same conditions. Thus, UV-VIS absorption, mass, and 1H NMR spectral data strongly support the structure of 3ox shown in Figs. 2 and 3.
Figure 3.

(Upper) Proposed alternative pathway of porphyrin biosynthesis from uroporphyrinogen III to coproporphyrinogen III in D. vulgaris. (Lower) Porphyrin esters shown are the oxidized and methyl-esterified forms of the corresponding porphyrinogens. A, M, P and Me stand for acetate group, methyl group, propionyl group, and methyl ester, respectively.
The presence of this new intermediate suggests that decarboxylation of the acetate groups at the rings C and D of precorrin-2 is followed by elimination of the acetate groups from the rings A and B to form coproporphyrinogen III in the biosynthetic pathway. Because 2,7-didecarboxymethylsirohydrochlorin hexamethyl ester (Mr 827) was not detected, even in the mass spectrum of the bulk porphyrin fraction, it is unlikely that the loss of the acetate groups from the rings A and B occurs before the decarboxylation at the rings C and D. Thus, heme c biosynthesis in D. vulgaris takes the following pathway (Fig. 3). (i) Formation of uroporphyrinogen III from ALA as for heme, chlorophyll a, and vitamin B12. (ii) Methyl transfer from l-methionine to the C-2 and C-7 positions of uroporphyrinogen III to form precorrin-2, which is also a precursor of vitamin B12 (13–17) and branches off from the known heme c biosynthesis pathway. (iii) Coproporphyrinogen III is formed, not by the direct decarboxylation of uroporphyrinogen III, but via precorrin-2 with sequential decarboxylation of acetic acid of rings C and D of precorrin-2, followed by elimination of acetate groups from rings A and B. (iv) Coproporphyrinogen III is then transformed into protoporphyrinogen IX, oxidized to protoporphyrin IX, and chelated with Fe(II).
To confirm the proposed pathway of porphyrin biosynthesis in D. vulgaris in terms of enzymology, soluble proteins extracted from the cells were fractionated by anion-exchange chromatography (Fig. 4) and enzyme activities possibly involved in the pathway (Fig. 3) were examined. The following experiments (experiments 1–3) were done more than 50 times.
Figure 4.

Elution profile of the activities of SUMT, precorrin-2 decarboxylase, and acetate eliminase on a DEAE-Toyopearl column. Chromatography was performed as described in the text. I and II, fractions containing SUMT activity and precorrin-2 decarboxylase activity, respectively. Acetate eliminase activity (III) was eluted following precorrin-2 decarboxylase.
Biosynthetic Formation of Precorrin-2 from Uroporphyrinogen III (Experiment 1).
The DEAE-Toyopearl fractions eluted with 0.1 M NaCl (fractions 5–7, I in Fig. 4) were incubated with 5 μM uroporphyrinogen III in the presence of 0.5 mM SAM and 5 mM DTT for 2 h at 37°C. The autooxidized and methyl-esterified product was isolated in a pure form and it was confirmed to be sirohydrochlorin octamethyl ester (2ox in Fig. 3) by mass spectra, UV-VIS absorption spectrum, and TLC and HPLC behaviors. The amount of uroporphyrin III octamethyl ester was concomitantly reduced. In the absence of SAM, only a trace amount of sirohydrochlorin octamethyl ester was detected, and uroporphyrin III octamethyl ester was recovered in a large amount. In the following experiments, these fractions were combined (fraction I) and used as partially purified SUMT.
Biosynthetic Formation of 12/18-Monodecarboxy- and 12,18-Didecarboxyprecorrin-2 (Experiment 2).
The DEAE-Toyopearl fractions eluted with 0.3 M NaCl (fractions 39–41, II in Fig. 4) were incubated with 5 μM uroporphyrinogen III, 0.5 mM SAM, 0.5 mM NADH, 0.5 mM NADPH, and 5 mM DTT, in the presence of fraction I (I in Fig. 4) for 3–4 h at 37°C. The TLC profile of the methyl esters of the autooxidized products extracted from the reaction mixture is shown in Fig. 5 (lane 1). The porphyrin esters were purified by repeated TLC or HPLC and the mass spectrum of each pigment was obtained. The pigments corresponding to bands a and b (Fig. 5, lane 1) gave molecular ion peaks of 859 and 916, respectively. These values of Mr were in good agreement with those of 12,18-didecarboxysirohydrochlorin hexamethyl ester and 12/18-monodecarboxysirohydrochlorin heptamethyl ester, respectively. The former compound was identical to pigment X isolated from the cultured bacterium. Thus, fraction II (Fig. 4) contained precorrin-2 decarboxylase activity, and we used this fraction as partially purified decarboxylase in the following experiment. The enzyme activity was inhibited by 10 mM N-ethylmaleimide or 5 mM EDTA but not by 1 mM α,α′-dipyridyl.
Figure 5.

TLC analysis of methyl esters of porphyrins derived from reaction mixtures containing fractions I and II (lane 1) or fractions I–III (lanes 2 and 3) as enzyme solutions. Fractions I, II, and III were prepared from the soluble fraction of D. vulgaris by DEAE-Toyopearl chromatography (see Fig. 4). Porphyrin esters were detected by their fluorescence with excitation at 366 nm using a Mineralight UVGL-58 (Upland, CA). Standard porphyrin esters: P, protoporphyrin IX dimethyl ester; C, coproporphyrin III tetramethyl ester; U, uroporphyrin III octamethyl ester. Lane 1, experiment 2; a, 12,18-didecarboxysirohydrochlorin hexamethyl ester; b, 12/18-monodecarboxysirohydrochlorin heptamethyl ester; c, sirohydrochlorin octamethyl ester; d, uroporphyrin III octamethyl ester. Lane 2, experiment 3; lane 3, experiment 3 without SAM.
Biosynthetic Formation of Coproporphyrinogen III (Experiment 3).
The DEAE-Toyopearl fractions 42–44 (III in Fig. 4) were incubated with 5 μM uroporphyrinogen III, 0.5 mM SAM, 0.5 mM NADH, 0.5 mM NADPH, 0.5 mM NAD+, 0.5 mM NADP+, and 5 mM DTT, in the presence of both fractions I and II for 3–4 h at 37°C under anaerobic and low-oxygen conditions (2–5 μM O2). A large amount of coproporphyrin III tetramethyl ester, with small amount of 2/7-monodecarboxymethyl-12,18-didecarboxysirohydrochlorin pentamethyl ester (Mr 785), was obtained with concomitant loss of 12,18-didecarboxysirohydrochlorin hexamethyl ester. A substantial amount of protoporphyrin IX dimethyl ester was also formed under low-oxygen conditions (Fig. 5, lane 2). Experiments without SAM gave uroporphyrin III octamethyl ester as the main recovered porphyrin and no evidence of coproporphyrin III tetramethyl ester (Fig. 5, lane 3).
Under strictly anaerobic condition (less than 0.3 μM O2), the reaction stopped at the stage of 12,18-didecarboxyprecorrin-2, and no coproporphyrinogen III was formed (data not shown; the TLC profile was similar to that shown in lane 1 of Fig. 5). Because it was difficult to isolate acetate eliminase activity in a stable form, low reproducibility was observed in experiment 3. In some experiments, 2/7-monodecarboxymethyl-12,18-didecarboxysirohydrochlorin pentamethyl ester was mainly obtained, but coproporphyrin III tetramethyl ester and protoporphyrin IX dimethyl ester were obtained in small amounts.
These results (experiments 1–3) indicate that the bacteria contain enzymic activity that decarboxylates precorrin-2 (2, Fig. 3) at the 12 and 18 positions to give 3 (Fig. 3) and that there is a separate enzymic activity that removes acetate side chains from 3 or a certain metabolite of 3 to give coproporphyrinogen III (4, Fig. 3) or some product that is eventually isolated as coproporphyrin III tetramethyl ester. Further investigation is necessary to determine whether protoporphyrinogen IX is biosynthesized via coproporphyrinogen IX.
Characterization of Purified SUMT.
SUMT was purified to homogeneity as judged by SDS/PAGE, NH2-terminal amino acid sequence, and matrix-assisted LDIMS spectrum. The sequence of the NH2-terminal 10 residues of SUMT was determined to be MNVLVINSGS. The subunit molecular mass was determined to be 44,286 Da by mass spectrometry.
DISCUSSION
This work establishes that a primitive pathway of porphyrin biosynthesis occurs in the obligate anaerobic bacterium D. vulgaris. Like the biosynthesis of siroheme (6, 7), vitamin B12 (13–17), and factor F430 (1), porphyrin biosynthesis in D. vulgaris deviates from the known pathway at uroporphyrinogen III (1 in Fig. 3) into isobacteriochlorin (precorrin-2) and reenters again into porphyrinogen (coproporphyrinogen III):
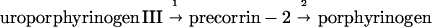 |
The first reaction, 1, occurs in the pathways for formation of siroheme, vitamin B12, and factor F430. The second step, 2, is revealed in the present study. Precorrin-2 is particularly important in sulfate-reducing bacteria. It undergoes oxidation to sirohydrochlorin and insertion of iron to form siroheme, a cofactor of a sulfate reductase (desulfoviridin). In this paper we show that it can also be an intermediate in heme biosynthesis.
We isolated a pigment from the culture of D. vulgaris, identified it as 12,18-didecarboxyprecorrin-2, and demonstrated that it is formed from precorrin-2 by a precorrin-2 decarboxylase. The further enzymic conversion of 12,18-didecarboxyprecorrin-2 to coproporphyrinogen III is intriguing. Submicromolar amounts of molecular oxygen may be involved in the elimination of the acetate groups from the C-2 and C-7 positions, but it is unknown what kind of two-carbon compound is extruded (glyoxylate?). The ambivalent relations of sulfate-reducing bacteria to molecular oxygen have been discussed recently (18). In general, oxygen is reduced prior to sulfate reduction. The present work suggests that molecular oxygen may be involved in some steps of heme formation.
Isolation of SUMT and precorrin-2 decarboxylase in active form by DEAE-Toyopearl chromatography was easy compared with isolation of coproporphyrinogen synthase, which is unstable, causing poor reproducibility in assays. This difficulty may be related to the technical difficulty of maintaining suitably low oxygen concentrations during the assay.
The generality of this detour in the biosynthetic pathway is unknown. Similar experiments using the facultative anaerobe Bacillus licheniformis, which is not related phylogenetically to D. vulgaris, indicated the presence of sirohydrochlorin but showed normal formation of coproporphyrinogen III from uroporphyrinogen III by uroporphyrinogen decarboxylase rather than from precorrin-2 as in D. vulgaris (A.S. and S.S., unpublished data). Recent studies by Bollivar et al. (19), using the photosynthetic bacteria Chlorobium vibrioforme and Rhodobacter capsulatus, also indicated that anaerobic protoporphyrin biosynthesis did not require the incorporation into tetrapyrroles of methyl groups from l-methionine. Although the phylogenetic relationships of these bacteria to D. vulgaris are unclear (20), it seems probable that the primitive pathway in D. vulgaris is conserved in only a limited group of modern-day bacteria. The evolutionary origin of such enzymes as SUMT and precorrin-2 decarboxylase and the date of replacement of the complicated pathway found in D. vulgaris by the single enzymic process seen in other (younger) organisms are intriguing questions which remain to be answered.
Acknowledgments
S.S. thanks Mr. S. Ohnishi and Miss H. Kubo (Shimadzu Company, Kyoto) for recording high-resolution LSIMS, thanks Prof. K. Horiike for the generous use of the equipment in his laboratory (Department of Biochemistry, Shiga University of Medical Science), and expresses sincere thanks to Mr. M. Ohiso, Mr. N. Sakamoto, Mr. T. Tuji, and Mr. S. Saito for their great help in the early stage of this work. We thank the Ministry of Education, Science, Sports, and Culture of Japan for financial support (no. 08457036).
ABBREVIATIONS
- ALA
5-aminolevulinic acid
- LDIMS
laser desorption ionization mass spectrometry
- LSIMS
liquid secondary ion mass spectrometry
- UV-VIS
UV-visible
- SAM
S-adenosyl-l-methionine
- SUMT
S-adenosyl-l-methionine:uroporphyrinogen III methyltransferase
References
- 1.Jordan P M, Akhtar M. In: Biosynthesis of Tetrapyrroles, New Comprehensive Biochemistry. Neuberger A, van Deenen L L M, editors. Vol. 19. Amsterdam: Elsevier; 1991. pp. 1–99. [Google Scholar]
- 2.Akutsu H, Park J-S, Sano S. J Am Chem Soc. 1993;115:12185–12186. [Google Scholar]
- 3.Singleton R., Jr . In: The Sulfate-Reducing Bacteria; Contemporary Perspectives. Odom J M, Singleton R Jr, editors. New York: Springer; 1993. pp. 1–20. [Google Scholar]
- 4.Postgate J R. Sulfate-Reducing Bacteria. 2nd Ed. Cambridge, U.K.: Cambridge Univ. Press; 1984. [Google Scholar]
- 5.Murphy M J, Siegel L M, Kamin H, Rosenthal D. J Biol Chem. 1973;248:2801–2814. [PubMed] [Google Scholar]
- 6.Murphy M J, Siegel L M. J Biol Chem. 1973;248:6911–6919. [PubMed] [Google Scholar]
- 7.Sano S, Granick S. J Biol Chem. 1961;236:1173–1180. [PubMed] [Google Scholar]
- 8.Kobayashi T, Ishida T, Horiike K, Takahara Y, Numao N, Nakazawa T, Nakazawa T, Nozaki M. J Biochem (Tokyo) 1995;117:614–622. doi: 10.1093/oxfordjournals.jbchem.a124753. [DOI] [PubMed] [Google Scholar]
- 9.Battersby A R, Jones K, McDonald E, Robinson J A, Morris H R. Tetrahedron Lett. 1977;25:2213–2216. [Google Scholar]
- 10.Battersby A R, McDonald E, Morris H R, Thompson M, Williams D C, Bykvosky V Y, Zaitseva N I, Bukin N V. Tetrahedron Lett. 1977;25:2217–2220. [Google Scholar]
- 11.Solouki T, Russell D H. Appl Spectrosc. 1993;47:211–217. [Google Scholar]
- 12.Scott A I, Irwin A J, Siegel L M, Shoolery J N. J Am Chem Soc. 1978;100:7987–7994. doi: 10.1021/ja00469a071. [DOI] [PubMed] [Google Scholar]
- 13.Bykvosky V Y, Zaitseva N I, Bukin N V. Dokl Akad Nauk SSSR. 1975;224:1431–1434. [PubMed] [Google Scholar]
- 14.Bykvosky V Y, Zaitseva N I. Prikl Biokhim Mikrobiol. 1976;12:365–370. [PubMed] [Google Scholar]
- 15.Bykvosky V Y, Zaitseva N I, Umrikhina A V, Yavorskaya A N. Prikl Biokhim Mikrobiol. 1976;12:825–833. [Google Scholar]
- 16.Scott A I. Angew Chem. 1993;32:1223–1243. [Google Scholar]
- 17.Battersby A R. Science. 1994;264:1551–1557. doi: 10.1126/science.8202709. [DOI] [PubMed] [Google Scholar]
- 18.Marschall C, Frenzel P, Cypionka H. Arch Microbiol. 1993;159:168–173. [Google Scholar]
- 19.Bollivar D W, Elliott T, Beale S I. J Bacteriol. 1995;177:5778–5783. doi: 10.1128/jb.177.20.5778-5783.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Woese C R. Microbiol Rev. 1987;51:221–271. doi: 10.1128/mr.51.2.221-271.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]