

Abstract
RNA molecules are now known to be involved in the processing of genetic information at all levels, taking on a wide variety of central roles in the cell. Understanding how RNA molecules carry out their biological functions will require an understanding of structure and dynamics at the atomistic level, which can be significantly improved by combining computational simulation with experiment. This review provides a critical survey of the state of molecular dynamics (MD) simulations of RNA, including a discussion of important current limitations of the technique and examples of its successful application. Several types of simulations are discussed in detail, including those of structured RNA molecules and their interactions with the surrounding solvent and ions, catalytic RNAs, and RNA–small molecule and RNA–protein complexes. Increased cooperation between theorists and experimentalists will allow expanded judicious use of MD simulations to complement conceptually related single molecule experiments. Such cooperation will open the door to a fundamental understanding of the structure–function relationships in diverse and complex RNA molecules.
Keywords: hydration and cation binding, molecular dynamics, nucleic acid-protein complex, RNA, structure-activity relationship
INTRODUCTION
The fundamental importance of RNA in a wide variety of biological contexts is becoming increasingly apparent. Since the discovery that RNA can catalyze chemical reactions in addition to its ability to carry genetic information,1,2 it has become clear that RNA takes on a wide variety of roles in the cell and is involved in all cellular processing of genetic information, from splicing to translation to modification, and particularly regulation, via RNA interference in eukaryotes and riboswitches in bacteria (for review please see, e.g., Ref. 3 - Ref. 21). Understanding how this central molecule of life is able to accomplish such a variety of functions will necessarily involve understanding RNA structure and dynamics at the atomistic level, a challenge that can be addressed only using a combination of experimental studies and molecular dynamics (MD) simulations.22
When planning and conducting MD simulations, the challenges that must be taken into account are conceptually very similar to those associated with single molecule experiments. One of the primary limitations of MD simulations is the insufficient sampling, stemming from the 1 to 100+ ns time scale of the technique. In single molecule experiments, likewise, there is concern about collecting large enough data sets to ensure that the conclusions drawn are generally valid, rather than true only for a particular molecule. The solutions to the sampling problem are also similar for in silico and in vitro approache: increase the time window of interrogation of a single molecule or perform short observations on many molecules in parallel. In addition, by combining theoretical and experimental approaches, each can help to synergistically explain and verify results from the other. Thus, MD simulation and single molecule techniques are inherently complementary, and direct cooperation between theorists and experimentalists working on related issues will benefit both groups and promote valuable, widely applicable solutions.
Opinions about MD simulations range from a flat rejection of everything that is calculated to the indiscriminate application of computational methods to problems that are far beyond the applicability of the technique. Both extreme views are equally unjustified. Modeling, as any other research method, has its scope, limitations, and error margins. Limitations are significant and need to be carefully evaluated when planning simulations and interpreting the results. Computers readily provide numbers and structures, but are not responsible for the reliability of the results and their interpretation. Unjustified application of simulations therefore provides data of limited value and can be misleading. Wisely applied simulations, by contrast, can considerably complement and aid in understanding of experiments and provide qualified predictions. In fact, experiments often can only be understood with substantial molecular modeling. In general, qualitative applications of computational methods are more appropriate than quantitative studies. This review provides an overview of MD simulations of RNA, with examples of successful applications and suggestions for future use.
GENERAL CONSIDERATIONS
The first simulations on RNA were limited by the resources available at the time. The force field parameters describing RNA had not been sufficiently optimized, and the simulation protocols before 1995 (particularly the treatment of long-range electrostatic forces) were unable to provide stable trajectories even on the 500 ps time scale due to a drastic accumulation of errors. Another major problem was the scarcity of crystal structures of RNA, and thus a lack of reliable high-resolution structures from which to start MD simulations. In the early 1990s, only a few tRNA crystal structures were available. By the mid-1990s, several more RNA structures had been solved, but some turned out to have limited relevance.23 This limitation left early simulations at a distinct disadvantage, since the typically limited sampling of MD means that a simulated structure may be unable to diverge substantially from its starting structure. (Assuming a Boltzmann distribution and standard Arrhenius kinetics, 10-ns scale simulations are only able to overcome free energy barriers that are on the order of ∼5–6 kcal/mol.) Thus, a realistic assessment of dynamics from a simulation depends on a realistic and accurate starting structure.
Some of the controversial results from the earliest RNA simulations were likely due to inadequate or less relevant starting structures.24,25 For example, several MD simulations were based on the crystal structure of the minimal hammerhead ribozyme,26 which was recently shown to be quite different from the active structure.27 Over the last 10 years RNA crystallography has improved substantially. For example, Doudna28 discusses the recent successful use of hexammines to aid in phasing and the importance of chemical modifications such as 2′-O-methyl, 2′H, and vanadate in “trapping” various states of ribozyme cleavage reactions. There are now almost 500 RNA crystal structures in the PDB,29 vastly increasing the possibilities for both the variety and the size of simulations. We have reached the point where even all-atom simulations of the ribosome are becoming feasible,30,31 but we must keep in mind that simulations are still limited by the crystal structures they are based on, and that low resolution in loop regions or uncertainty regarding metal binding can still contribute to error in simulations.
The force fields for simulating RNA have been refined and stable simulation protocols for polyanionic RNA molecules have been introduced, primarily the Particle-Mesh Ewald (PME) treatment of the strong, long-range electrostatic forces.32 A broad set of RNA applications has been tested using the AMBER code and associated force field. This method matches quantum chemical data for stacking33 and base pairing34 (including the complex non-Watson Crick interaction patterns) and, perhaps more importantly, has provided long stable simulations of numerous complex RNA molecules. We have tested the AMBER force field in a total of ∼4 μs of RNA simulations (each 10–200 ns long) and so far have observed rather satisfactory performance. To the best of our knowledge, no other force field has been validated in long stable simulations of complex folded RNA molecules. By comparison, the CHARMM force field, when applied to simple duplexes, showed accelerated opening (or breathing) events of A-helical RNA base pairs that were not observed in AMBER simulations or in imino proton exchange experiments by 1H-NMR.35,36 This strong foundation, however, does not imply that AMBER-based MD simulations are entirely accurate, and it is always important that MD studies are executed with careful consideration and discussion of possible simulation artifacts as detailed here.
The computing power available for MD calculations has increased dramatically in recent years, and strategies for distributed computing, such as Pande's Folding@Home,37,38 allow computations that use tens of thousands of processors in parallel to reach hundreds of microseconds of total simulation time.38 Yet, even if future computers are fast enough to allow in silico folding of RNAs of unknown structure, there is no guarantee that the force field will find the correct global minimum. Thus, MD applications will continue to primarily provide analytical insights about known RNA structures and their modest sequence and structural variants, with specific in-depth insights into the complex roles of dynamic molecular interactions in nucleic acids. The combination of more reliable starting structures, improved force fields, and increased computational power make MD a very feasible and promising technique to study the structure and dynamics of RNA, especially when MD is used in conjunction with (single molecule) experiments.
METHODS
The goal of MD simulations is to mimic the real-time dynamics of single solvated biomolecules in order to elucidate atomic-resolution details of their structural dynamics and eventually estimate the free energies of specific conformations. Simulated RNA molecules are immersed in a sufficiently large (extending at least 10 Å from the RNA) box of water molecules and ions that is periodically extended in all directions (periodic boundary condition). The molecules are described by simple pair additive atomistic potentials (force fields) that treat atoms as van der Waals spheres with partial, constant, point charges localized at the individual atomic centers, linked by harmonic springs mimicking covalent structure and supplemented by simple torsion profiles.
The main limitations of MD simulations stem from: (i) the time-scale of simulations (trajectories are typically 1−100+ ns) and (ii) force field inaccuracies. The calculation of long-range electrostatics has previously been described as a major limitation,39 but introduction of the PME treatment of electrostatics appears to remove all major drawbacks in these calculations.32 It has been suggested that PME can overstabilize simulated systems,40,41 but this has not yet been clearly shown, and the problem should be rather marginal compared to sampling and force field limitations.
Sampling Limitations
The short timescale and resulting limited conformational sampling are often discussed, frequently with the conclusion that faster computers would lead to better results. However, longer simulations may also expose force field deficiencies that have cumulative effects over time, which will become a significant problem once computers can routinely produce microsecond-long trajectories. Additionally, the timescale of real events is of course still much longer than affordable simulation timescales. However, this limitation can sometimes be overcome by running multiple simulations with distinct starting structures or by using enhanced sampling MD methods (see Enhanced Sampling section). Furthermore, a simulated RNA conformation is typically not fully equilibrated as the simulation begins so that the probability to capture structural changes is initially higher than for real systems, facilitating sampling.
Force Field Approximations
Force field deficiencies are often not discussed, although they are potentially more serious than sampling limitations. The force field is (remain in the future for a variety of reasons) so simple that it cannot capture accurately all force contributions simultaneously, especially when using multipurpose biomolecular force fields. One can tune the force field to reproduce experimental data for one aspect of the simulated system, but this tends to increase errors elsewhere. For example, polar hydrogens have small van der Waals radii when interacting with polar groups but quite large ones when they are in contact with nonpolar groups, and so the fixed radii in a simulation must be a compromise.42
Force field deviations from reality are not random. For the AMBER force field, rigorous comparison with quantum mechanical electronic structure calculations has revealed that base stacking and hydrogen bonding parameters are described surprisingly well, including those of base pairs that utilize the 2′-OH group.33,34 AMBER uses atomic charges that were derived to reproduce the electrostatic field around the monomers, which turns out to describe the molecular interactions of nucleobases quite well. The van der Waals term derived from Lennard–Jones potentials is also well balanced. The sugar–phosphate backbone, however, is considerably more difficult to deal with for two reasons: (i) it is flexible and thus the constant point charges do not reproduce the electrostatic potential equally well for distinct backbone conformational classes43,44 and; (ii) the phosphodiester represents a highly polarizable anion, with a complicated electronic structure that changes with solvation. These contributions are neglected by the force field. Despite these problems, the behavior of the backbone in RNA simulations has been quite realistic (see, for example, Refs. 45 and 46).
A significant deficiency of the force field is the description of divalent cations. The Mg2+…N7(guanine) interaction energy term is ∼210 kcal/mol, while the force field accounts for only ∼150 kcal/mol. This discrepancy is due to an interaction energy of ∼70 kcal/mol contributed by nonadditive effects (neglected by the purely additive force field) in the first ligand shell of Mg2+, mainly inter-ligand polarization repulsion.47 In reality, the first-shell water molecules are heavily polarized by the ion, and their properties are very different from those of bulk water molecules (for example, they have the capacity to form strong, low-barrier hydrogen bonds with large bathochromic infrared absorption shifts, where the hydrogen can easily jump between heteroatoms or may even be delocalized48). Additionally, simulations are unable to provide a well-equilibrated distribution for multivalent ions. Present force fields are biased towards direct (inner-shell) binding of Mg2+ to solute and require careful initial equilibration, since divalent cations consequently sample very poorly.49 Although the overall artifacts in simulations that include divalents are reduced through partial error compensation, divalent ions should be included in simulations only when absolutely necessary. It is not necessary to include divalents in simulations even when complex tertiary structured RNAs require them for folding in experiments, since the timescale of MD is typically too short to result in unfolding due to the absence of divalent ions.50
The description of anions is also quite challenging, since they have electrons localized far from their atomic centers and are typically highly polarizable. A comprehensive description would require the use of a polarizable force field.51 In addition, high-salt simulations that include anions can cause new artifacts, such as the formation of KCl clusters when using standard AMBER force field parameters.52,53
Force field errors are considerably smaller for monovalent cations, which also sample quite well in 25+ ns simulations.45,54-56 Analysis of the residence times and locations of monovalent cations can confidently predict binding sites with occupancies of >50%, though studies of weaker binding sites, such as those in B-DNA minor grooves, are less straightforward. Taking all these observations into consideration, the common approach of running simulations containing only neutralizing monovalent cations (∼0.2M) is well justified.
Currently, MD simulations are not capable of predicting three-dimensional structures of RNA molecules on their own but rather require meaningful starting geometries, preferably high-resolution X-ray crystal structures. If major structural rearrangements occur during the equilibration or the initial stages of a simulation (which is commonly observed when starting from modeled and some NMR-derived structures) they almost always indicate inaccuracies of the starting conformations and not of the force field.57
When assessing the outcome of simulations, it is important to consider all of the aforementioned limitations. The force field may be sufficient for some applications and fail for others. It is prudent to study scientific questions where stacking and base pairing are important and complementary experimental data are available for reference, as such problems are more likely to be properly addressed by contemporary MD simulations. It may even happen that, in a given simulation, different aspects of the simulated molecule(s) are described at different levels of accuracy. For example, simulations have been found to realistically reflect the overall dynamics of guanine quadruplex (G-DNA) stems and the overall electrostatic role of cations in their stabilization, yet the direct, local interactions of the G-quartets with cations are imperfectly described.58,59 In the latter studies, minor distortions of quartet geometries were observed and the radius of the cations appeared oversized (see Figure 1). The force field also fails to properly describe some properties of the flexible single-stranded connecting loops of the G-DNA (i.e., the predicted global minimum does not conform to experiment), and it falls short of predicting the experimentally observed cation binding sites at the stem–loop junctions.58 These observations should be taken into consideration for RNA simulations as well, where it is likely that simulations of, for example, the structural dynamics of hairpin loops or bulged bases are more susceptible to force field imbalances than are simulations of compact stem structures.
FIGURE 1.
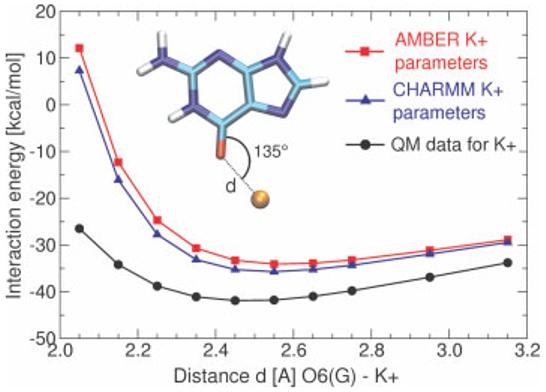
Limited accuracy of a nonpolarizable force field for cation–solute interactions. The dependence of the interaction energy between O6(G) and K+ in a G-DNA like geometry. Black, reference QM data; blue and red, standard CHARMM (radius 1.76375 A and well depth 0.087 kcal/mol−1) and AMBER (radius 2.6580 A and well depth 0.000328 kcal/mol−1) parameters, respectively. The cation in this particular interaction appears too large while the binding energy is clearly underestimated. Note that the discrepancy originates from the lack of polarization in the force field and cannot be fully overcome within the framework of pair additive (nonpolarizable) potentials. For example, reduction of the cation radius could worsen the calculated ion solvation energy.
Enhanced Sampling
Explicit solvent MD simulations represent the gold standard in contemporary modeling of the structural dynamics of nucleic acids in the context of their aqueous environment, which often plays important structural and functional roles. These studies can be further complemented by enhanced sampling techniques. However, such enhancement necessarily introduces additional, quite substantial, approximations. Locally Enhanced Sampling (LES), replica exchange MD (REMD) and targeted MD techniques aim to address the limited sampling problem.
LES splits a selected small part of the molecule (e.g., a loop) into N (3–5) copies that move independently. Although the method does not use a proper Boltzmann distribution, the height of conformational free energy barriers between distinct substates is reduced roughly by 1/N.60 We have successfully applied LES to studies of the single-stranded loops of G-DNA.58 During our studies of the hepatitis delta virus (HDV) ribozyme, however, LES applied to loop 3 and the catalytically involved trefoil turn motif did not converge.61 LES has also been applied to different regions of RNA Kink-turns, although the results were clearly dependent on the number of simulated copies.62
In REMD, several noninteracting copies (replicas) are simulated independently at different temperatures (e.g., 300–500 K). In contrast to LES, each replica is a copy of the whole system. Conformations of the individual simulated systems are exchanged during simulations using Metropolis-like algorithms that consider the probability of sampling each conformation at the alternate temperature.38,63
In targeted MD, force restraints drive the simulated molecule swiftly from a starting to a final conformation. This approach accelerates the simulation timescale, but also introduces a major sampling bias along the route.31 Finally, elevating the temperature (to, e.g., 400 K instead of 300 K) is another crude way to destabilize the simulated molecules and overcome conformational free energy barriers. However, raising the temperature will not produce sampling equivalent to that of a prolonged simulation, and since the pair additive potential is tuned for room temperature simulations, adverse effects such as empty cavities in the bulk solvent may occur at higher temperatures.
Continuum Solvent Methods
The costly explicit solvent can be replaced by a continuum solvent using Poisson Boltzmann (PB) or more approximate Generalized Born (GB) approaches, along with a Surface Area (SA) term. A fundamental advantage of continuum solvent methods is the assessment of hydration and free energies that cannot be derived from explicit solvent models. However, these approaches are based on additional substantial approximations, require parameterization, and their results are very sensitive to the atomic radii used to define the solute cavity. The GB/SA approach can be used to run MD simulations, but there is limited experience with this approach on RNA, and the range of its applicability has yet to be established. GB/SA dynamics show faster sampling because of the absence of friction between solute and solvent, but the method may significantly overstabilize folded structures. Continuum solvent methods can also be applied to systematic molecular mechanical conformational searches (as done recently for kink-turns64). It should be noted that the simplest electrostatic models in vacuo or based on a distance dependent permittivity provide an incorrect (reversed) order of stability for different conformations of a given molecule, while GB/SA is thought to perform better.65
A more accurate alternative is to perform standard explicit solvent MD simulations and subsequently apply the continuum solvent method, by stripping the explicit solvent and averaging free energies over a sufficient number of snapshots (referred to as MM-GBSA or MM-PBSA methods). Since free energy is a state function, one can evaluate free energy differences between distinct substates without simulating the transition. We found this MM-PBSA method to be very instructive in comparing different substates of guanine quadruplex DNA,66 while it failed to predict the absolute values of DAPI binding to B-DNA. In the latter case, the computed results were ∼0 or −20 kcal/mol, depending on whether the solute entropy was considered, while the experimental values are −10 to −12 kcal.67 This study also demonstrated that careful parameterization with the aid of quantum chemical calculations is required to obtain a meaningful force field for a drug, while ad hoc force fields (based on analogy with existing atom types or obtained via automated parameterization procedures) can be quite inaccurate, especially regarding torsion profiles between distinct ligand segments. Unfortunately, few MD studies of nucleic acid–drug complexes pay sufficiently close attention to the drug force field parameterization.
We have recently applied the MM-GBSA and MM-PBSA methods to estimate binding energies of ribosomal packing interactions,68 RNA kissing complexes (unpublished), and to monitor the free energy changes along Kink-turn simulation trajectories.64 The calculations, however, were not sufficiently accurate to achieve conclusive results. Zacharias applied such calculations quite successfully to investigate the context dependence of the structure of G/A base pairs in internal RNA loops, though large error margins in the calculations were reported.69
Among the other applications of continuum solvent methods, PB theory was used to roughly estimate binding affinities for several antibiotics to the ribosome,70 and Brooks and coworkers used PB methods to qualitatively look at the assembly of the ribosome. As expected, the results are quite sensitive to parameters such as dielectric constant, dielectric boundaries, and partial charges.71 GB/SA dynamics were also applied to assess the dynamic behavior of the 16S rRNA central domain in the presence and absence of the ribosomal protein S15.72 Finally, Barthel and Zacharias found qualitative (but not quantitative) correspondence between potential-of-mean-force explicit solvent free energy calculation and implicit solvent GB model conformational transitions of single nucleotide RNA bulges.73
In summary, continuum solvent approaches can provide useful insights into the molecular interactions but one should keep in mind that the accuracy of such calculations is rather modest.
REPRESENTATIVE MD SIMULATIONS OF RNA AND THEIR RESULTS
In the following sections we describe recent representative simulations of single- and double-stranded RNAs, catalytic RNA molecules, small molecules binding to RNA, and increasingly large RNA–protein complexes. Some studies represent a synergistic interaction between simulation and experiment. While we cannot possibly discuss every publication describing RNA MD simulations (of which there are now several hundred), these examples give a sense of the variety in systems and techniques, and the vast scope of information available from MD simulations.
Structured RNA Molecules, Isostericity, Long-Residency Waters, and Ion Binding
Much work has been done to model the behavior of various configurations of single-74 and double-stranded75,76 RNA molecules, including simulations focusing on the flexibility of the RNA,35,49,77-81 and the effects that noncanonical base pairs54,68,69,82-84 and modifications85-90 have on RNA structure and dynamics. The stability and dynamics of various RNA structures have been studied, including bulges73 and stem-loop structures,87,90-98 which are often used as model systems to test enhanced MD methods,99,100 as well as more complex structures such as entire tRNA molecules.101 Specific RNA motifs observed in the ribosome such as kink turns,50,62,64,102 the sarcin-ricin loop,103 and loop E54,104-106 have been studied separately, assuming that the modular organization of complex RNA molecules makes an isolated module a good model. Additionally, MD has been used to investigate RNA–RNA interactions, such as HIV-1 dimerization,57,107-112 as well as the interaction of RNA with solvent and ions.45,46,54,57,64,84,104,105,113,114 MD simulation is even being used to study the diffusion and electrophoretic mobility of single-stranded RNA115,116 and the folding of a small RNA hairpin.37
tRNA has been studied using MD techniques for more than 20 years. It was the first RNA molecule to have an X-ray structure and consequently was the first RNA molecule to be studied computationally.117 The MD techniques available for this first simulation of tRNAPhe were very limited, with no explicit treatment of solvent, counterions, or even the hydrogen atoms of the RNA molecule, and a poor treatment of electrostatics. Thus, many tertiary interactions seen in the crystal structure were lost during the simulation. Nevertheless, the simulation maintained important features of the secondary structure, and represented a first step toward using MD techniques to study the motions of RNA molecules. More recent studies have demonstrated stable simulations of fully neutralized and solvated tRNA molecules101 (see Figure 2), and have continued to use tRNA as a model system for MD simulations on large structured RNA molecules.118
FIGURE 2.
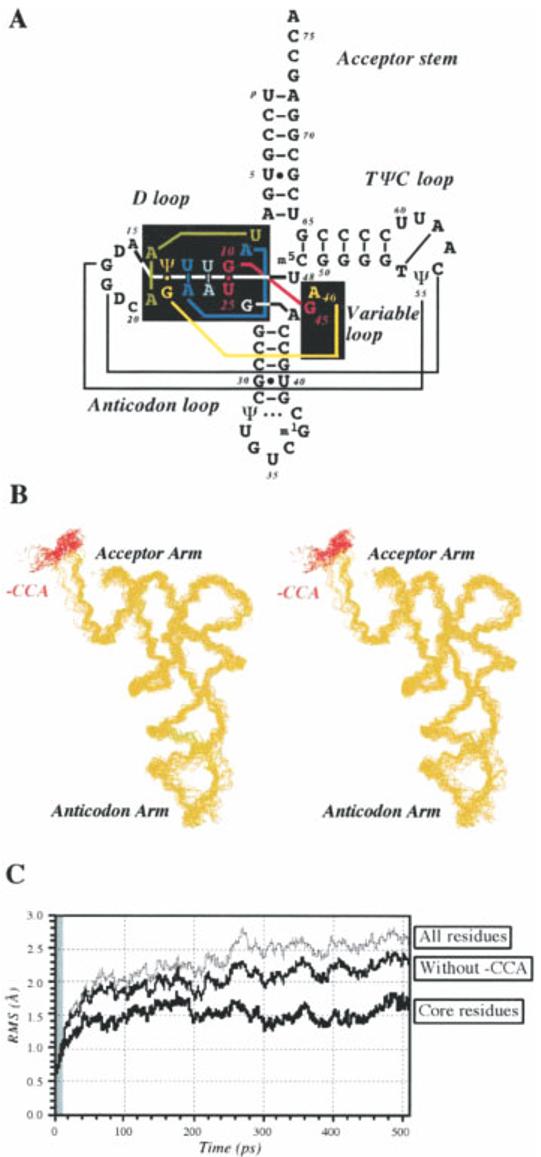
Stability of a 500 ps simulation of a fully neutralized and solvated tRNA molecule. (A) Yeast tRNAAsp secondary structure. (B) Stereo view of snapshots of the tRNA backbone, taken at 20 ps intervals. (C) Time course of RMS deviations from the starting structure. Reproduced with permission from Auffinger, P.; Louise-May, S.; Westhof, E. Biophys J 1999, 76, 50–64, © Biophysical Society.
Loop E of 5S rRNA is a unique RNA motif that consists of seven consecutive non-Watson–Crick base pairs and binds to the L25 protein in the E. coli ribosome (see Figure 3). MD simulations, along with phylogenetic analyses and isostericity considerations, were used to predict the structure of spinach chloroplast loop E, based on the structure of loop E from E. coli.54 The sequences have only 60% identity, but simulations predicted a nearly identical three-dimensional structure, since changes to the non-Watson–Crick base pairs (including four G-to-A substitutions) preserved the overall shape and unique pockets of negative electrostatic potential. This prediction was confirmed by solution NMR experiments.123 MD simulations of the loop E motif also showed long residency water molecules bound to specific hydration sites in the RNA molecule for up to 5 ns,54 much longer than the 50–500 ps water binding times typically observed in nucleic acid simulations. Some static long residency water molecules remain bound even longer (up to 25 ns, the entire simulation) in the loop E–L25 protein complex, suggesting that specific hydration sites are probably important in structural stabilization of RNA–protein interactions in the ribosome.122 MD simulation is one of the only methods able to probe detailed solvent dynamics and identify specific long-residency hydration sites.
FIGURE 3.
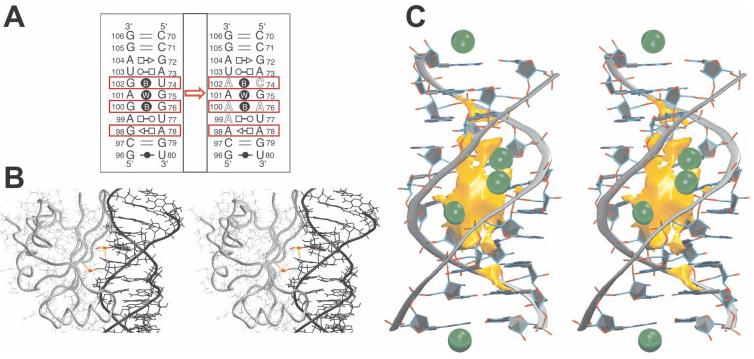
Structural and dynamic signatures of 5S ribosomal (r)RNA loop E. (A) Base pairing pattern (using classification from Ref. 119) of (left) the bacterial Loop E X-ray structure120 and (right) the spinach chloroplast Loop E architecture predicted from isostericity rules121 and MD simulations.54 The mutations from one sequence to the other are highlighted by red boxes. (B) MD simulations of Helix IV Loop E 5S rRNA in complex with ribosomal protein L25 reveal unique hydration sites with single water molecules bound throughout 25 ns trajectories.122 (C) The deep major groove of Loop E forms a unique pocket of negative electrostatic potential heavily occupied by monovalent cations (green balls).54 In summary, MD characterizes Loop E as a rigid RNA segment with unique shape and prominent ion-binding properties.
An entirely different, dynamical long-residency hydration site was identified in ribosomal Kink-turns,50,64 where it participates in the A-minor interaction mediating the tertiary contact between the C and NC stems of these recurrent structural motifs. This hydration site provides an unprecedented flexibility to Kink-turns, allowing them to act as unique molecular elbows during, for example, the elongation cycle. Such dynamics of Kink-turns have been implicated in large scale motions in the ribosome during protein synthesis (see Figure 4).50,64,102
FIGURE 4.
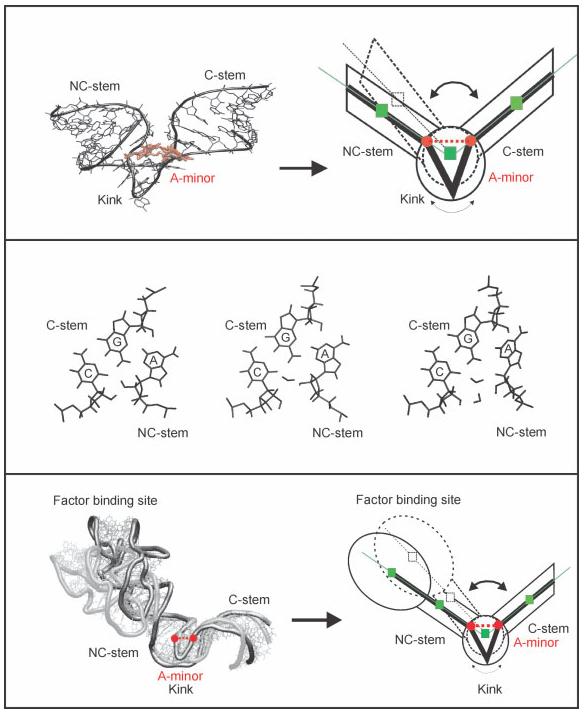
V-shaped Kink-turns (K-turns) are among the most recurrent RNA motifs.124 MD simulations predict (top) that K-turns are uniquely flexible elbow-like RNA building blocks, where subtle local conformational changes in the kink area propagate as large scale motions towards the attached helical stems.102 The local dynamics associated with K-turn flexibility are due to dynamical insertion of long-residency waters between the C and A nucleotides of the A-minor type I interaction between the two helical arms (middle).50 Large-scale elbow-like dynamics are observed in a simulation of the Helix 42–44 RNA portion of the large ribosomal subunit upon dynamical water insertion into the A-minor interaction in the universally conserved K-turn 42 of Helix 42 (bottom).64 The MD results appear to agree with cryo-EM data that show large-scale dynamics of this rRNA segment (the RNA part of the L7/L12 stalk) during the elongation cycle.125 Reproduced with permission from Razga, F.; Zacharias, M.; Reblova, K.; Koca, J.; Sponer, J. Structure 2006, 14, 825–835, © Cell Press.
Complex RNA folds are also associated with major cation binding sites that are not typically found around standard A-form RNA helices. For example, the loop E motif and RNA kissing complexes form unique ion binding pockets that are continuously occupied by 2–3 monovalent cations in simulations.54,57,104,105 Despite this high occupancy, the kissing complex ions are delocalized and smoothly exchange with bulk solvent (see Figure 5). Such dynamic monovalent ion binding pockets are unlikely to be detected in X-ray diffraction experiments due to dynamical disorder.
FIGURE 5.
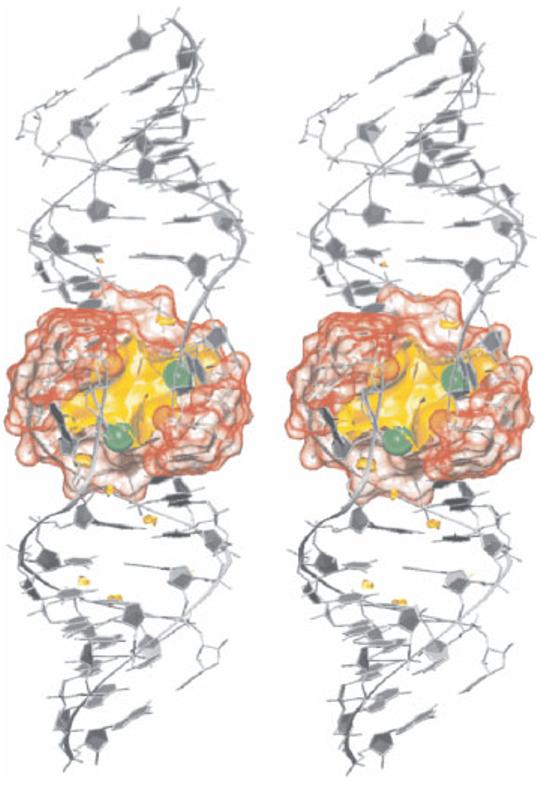
The central pocket of the HIV-1 DIS RNA kissing complex is permanently occupied by 2–3 delocalized monovalent cations (green), smoothly exchanging with bulk solvent on a timescale of a few nanoseconds.57
Finally, with recent advances in distributed computing, MD simulations can be used to examine not only dynamics in a defined fold, but also the folding of RNA molecules into known structures. Pande and coworkers have developed a Folding@Home distributed computing network, which approximates the computational power of a 150,000 CPU cluster, allowing the cumulative simulation of hundreds of microseconds of explicit solvent MD on small RNA molecules. A recent study investigating the role of solvent molecules in the folding of a 12-nucleotide RNA hairpin (see Figure 6) found an extrapolated folding time of 8.8 μs that matches experimental values.37 Additionally, a comparison of this study with a previous study using implicit solvent showed that water-mediated interactions were important in hydrophobic collapse of the RNA and in mediating long-range nucleation events, suggesting that water molecules, more so than counterions, play a crucial role in RNA folding.
FIGURE 6.
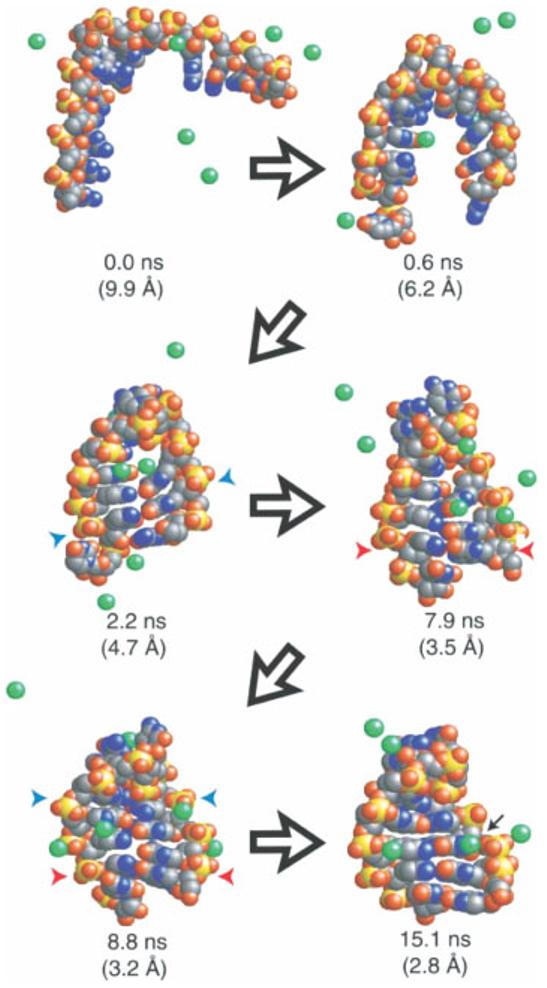
An example trajectory showing diverse conformational sampling in the folding of a small RNA hairpin. Na+ ions are shown in green, blue and red arrows indicate native and non-native base pairing. The black arrow in the last frame shows a hydrated ion bound to a site also seen in simulations of the native structure. Reproduced with permission from Sorin, E. J.; Rhee, Y. M.; Pande, V. S. Biophys J 2005, 88, 2516–2524, © Biophysical Society.
Catalytic RNAs
MD simulations have been applied to ribozymes—small RNA molecules that catalyze chemical reactions. While an accurate view of the chemistry itself requires quantum mechanical calculations, standard MD techniques can shed light on molecular interactions that affect the structure and dynamics globally as well as locally at the active sites of ribozymes. The hepatitis delta virus (HDV),45,61,126 hairpin,46,127 and hammerhead ribozymes128,129 have all been studied using MD.
The HDV ribozyme catalyzes the self-cleavage of a specific phosphodiester bond by a transesterification reaction. This activity requires the presence of the C75 nucleotide, which is in close proximity to the active site and cannot be mutated without decreasing activity by more than 100-fold, suggesting its direct involvement in chemistry. The exact role of C75 remains unclear, however, with biochemical and structural evidence supporting both general acid and general base models.130-135 MD simulations of pre- and post-cleavage HDV ribozyme constructs show hydrogen bonding patterns consistent with a general base mechanism.126 An unprotonated C75 transiently forms a hydrogen bond necessary for general base activity, while a protonated C75 does not form the hydrogen bond required in the general acid mechanism. Thus, assuming that the precursor X-ray crystal structure is sufficiently close to the transition state, the results from MD simulations suggest that C75 acts as the general base rather than the general acid in catalysis (see Figure 7).
FIGURE 7.
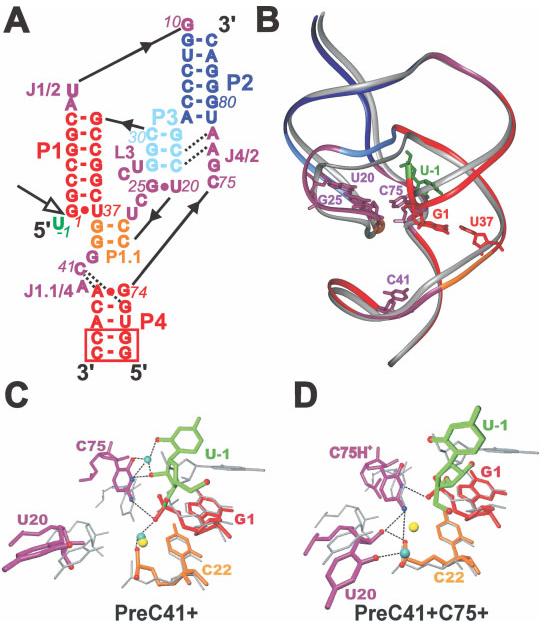
MD simulations of pre- and post-cleavage HDV ribozyme constructs showing hydrogen bonding patterns consistent with a general base mechanism. (A) Sequence and secondary structure of the simulated genomic HDV ribozyme with structural elements color-coded. The product form lacks U-1. The open arrow indicates the cleavage site. (B) Overlap of the crystal structures of the precursor134 (color-coded as in A) and product136 (silver) with key nucleotides indicated. Bottom panels: Overlay of representative averaged structures of the catalytic pocket from precursor simulations (color-coded as in A; cyan spheres, Na+ ions; broken lines, hydrogen bonds and inner-sphere ion contacts) with the precursor crystal structure (gray; yellow sphere, crystallographically resolved Mg ion). (C) Simulation with an unprotonated C75, where the U-1(O2′)–C75(N3) hydrogen bond necessary for general base activity forms. (D) Simulation with a protonated C75, where the C75H+(N3)–G1(O5′) hydrogen bond necessary for general acid activity does not form. Reproduced with permission from Krasovska, M. V.; Sefcikova, J.; Spackova, N.; Sponer, J.; Walter, N. G. J Mol Biol 2005, 351, 731–748, © Academic Press.
The hairpin ribozyme also catalyzes self-cleavage via a transesterification reaction where the details of the catalytic mechanism remain unclear. Our recent study combined MD and single molecule fluorescence techniques to explore the role of water molecules in the catalytic core of the hairpin ribozyme.46 When comparing the wild-type ribozyme to several mutants, a linear relationship was observed between the number of hydrogen bonds lost in the MD simulations and the loss of docking free energy calculated from single molecule fluorescence data, revealing a quantitative agreement between simulation and experiment. Analysis of the MD trajectories showed that a network of hydrogen bonds in the catalytic core of the ribozyme involves several long-residency, trapped water molecules. These water molecules are in a position where they could plausibly be involved in reaction chemistry (see Figure 8), suggesting that interactions with solvent are important not only for structure, flexibility, and folding of RNA molecules as described earlier, but also potentially for catalytic activity. These observations underscore that an analysis of specific hydration sites should probably be done for all explicit solvent MD simulations of RNA.
FIGURE 8.
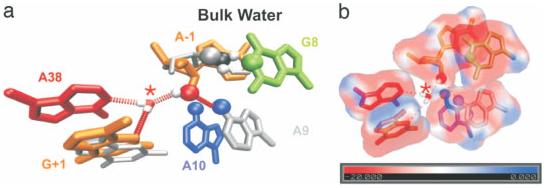
Possible catalytic role of intracavity water in the hairpin ribozyme. (a) The initial active site geometry is that of the crystal structure (gray), but over the course of the MD simulations, the 2′OH shifts from bulk solvent to the site of chemistry and forms a hydrogen bond with the asterisked water molecule (color). (b) The electrostatic potential map shows a minimum of −51 kT/e near the asterisked water molecule. Reproduced with permission from Rhodes, M.M., Reblova, K., Sponer, J., Walter, N.G. Proc Natl Acad Sci USA 103, 2006, 13381–13385, © National Academy of Sciences.
RNA Binding to Small Molecules
Understanding how RNA interacts with small molecules is crucial in drug design, where small molecules are optimized to bind a target RNA, and in characterizing natural and in vitro selected RNA riboswitches (aptamers) that bind specific small molecules. Obtaining a high-quality force field for small molecules can be difficult, since specialized parameterization based on quantum chemical data is necessary unless the ligand is rigid.67 Nevertheless, these types of MD simulations can elucidate details of small molecule binding to RNA, and can provide information, often missing from other techniques, on the role(s) of dynamics and solvent molecules in these interactions. Several studies have examined the binding of antibiotics to the hammerhead ribozyme137 and to various sites in the ribosome,53,138 while others have examined small molecule–RNA interactions such as pyrene binding to RNA duplexes139 and ligands such as FMN140 binding to their RNA aptamers.
The interaction between aminoglycoside antibiotics and the A-site of the ribosome has been well studied as a model system for small molecule binding to RNA, with a wealth of available crystal structures and biochemical data. A recent MD study53 on a paromomycin–A-site complex analyzed the hydrogen bonding network connecting the antibiotic to the RNA and verified that the conserved neamine portion of paromomycin was most stably bound to the RNA, with both more numerous and longer-lived hydrogen bonds than are established by rings III and IV of paromomycin. An analysis of hydration patterns reproduced crystallographic water binding sites and suggested additional long-residency sites that are thought to play a key role in mediating antibiotic–RNA interactions (see Figure 9).
FIGURE 9.
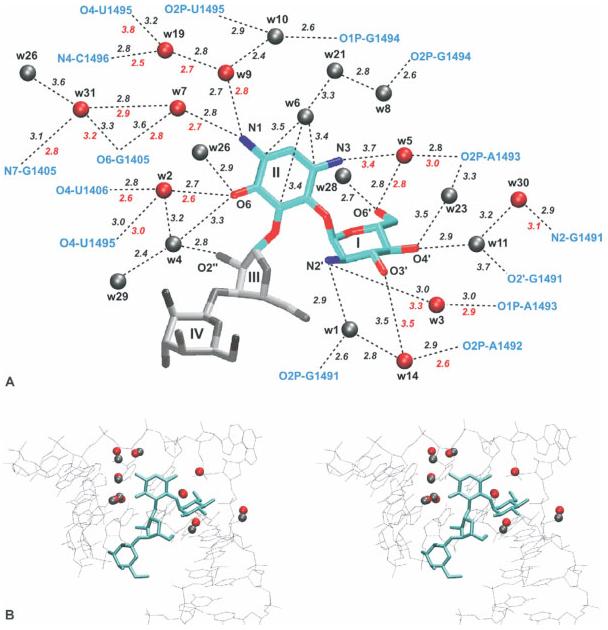
(A) Experimental and calculated hydration sites in the binding of paromomycin to the rRNA A-site. Red sites are found in both the crystal structure and the simulation, while gray sites are found only in the simulation. Sites are numbered according to the peak height of the calculated density, with 1 as the highest peak. (B) Stereo view of the experimental hydration sites (red) and the closest corresponding sites from the simulation (gray). Reproduced with permission from Vaiana, A. C.; Westhof, E.; Auffinger, P. Biochimie 2006, 88, 1061–1073, © Editions Scientifiques Elsevier.
RNA–Protein Complexes
RNA–protein interactions are ubiquitous and play a key role in cellular regulation. MD simulations of RNA–protein interactions have dramatically increased in recent years, especially since high-resolution crystal structures of the ribosome became available.141 The large variety in RNA–protein simulations now includes proteins binding RNA fragments,142-147 HIV RNA–protein interactions,148-151 RNase–RNA interactions,144 various ribosomal protein–ribosomal RNA interactions,72,122,152,153 simulations of peptide bond formation,154 coarse-grained155 and all-atom30,31 simulations of the complete ribosome, and even an all-atom simulation of the entire satellite tobacco mosaic virus.156 One protein–RNA interaction that has been studied in great detail using MD simulations is the U1A–RNA complex,157-164 discussed later.
The U1A protein, part of the U1 small nuclear ribonucleoprotein (snRNP) complex, is a well-studied RNA-binding protein that contains RNA recognition motifs (RRMs) and binds to both a hairpin in U1 RNA and its own 3′-UTR with picomolar affinities. Such U1A–RNA complexes were among the first protein–RNA interactions to be studied using MD techniques, and simulations of U1A–RNA complexes continue to enrich understanding of RNA binding proteins. Initial studies of the U1A protein bound to either RNA binding site158 were consistent with experimental data, showing stable hydrogen bonding between the protein and a loop contained in both RNAs, while the limited flexibility of the RNA–protein interface agreed with experimental thermodynamic data. This agreement established the validity of using MD to study protein–RNA complexes. Since then, this technique has been used alongside experiment to probe the U1A–RNA interaction in much greater detail, with recent studies focusing on the roles of specific residues. One simulation, for example, showed substantial variation in the time three lysine residues spend near the RNA backbone,163 explaining differences in their experimentally observed effects on complex stability. A second study, probing the effects of compensatory mutations in RNA and protein residues,164 used free energy and hydrogen bonding analyses to show that two induced fit processes affect binding. This approach explained experimental affinity data on several mutants (see Figure 10), providing information important for future rational design of RNA–protein interactions. Additionally, recent cross-correlation analysis of a U1A–RNA MD simulation162 was consistent with experimentally observed cooperativity and a statistical covariance analysis, and also made predictions about the roles of individual residues that can be further tested experimentally.
FIGURE 10.
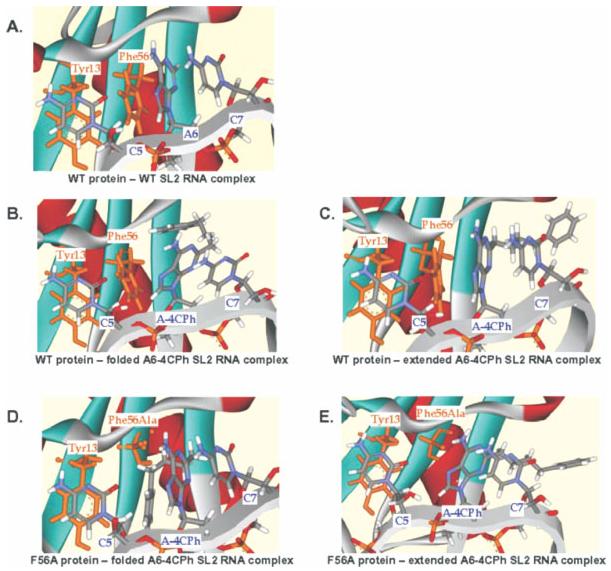
Structures from MD simulations investigating the role of conserved aromatic amino acids in U1A–RNA binding. These simulations provide a model to explain the stabilization of a phenylalanine to alanine mutant protein by a modified base (A-4CPh). This study predicts an extended conformation for the modified base when bound to the WT protein, and a folded conformation when bound to the mutant protein. (A) WT protein and WT RNA show stacking. (B) WT protein with a folded modified base interrupts stacking. (C) WT protein with an extended modified base preserves stacking. (D) Mutant protein with a folded modified RNA preserves stacking. (E) Mutant protein with an extended modified base interrupts stacking. Reproduced with permission from Zhao, Y.; Kormos, B. L.; Beveridge, D. L.; Baranger, A. M. Biopolymers 2006, 81, 256–269, © Wiley Interscience.
The range of RNA–protein complexes accessible to MD simulations is continually increasing, and now even all-atom simulations of the ribosome are becoming possible. Sanbonmatsu has used targeted MD techniques on the entire ribosome31 to investigate the process of message decoding, where the tRNA molecule moves from the “A/T” to the “A/A” site in response to a match between mRNA codon and tRNA anticodon. While targeted MD simulations represent unrealistic sampling, they do suggest possible pathways between an initial and final structure than can be tested experimentally. The ribosome study suggested that highly conserved residues in the A loop and Helix 89 interact with the tRNA molecule as it moves to the A/A state (see Figure 11). This observation generally agrees with biochemical data, providing evidence that MD simulations on large systems can aid in understanding the biological function of very complex RNA–protein complexes.
FIGURE 11.
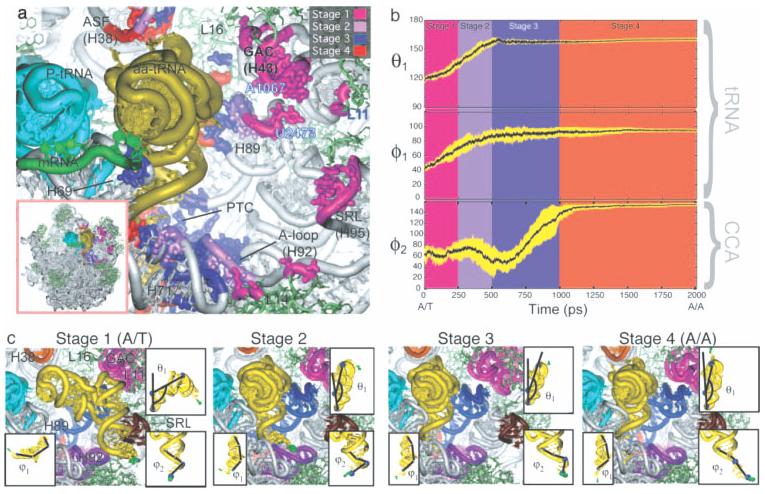
Four stages of accommodation during tRNA movement from the A/T to the A/A site in a targeted all-atom MD simulation. (a) tRNA interaction regions colored by accommodation stage. (b) Time evolution of parameters describing tRNA deformation. (c) Snapshots from each stage of accommodation. Reproduced with permission from Sanbonmatsu, K. Y.; Joseph, S.; Tung, C. S. Proc Natl Acad Sci USA 2005, 102, 15854–15859, © National Academy of Sciences.
SUMMARY, CONCLUSIONS, OUTLOOK
MD simulations of RNA have reached a point where they can be used in a wide variety of systems to complement experiment and verify or explain experimental results. There are a number of limitations in the use and interpretation of simulation data, including the short time scale and force field inaccuracies of MD simulations. However, the timescales of experiment and simulation are getting closer, with experiments able to access faster and faster dynamics and with more computational power leading to longer simulations. The future progress of MD simulations will critically depend on further tuning of the force fields (including development of polarization potentials), development of better methods to estimate free energies (in order to directly link molecular structures and free energies), and integration of molecular mechanics with quantum mechanical treatments (in order to deal with enzymatic reactions). All three tasks are formidable, but will allow MD simulations to address an even wider variety of biologically relevant questions in the future. The gap between theory and experiment is closing, and it is becoming clear that computational methods and particularly single molecule experiments are often highly complementary and synergistic. Collaborations between theorists and experimentalists can therefore be very productive and mutually beneficial. Increased cooperation, facilitated by learning each other's scientific language, will greatly aid in understanding the structure–function relationships of diverse and complex RNA molecules in our newly discovered post-genomic RNA World. This review shares a snapshot of the current state-of-the-art and hopes to inspire increased, yet prudent, use of MD tools in the ever-expanding RNA field.
Acknowledgments
We thank Jana Sefcikova for critical reading of the manuscript and Martin Zacharias for helpful discussion.
Footnotes
Contract grant sponsor: NSF predoctoral fellowship to S.E.M.
Contract grant sponsors: National Institutes of Health
Contract grant number: GM62357
Contract grant sponsor: Wellcome Trust
Contract grant number: GR067507
Contract grant sponsor: Grant Agency of Czech Republic
Contract grant numbers: GA203_05_0009, GA203_05_0388
Contract grant sponsor: Internal Grant Agency, Academy of Sciences
Contract grant number: 1QS500040581
Contract grant sponsor: Ministry of Education, Youth, and Sports
Contract grant numbers: LC06030, LC512, MSM0021622413, AVOZ50040507, AVOZ40550506
REFERENCES
- 1.Kruger K, Grabowski PJ, Zaug AJ, Sands J, Gottschling DE, Cech TR. Cell. 1982;31:147–158. doi: 10.1016/0092-8674(82)90414-7. [DOI] [PubMed] [Google Scholar]
- 2.Guerrier-Takada C, Gardiner K, Marsh T, Pace N, Altman S. Cell. 1983;35:849–857. doi: 10.1016/0092-8674(83)90117-4. [DOI] [PubMed] [Google Scholar]
- 3.Flores R, Delgado S, Gas ME, Carbonell A, Molina D, Gago S, De la Pena M. FEBS Lett. 2004;567:42–48. doi: 10.1016/j.febslet.2004.03.118. [DOI] [PubMed] [Google Scholar]
- 4.Dorsett Y, Tuschl T. Nat Rev Drug Discov. 2004;3:318–329. doi: 10.1038/nrd1345. [DOI] [PubMed] [Google Scholar]
- 5.Hannon GJ, Rossi JJ. Nature. 2004;431:371–378. doi: 10.1038/nature02870. [DOI] [PubMed] [Google Scholar]
- 6.He L, Hannon GJ. Nat Rev Genet. 2004;5:522–531. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 7.Meister G, Tuschl T. Nature. 2004;431:343–349. doi: 10.1038/nature02873. [DOI] [PubMed] [Google Scholar]
- 8.Storz G, Altuvia S, Wassarman KM. Annu Rev Biochem. 2005;74:199–217. doi: 10.1146/annurev.biochem.74.082803.133136. [DOI] [PubMed] [Google Scholar]
- 9.Ryther RC, Flynt AS, Phillips JA, III, Patton JG. Gene Ther. 2005;12:5–11. doi: 10.1038/sj.gt.3302356. [DOI] [PubMed] [Google Scholar]
- 10.Zamore PD, Haley B. Science. 2005;309:1519–1524. doi: 10.1126/science.1111444. [DOI] [PubMed] [Google Scholar]
- 11.Lu PY, Xie F, Woodle MC. Adv Genet. 2005;54:117–142. doi: 10.1016/S0065-2660(05)54006-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sontheimer EJ, Carthew RW. Cell. 2005;122:9–12. doi: 10.1016/j.cell.2005.06.030. [DOI] [PubMed] [Google Scholar]
- 13.Filipowicz W, Jaskiewicz L, Kolb FA, Pillai RS. Curr Opin Struct Biol. 2005;15:331–341. doi: 10.1016/j.sbi.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 14.Huttenhofer A, Schattner P. Nat Rev Genet. 2006;7:475–482. doi: 10.1038/nrg1855. [DOI] [PubMed] [Google Scholar]
- 15.Mattick JS, Makunin IV. Hum Mol Genet. 2006;15:R17–R29. doi: 10.1093/hmg/ddl046. [DOI] [PubMed] [Google Scholar]
- 16.Hammond SM. Curr Opin Genet Dev. 2006;16:4–9. doi: 10.1016/j.gde.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 17.Chang SC, Tucker T, Thorogood NP, Brown CJ. Front Biosci. 2006;11:852–866. doi: 10.2741/1842. [DOI] [PubMed] [Google Scholar]
- 18.Lau NC, Seto AG, Kim J, Kuramochi-Miyagawa S, Nakano T, Bartel DP, Kingston RE. Science. 2006;313:363–367. doi: 10.1126/science.1130164. [DOI] [PubMed] [Google Scholar]
- 19.Pollard KS, Salama SR, Lambert N, Lambot MA, Coppens S, Pedersen JS, Katzman S, King B, Onodera C, Siepel A, Kern AD, Dehay C, Igel H, Ares M, Jr., Vanderhaeghen P, Haussler D. Nature. 2006;443:167–172. doi: 10.1038/nature05113. [DOI] [PubMed] [Google Scholar]
- 20.Willingham AT, Orth AP, Batalov S, Peters EC, Wen BG, Aza-Blanc P, Hogenesch JB, Schultz PG. Science. 2005;309:1570–1573. doi: 10.1126/science.1115901. [DOI] [PubMed] [Google Scholar]
- 21.Gesteland RF, Cech T, Atkins JF. The RNA World: The Nature of Modern RNA Suggests a Prebiotic RNA World. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 2006. [Google Scholar]
- 22.Sponer J, Lankas F, editors. Computational Studies of RNA and DNA. The Netherlands: Springer; 2006. [Google Scholar]
- 23.Nelson JA, Uhlenbeck OC. Mol Cell. 2006;23:447–450. doi: 10.1016/j.molcel.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 24.Mei HY, Kaaret TW, Bruice TC. Proc Natl Acad Sci USA. 1989;86:9727–9731. doi: 10.1073/pnas.86.24.9727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nilsson L, Ahgren-Stalhandske A, Sjogren AS, Hahne S, Sjoberg BM. Biochemistry. 1990;29:10317–10322. doi: 10.1021/bi00497a005. [DOI] [PubMed] [Google Scholar]
- 26.Scott WG, Finch JT, Klug A. Cell. 1995;81:991–1002. doi: 10.1016/s0092-8674(05)80004-2. [DOI] [PubMed] [Google Scholar]
- 27.Martick M, Scott WG. Cell. 2006;126:309–320. doi: 10.1016/j.cell.2006.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doudna JA. Nat Chem Biol. 2005;1:300–303. doi: 10.1038/nchembio1105-300. [DOI] [PubMed] [Google Scholar]
- 29.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanbonmatsu KY, Joseph S. J Mol Biol. 2003;328:33–47. doi: 10.1016/s0022-2836(03)00236-5. [DOI] [PubMed] [Google Scholar]
- 31.Sanbonmatsu KY, Joseph S, Tung CS. Proc Natl Acad Sci USA. 2005;102:15854–15859. doi: 10.1073/pnas.0503456102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheatham TE, Miller JL, Fox T, Darden TA, Kollman PA. J Am Chem Soc. 1995;117:4193–4194. [Google Scholar]
- 33.Sponer J, Jurecka P, Marchan I, Luque FJ, Orozco M, Hobza P. Chem Eur J. 2006;12:2854–2865. doi: 10.1002/chem.200501239. [DOI] [PubMed] [Google Scholar]
- 34.Sponer JE, Spackova N, Leszczynski J, Sponer J. J Phys Chem B. 2005;109:11399–11410. doi: 10.1021/jp051126r. [DOI] [PubMed] [Google Scholar]
- 35.Pan Y, MacKerell AD., Jr. Nucleic Acids Res. 2003;31:7131–7140. doi: 10.1093/nar/gkg941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Snoussi K, Leroy JL. Biochemistry. 2001;40:8898–8904. doi: 10.1021/bi010385d. [DOI] [PubMed] [Google Scholar]
- 37.Sorin EJ, Rhee YM, Pande VS. Biophys J. 2005;88:2516–2524. doi: 10.1529/biophysj.104.055087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sorin EJ, Rhee YM, Nakatani BJ, Pande VS. Biophys J. 2003;85:790–803. doi: 10.1016/S0006-3495(03)74520-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.York DM, Darden TA, Pedersen LG. J Chem Phys. 1993;99:8345–8348. [Google Scholar]
- 40.Hunenberger PH, McCammon JA. J Chem Phys. 1999;110:1856–1872. [Google Scholar]
- 41.Kastenholz MA, Hunenberger PH. J Phys Chem B. 2004;108:774–788. [Google Scholar]
- 42.Halgren TA. J Comput Chem. 1996;17:520–552. [Google Scholar]
- 43.Cieplak P, Cornell WD, Bayly C, Kollman PA. J Comput Chem. 1995;16:1357–1377. [Google Scholar]
- 44.Reynolds CA, Essex JW, Richards WG. J Am Chem Soc. 1992;114:9075–9079. [Google Scholar]
- 45.Krasovska MV, Sefcikova J, Reblova K, Schneider B, Walter NG, Sponer J. Biophys J. 2006;91:626–638. doi: 10.1529/biophysj.105.079368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rhodes MM, Reblova K, Sponer J, Walter NG. Proc Natl Acad Sci USA. 2006;103:13381–13385. doi: 10.1073/pnas.0605090103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sponer J, Burda JV, Sabat M, Leszczynski J, Hobza P. J Phys Chem A. 1998;102:5951–5957. [Google Scholar]
- 48.Sponer J, Sabat M, Gorb L, Leszczynski J, Lippert B, Hobza P. J Phys Chem B. 2000;104:7535–7544. [Google Scholar]
- 49.Reblova K, Lankas F, Razga F, Krasovska MV, Koca J, Sponer J. Biopolymers. 2006;82:504–520. doi: 10.1002/bip.20503. [DOI] [PubMed] [Google Scholar]
- 50.Razga F, Koca J, Sponer J, Leontis NB. Biophys J. 2005;88:3466–3485. doi: 10.1529/biophysj.104.054916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dang LX, Chang TM. J Phys Chem B. 2002;106:235–238. [Google Scholar]
- 52.Cheatham TE. Curr Opin Struct Biol. 2004;14:360–367. doi: 10.1016/j.sbi.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 53.Vaiana AC, Westhof E, Auffinger P. Biochimie. 2006;88:1061–1073. doi: 10.1016/j.biochi.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 54.Reblova K, Spackova N, Stefl R, Csaszar K, Koca J, Leontis NB, Sponer J. Biophys J. 2003;84:3564–3582. doi: 10.1016/S0006-3495(03)75089-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Varnai P, Zakrzewska K. Nucleic Acids Res. 2004;32:4269–4280. doi: 10.1093/nar/gkh765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ponomarev SY, Thayer KM, Beveridge DL. Proc Natl Acad Sci USA. 2004;101:14771–14775. doi: 10.1073/pnas.0406435101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Reblova K, Spackova N, Sponer JE, Koca J, Sponer J. Nucleic Acids Res. 2003;31:6942–6952. doi: 10.1093/nar/gkg880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fadrna E, Spackova N, Stefl R, Koca J, Cheatham TE, Sponer J. Biophys J. 2004;87:227–242. doi: 10.1529/biophysj.103.034751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Spackova N, Berger I, Sponer J. J Am Chem Soc. 1999;121:5519–5534. doi: 10.1021/ja002656y. [DOI] [PubMed] [Google Scholar]
- 60.Simmerling C, Miller JL, Kollman PA. J Am Chem Soc. 1998;120:7149–7155. [Google Scholar]
- 61.Sefcikova J, Krasovska MV, Sponer J, Walter NG. Nucleic Acids Res. doi: 10.1093/nar/gkl1104. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cojocaru V, Klement R, Jovin TM. Nucleic Acids Res. 2005;33:3435–3446. doi: 10.1093/nar/gki664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cheng XL, Hornak V, Simmerling C. J Phys Chem B. 2004;108:426–437. [Google Scholar]
- 64.Razga F, Zacharias M, Reblova K, Koca J, Sponer J. Structure. 2006;14:825–835. doi: 10.1016/j.str.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 65.Zacharias M. Curr Opin Struct Biol. 2000;10:311–317. doi: 10.1016/s0959-440x(00)00089-0. [DOI] [PubMed] [Google Scholar]
- 66.Stefl R, Cheatham TE, Spackova N, Fadrna E, Berger I, Koca J, Sponer J. Biophys J. 2003;85:1787–1804. doi: 10.1016/S0006-3495(03)74608-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Spackova N, Cheatham TE, Ryjacek F, Lankas F, van Meervelt L, Hobza P, Sponer J. J Am Chem Soc. 2003;125:1759–1769. doi: 10.1021/ja025660d. [DOI] [PubMed] [Google Scholar]
- 68.Mokdad A, Krasovska MV, Sponer J, Leontis NB. Nucleic Acids Res. 2006;34:1326–1341. doi: 10.1093/nar/gkl025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Villescas-Diaz G, Zacharias M. Biophys J. 2003;85:416–425. doi: 10.1016/S0006-3495(03)74486-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yang G, Trylska J, Tor Y, McCammon JA. J Med Chem. 2006;49:5478–5490. doi: 10.1021/jm060288o. [DOI] [PubMed] [Google Scholar]
- 71.Trylska J, McCammon JA, Brooks CL. J Am Chem Soc. 2005;127:11125–11133. doi: 10.1021/ja052639e. [DOI] [PubMed] [Google Scholar]
- 72.Li W, Ma BY, Shapiro BA. Nucleic Acids Res. 2003;31:629–638. doi: 10.1093/nar/gkg149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Barthel A, Zacharias M. Biophys J. 2006;90:2450–2462. doi: 10.1529/biophysj.105.076158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Isaksson J, Acharya S, Barman J, Cheruku P, Chattopadhyaya J. Biochemistry. 2004;43:15996–16010. doi: 10.1021/bi048221v. [DOI] [PubMed] [Google Scholar]
- 75.Hart K, Nystrom B, Ohman M, Nilsson L. RNA. 2005;11:609–618. doi: 10.1261/rna.7147805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schneider C, Brandl M, Suhnel J. J Mol Biol. 2001;305:659–667. doi: 10.1006/jmbi.2000.4338. [DOI] [PubMed] [Google Scholar]
- 77.Fadrna E, Koca J. J Biomol Struct Dyn. 2003;20:715–732. doi: 10.1080/07391102.2003.10506888. [DOI] [PubMed] [Google Scholar]
- 78.Kaukinen U, Venalainen T, Lonnberg H, Perakyla M. Org Biomol Chem. 2003;1:2439–2447. doi: 10.1039/b302751a. [DOI] [PubMed] [Google Scholar]
- 79.Noy A, Perez A, Lankas F, Javier Luque F, Orozco M. J Mol Biol. 2004;343:627–638. doi: 10.1016/j.jmb.2004.07.048. [DOI] [PubMed] [Google Scholar]
- 80.Noy A, Perez A, Marquez M, Luque FJ, Orozco M. J Am Chem Soc. 2005;127:4910–4920. doi: 10.1021/ja043293v. [DOI] [PubMed] [Google Scholar]
- 81.Joli F, Hantz E, Hartmann B. Biophys J. 2006;90:1480–1488. doi: 10.1529/biophysj.105.070862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Varnai P, Canalia M, Leroy J-L. J Am Chem Soc. 2004;126:14659–14667. doi: 10.1021/ja0470721. [DOI] [PubMed] [Google Scholar]
- 83.Pan Y, Priyakumar UD, MacKerell AD., Jr. Biochemistry. 2005;44:1433–1443. doi: 10.1021/bi047932q. [DOI] [PubMed] [Google Scholar]
- 84.Csaszar K, Spackova N, Stefl R, Sponer J, Leontis NB. J Mol Biol. 2001;313:1073–1091. doi: 10.1006/jmbi.2001.5100. [DOI] [PubMed] [Google Scholar]
- 85.Premraj BJ, Raja S, Yathindra N. Biophys Chem. 2002;95:253–272. doi: 10.1016/s0301-4622(02)00040-6. [DOI] [PubMed] [Google Scholar]
- 86.Gyi JI, Gao D, Conn GL, Trent JO, Brown T, Lane AN. Nucleic Acids Res. 2003;31:2683–2693. doi: 10.1093/nar/gkg356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sarzynska J, Nilsson L, Kulinski T. Biophys J. 2003;85:3445–3459. doi: 10.1016/S0006-3495(03)74766-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zacharias M, Engels JW. Nucleic Acids Res. 2004;32:6304–6311. doi: 10.1093/nar/gkh971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nina M, Fonne-Pfister R, Beaudegnies R, Chekatt H, Jung PMJ, Murphy-Kessabi F, De Mesmaeker A, Wendeborn S. J Am Chem Soc. 2005;127:6027–6038. doi: 10.1021/ja0486566. [DOI] [PubMed] [Google Scholar]
- 90.Sarzynska J, Kulinski T. J Biomol Struct Dyn. 2005;22:425–439. doi: 10.1080/07391102.2005.10507014. [DOI] [PubMed] [Google Scholar]
- 91.Kulinski T, Olejniczak M, Huthoff H, Bielecki L, Pachulska-Wieczorek K, Das AT, Berkhout B, Adamiak RW. J Biol Chem. 2003;278:38892–38901. doi: 10.1074/jbc.M301939200. [DOI] [PubMed] [Google Scholar]
- 92.Seetharaman M, Williams C, Cramer CJ, Musier-Forsyth K. Nucleic Acids Res. 2003;31:7311–7321. doi: 10.1093/nar/gkg930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Aci S, Ramstein J, Genest D. J Biomol Struct Dyn. 2004;21:833–840. doi: 10.1080/07391102.2004.10506973. [DOI] [PubMed] [Google Scholar]
- 94.Koplin J, Mu Y, Richter C, Schwalbe H, Stock G. Structure. 2005;13:1255–1267. doi: 10.1016/j.str.2005.05.015. [DOI] [PubMed] [Google Scholar]
- 95.Yingling YG, Shapiro BA. J Mol Biol. 2005;348:27–42. doi: 10.1016/j.jmb.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 96.Nagan MC, Beuning P, Musier-Forsyth K, Cramer CJ. Nucleic Acids Res. 2000;28:2527–2534. doi: 10.1093/nar/28.13.2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nagan MC, Kerimo SS, Musier-Forsyth K, Cramer CJ. J Am Chem Soc. 1999;121:7310–7317. [Google Scholar]
- 98.Villa A, Stock G. J Chem Theory Comput. 2006;2:1228–1236. doi: 10.1021/ct600160z. [DOI] [PubMed] [Google Scholar]
- 99.Bielecki L, Popenda M, Adamiak RW. Nucleic Acids Symp Ser. 2002;2:57–58. doi: 10.1093/nass/2.1.57. [DOI] [PubMed] [Google Scholar]
- 100.Cheng X, Cui G, Hornak V, Simmerling C. J Phys Chem B. 2005;109:8220–8230. doi: 10.1021/jp045437y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Auffinger P, Louise-May S, Westhof E. Biophys J. 1999;76:50–64. doi: 10.1016/S0006-3495(99)77177-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Razga F, Spackova N, Reblova K, Koca J, Leontis NB, Sponer J. J Biomol Struct Dyn. 2004;22:183–194. doi: 10.1080/07391102.2004.10506994. [DOI] [PubMed] [Google Scholar]
- 103.Spackova N, Sponer J. Nucleic Acids Res. 2006;34:697–708. doi: 10.1093/nar/gkj470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Auffinger P, Bielecki L, Westhof E. Chem Biol. 2003;10:551–561. doi: 10.1016/s1074-5521(03)00121-2. [DOI] [PubMed] [Google Scholar]
- 105.Auffinger P, Bielecki L, Westhof E. J Mol Biol. 2004;335:555–571. doi: 10.1016/j.jmb.2003.10.057. [DOI] [PubMed] [Google Scholar]
- 106.Golebiowski J, Antonczak S, Di-Giorgio A, Condom R, Cabrol-Bass D. J Mol Model. 2004;10:60–68. doi: 10.1007/s00894-003-0170-9. (Online) [DOI] [PubMed] [Google Scholar]
- 107.Kieken F, Arnoult E, Barbault F, Paquet F, Huynh-Dinh T, Paoletti J, Genest D, Lancelot G. Eur Biophys J. 2002;31:521–531. doi: 10.1007/s00249-002-0251-1. [DOI] [PubMed] [Google Scholar]
- 108.Beaurain F, Laguerre M. Oligonucleotides. 2003;13:501–514. doi: 10.1089/154545703322860816. [DOI] [PubMed] [Google Scholar]
- 109.Beaurain F, Di Primo C, Toulme JJ, Laguerre M. Nucleic Acids Res. 2003;31:4275–4284. doi: 10.1093/nar/gkg467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Aci S, Gangneux L, Paoletti J, Genest D. Biopolymers. 2004;74:177–188. doi: 10.1002/bip.20032. [DOI] [PubMed] [Google Scholar]
- 111.Golebiowski J, Antonczak S, Fernandez-Carmona J, Condom R, Cabrol-Bass D. J Mol Model. 2004;10:408–417. doi: 10.1007/s00894-004-0216-7. (Online) [DOI] [PubMed] [Google Scholar]
- 112.Aci S, Mazier S, Genest D. J Mol Biol. 2005;351:520–530. doi: 10.1016/j.jmb.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 113.Auffinger P, Westhof E. Biophys Chem. 2002;95:203–210. doi: 10.1016/s0301-4622(01)00257-5. [DOI] [PubMed] [Google Scholar]
- 114.Tsui V, Case DA. J Phys Chem B. 2001;105:11314–11325. [Google Scholar]
- 115.Yeh I-C, Hummer G. Biophys J. 2004;86:681–689. doi: 10.1016/S0006-3495(04)74147-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yeh I-C, Hummer G. Proc Natl Acad Sci USA. 2004;101:12177–12182. doi: 10.1073/pnas.0402699101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Harvey SC, Prabhakaran M, Mao B, McCammon JA. Science. 1984;223:1189–1191. doi: 10.1126/science.6560785. [DOI] [PubMed] [Google Scholar]
- 118.Nina M, Simonson T. J Phys Chem B. 2002;106:3696–3705. [Google Scholar]
- 119.Leontis NB, Stombaugh J, Westhof E. Nucleic Acids Res. 2002;30:3497–3531. doi: 10.1093/nar/gkf481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Correll CC, Freeborn B, Moore PB, Steitz TA. Cell. 1997;91:705–712. doi: 10.1016/s0092-8674(00)80457-2. [DOI] [PubMed] [Google Scholar]
- 121.Leontis NB, Westhof E. RNA. 1998;4:1134–1153. doi: 10.1017/s1355838298980566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Reblova K, Spackova N, Koca J, Leontis NB, Sponer J. Biophys J. 2004;87:3397–3412. doi: 10.1529/biophysj.104.047126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Vallurupalli P, Moore PB. J Mol Biol. 2003;325:843–856. doi: 10.1016/s0022-2836(02)01270-6. [DOI] [PubMed] [Google Scholar]
- 124.Klein DJ, Schmeing TM, Moore PB, Steitz TA. EMBO J. 2001;20:4214–4221. doi: 10.1093/emboj/20.15.4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Frank J, Spahn CMT. Rep Prog Phys. 2006;69:1383–1417. [Google Scholar]
- 126.Krasovska MV, Sefcikova J, Spackova N, Sponer J, Walter NG. J Mol Biol. 2005;351:731–748. doi: 10.1016/j.jmb.2005.06.016. [DOI] [PubMed] [Google Scholar]
- 127.Park H, Lee S. J Chem Theory Comput. 2006;2:858–862. doi: 10.1021/ct0503015. [DOI] [PubMed] [Google Scholar]
- 128.Torres RA, Bruice TC. J Am Chem Soc. 2000;122:781–791. [Google Scholar]
- 129.Van Wynsberghe AW, Cui Q. Biophys J. 2005;89:2939–2949. doi: 10.1529/biophysj.105.065664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Perrotta AT, Shih I, Been MD. Science. 1999;286:123–126. doi: 10.1126/science.286.5437.123. [DOI] [PubMed] [Google Scholar]
- 131.Nakano S, Chadalavada DM, Bevilacqua PC. Science. 2000;287:1493–1497. doi: 10.1126/science.287.5457.1493. [DOI] [PubMed] [Google Scholar]
- 132.Wadkins TS, Shih I, Perrotta AT, Been MD. J Mol Biol. 2001;305:1045–1055. doi: 10.1006/jmbi.2000.4368. [DOI] [PubMed] [Google Scholar]
- 133.Shih IH, Been MD. Annu Rev Biochem. 2002;71:887–917. doi: 10.1146/annurev.biochem.71.110601.135349. [DOI] [PubMed] [Google Scholar]
- 134.Ke A, Zhou K, Ding F, Cate JH, Doudna JA. Nature. 2004;429:201–205. doi: 10.1038/nature02522. [DOI] [PubMed] [Google Scholar]
- 135.Das S, Piccirilli J. Nat Chem Biol. 2005;1:45–52. doi: 10.1038/nchembio703. [DOI] [PubMed] [Google Scholar]
- 136.Ferre-D'Amare AR, Doudna JA. J Mol Biol. 2000;295:541–556. doi: 10.1006/jmbi.1999.3398. [DOI] [PubMed] [Google Scholar]
- 137.Hermann T, Westhof E. J Mol Biol. 1998;276:903–912. doi: 10.1006/jmbi.1997.1590. [DOI] [PubMed] [Google Scholar]
- 138.Murray JB, Meroueh SO, Russell RJM, Lentzen G, Haddad J, Mobashery S. Chem Biol. 2006;13:129–138. doi: 10.1016/j.chembiol.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 139.Nakamura M, Fukunaga Y, Sasa K, Ohtoshi Y, Kanaori K, Hayashi H, Nakano H, Yamana K. Nucleic Acids Res. 2005;33:5887–5895. doi: 10.1093/nar/gki889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Schneider C, Suhnel J. Biopolymers. 1999;50:287–302. doi: 10.1002/(SICI)1097-0282(199909)50:3<287::AID-BIP5>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 141.Yusupov MM, Yusupova GZ, Baucom A, Lieberman K, Earnest TN, Cate JHD, Noller HF. Science. 2001;292:883–896. doi: 10.1126/science.1060089. [DOI] [PubMed] [Google Scholar]
- 142.Guo JX, Gmeiner WH. Biophys J. 2001;81:630–642. doi: 10.1016/s0006-3495(01)75728-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Castrignano T, Chillemi G, Varani G, Desideri A. Biophys J. 2002;83:3542–3552. doi: 10.1016/S0006-3495(02)75354-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Barvik I., Jr. Nucleosides Nucleotides Nucleic Acids. 2005;24:435–441. doi: 10.1081/ncn-200059984. [DOI] [PubMed] [Google Scholar]
- 145.Sanjeev BS, Vishveshwara S. J Biomol Struct Dyn. 2005;22:657–672. doi: 10.1080/07391102.2005.10507033. [DOI] [PubMed] [Google Scholar]
- 146.Ziolkowska K, Derreumaux P, Folichon M, Pellegrini O, Regnier P, Boni IV, Hajnsdorf E. Nucleic Acids Res. 2006;34:709–720. doi: 10.1093/nar/gkj464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Suenaga A, Okimoto N, Futatsugi N, Hirano Y, Narumi T, Ohno Y, Yanai R, Hirokawa T, Ebisuzaki T, Konagaya A, Taiji M. Biochem Biophys Res Commun. 2006;343:90–98. doi: 10.1016/j.bbrc.2006.02.124. [DOI] [PubMed] [Google Scholar]
- 148.Reyes CM, Nifosi R, Frankel AD, Kollman PA. Biophys J. 2001;80:2833–2842. doi: 10.1016/S0006-3495(01)76250-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Fisher TS, Darden T, Prasad VR. J Virol. 2003;77:5837–5845. doi: 10.1128/JVI.77.10.5837-5845.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Mu Y, Stock G. Biophys J. 2006;90:391–399. doi: 10.1529/biophysj.105.069559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Pantano S, Marcello A, Ferrari A, Gaudiosi D, Sabo A, Pellegrini V, Beltram F, Giacca M, Carloni P. Proteins. 2006;62:1062–1073. doi: 10.1002/prot.20805. [DOI] [PubMed] [Google Scholar]
- 152.Li W, Sengupta J, Rath BK, Frank J. RNA. 2006;12:1240–1253. doi: 10.1261/rna.2294806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Li W, Ma BY, Shapiro BA. J Biomol Struct Dyn. 2001;19:381–396. doi: 10.1080/07391102.2001.10506748. [DOI] [PubMed] [Google Scholar]
- 154.Trobro S, Aqvist J. Proc Natl Acad Sci USA. 2005;102:12395–12400. doi: 10.1073/pnas.0504043102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Trylska J, Tozzini V, McCammon JA. Biophys J. 2005;89:1455–1463. doi: 10.1529/biophysj.104.058495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Freddolino PL, Arkhipov AS, Larson SB, McPherson A, Schulten K. Structure. 2006;14:437–449. doi: 10.1016/j.str.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 157.Blakaj DM, McConnell KJ, Beveridge DL, Baranger AM. J Am Chem Soc. 2001;123:2548–2551. doi: 10.1021/ja005538j. [DOI] [PubMed] [Google Scholar]
- 158.Reyes CM, Kollman PA. RNA. 1999;5:235–244. doi: 10.1017/s1355838299981657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Pitici F, Beveridge DL, Baranger AM. Biopolymers. 2002;65:424–435. doi: 10.1002/bip.10251. [DOI] [PubMed] [Google Scholar]
- 160.Showalter SA, Hall KB. J Mol Biol. 2004;335:465–480. doi: 10.1016/j.jmb.2003.10.055. [DOI] [PubMed] [Google Scholar]
- 161.Showalter SA, Hall KB. Biophys J. 2005;89:2046–2058. doi: 10.1529/biophysj.104.058032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kormos BL, Baranger AM, Beveridge DL. J Am Chem Soc. 2006;128:8992–8993. doi: 10.1021/ja0606071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Law MJ, Linde ME, Chambers EJ, Oubridge C, Katsamba PS, Nilsson L, Haworth IS, Laird-Offringa IA. Nucleic Acids Res. 2006;34:275–285. doi: 10.1093/nar/gkj436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Zhao Y, Kormos BL, Beveridge DL, Baranger AM. Biopolymers. 2006;81:256–269. doi: 10.1002/bip.20408. [DOI] [PubMed] [Google Scholar]