


Abstract
Alternative splicing constitutes a major mechanism creating protein diversity in humans. This diversity can result from the alternative skipping of entire exons or by alternative selection of the 5′ or 3′ splice sites that define the exon boundaries. In this study, we analyze the sequence and evolutionary characteristics of alternative 3′ splice sites conserved between human and mouse genomes for distances ranging from 3 to 100 nucleotides. We show that alternative splicing events can be distinguished from constitutive splicing by a combination of properties which vary depending on the distance between the splice sites. Among the unique features of alternative 3′ splice sites, we observed an unexpectedly high occurrence of events in which a polypyrimidine tract was found to overlap the upstream splice site. By applying a machine-learning approach, we show that we can successfully discriminate true alternative 3′ splice sites from constitutive 3′ splice sites. Finally, we propose that the unique features of the intron flanking alternative splice sites are indicative of a regulatory mechanism that is involved in splice site selection. We postulate that the process of splice site selection is influenced by the distance between the competitive splice sites.
INTRODUCTION
Alternative acceptors (AA) constitute ∼20% of all conserved alternative splicing events in humans and mice (1). During the second transesterification step of the splicing process, the acceptor site, namely the 3′ splice site (3′SS), is selected by the splicing machinery. In mammalians, the 3′SS is defined by a highly conserved AG dinucleotide, a polypyrimidine tract (PPT) located upstream of the splice site and an invariant adenine which is part of the consensus branch point (BP), normally found upstream of the PPT (2). Early genomics studies have observed that certain splice site compositions are more probable than others, and these probabilities were used to derive the Shapiro–Senapathy splice site scores (3). Other models were further developed to characterize the 3′SS, such as the MAXNET algorithm, which takes into account position dependencies (4). Despite the relative weak signal of the 3′SS, the splicing machinery can accurately recognize the authentic splice site from an array of potential sites. In many cases, more than one splice site can be identified, leading to AA which may be regulated in a tissue specific manner (5).
In vitro experimental studies have shown that when two AG sites are placed downstream of the BP and PPT; most often the AG site which is located proximal to the BP is preferred by the splicing machinery (6,7). Nevertheless, selection of a distal AG site both in constitutive or alternative splicing has routinely been observed (6–10). It was proposed that the selection of a distal splice site can be influenced by its closeness to the BP and the proximal AG site (6–8,11). Specifically, it has been shown that AGs which are relatively close to the BP can be bypassed and that close AG dinucleotides are highly competitive for binding to the spliceosome (6–8). In the far end, a number of alternatively spliced exons were found to be characterized by an extremely large distance between the BP and the 3′SS. This region is known as the AG exclusion zone (AGEZ), where AGs are recognized but not utilized by the splicing machinery, possibly repressing downstream splice sites (9). Further experiments demonstrated that a proximal AG dinucleotide can be recognized during the first transesterification step, leading to the selection of a neighboring distal splice site, even when the proximal AG is not functional (12). In agreement with the observed nucleotide preferences, competition experiments have confirmed that the nucleotide preceding the AG can influence the choice of 3′SS: CAG > TAG > AAG > GAG (6).
The composition of the 3′SS is thought to be an important component in the process of splice site selection. Recently it was shown that the identity of the nucleotide (N) preceding the invariant AG splice site is also associated with the observed splice site selection in the NAGNAG motif, where the two potential acceptor sites are placed in tandem (10,13,14). In addition, the recognition of the 3′ss during the first (15) or second (16) step of transesterification, as well as the tendency of a cryptic splice site to be selected or avoided (17), is dependent upon the length and composition of the PPT and the presence of splicing factors that bind cis-regulatory elements nearby the splice sites (2,15,18–21). Many regulatory factors have been shown to be involved in splice site selection, e.g. U2AF (15,20), hSlu7 (18), as well as splicing enhancers and silencers such as SF2/ASF and hnRNPA1 (21). Experimental studies have shown that AG dinucleotides which are appropriately positioned relative to the BP have an intrinsic potential to become active splice sites (6,7). What is less clear is how the selection or avoidance of alternative AG dinucleotides is regulated in order to prevent undesired transcripts from being produced.
In the last few years, global computational analyses of alternative splicing have been performed, focusing on exon skipping events (22–24) and alternative 3′/5′ splice sites (24,25). Generally, these automatic methods achieved good performance when classifying skipped exons (23) as well as distant (>50 nt) alternative 3′/5′ sites from constitutive splice sites (25), based on sequence features such as splice site strength (23,25), composition and position of the PPT (23,25), evolutionary conservation (23) and frame preservation (23,25). In addition, several unrelated studies have demonstrated an unusually high level of intronic conservation flanking skipped exons (1,26,27). This property was successfully used for automatic classification of alternative exons (23). High levels of intronic conservation were also observed upstream of NAGNAG 3′SS that undergo AS, while intron conservation upstream of constitutively spliced NAGNAGs was generally low (10). Though not fully understood, the sequence conservation at the intronic regions flanking alternative spliced events suggests the presence of regulatory elements which are under evolutionary selection. In addition, differences in the overabundance of exonic sequence enhancers (ESE) and exonic sequence silencers (ESS) in the vicinity of alternatively spliced exons compared to constitutively spliced exons were also observed, revealing a complex relationship between splice-site selection and presence of splice-factor binding sites (28,29).
Here we applied a genome-wide approach to analyze human–mouse conserved AA. In order to identify properties which are characteristics of AA, we have analyzed sequence features such as splice site strength, PPT and BP position and composition; intronic evolutionary conservation, ESE/ESS density, GC content and pseudo splice site distribution. We have divided the AA into subgroups according to the distance between the alternative splice sites and compared them to equivalent groups of constitutive acceptors. We have applied both classical statistical analyses on the individual features as well as a machine-learning approach [Support Vector Machine (SVM)] to study the effect of the different features on splicing selection. We show that different splicing patterns can be better differentiated when combining multiple features and that the contribution of the different features to SVM performance varies in relation to the distance between the splice site pairs. Furthermore, we observed an unexpectedly high occurrence of the alternative splicing events in which the PPT was found to overlap the upstream (or proximal) splice site. Overall, the occurrence of multiple PPTs as well as high intronic conservation in the vicinity of the splice sites, are unique properties of AA. Finally, we suggest that the observed differences between the sequence properties of alternative versus constitutive splice sites are indicative of a regulatory mechanism that is involved in the process of splice site selection. We postulate that the process of splice site selection depends on the distance between the competitive splice sites.
METHODS
Dataset construction
AA events derived from a database of human–mouse conserved alternative splice sites (27) were analyzed. In addition, a control set of pseudo acceptors separated from the constitutive splice sites by an AG-depleted region (CA/PA pairs) was extracted. The term ‘pseudo acceptors’ refers to HAG triplets (AAG, TAG or CAG), which were not identified as splice sites based on the existence of EST or mRNA. To avoid the inclusion of alternative splicing events that were undetected in the EST data we required that the PA site (specifically the AG) in each group was not conserved between the species. In addition GAG triplets were not accounted as pseudo acceptors since these rarely serve as splice sites (30). We also discarded events in which human–mouse alignments (hg17/mm7) for at least 30 nt upstream to the splice site were not available in the UCSC Genome Browser (ending up with a total of 396 AA pairs and 55,606 CA/PA pairs).
The AA dataset was divided into four groups according to the distance between the splice site pairs: 197 NAGNAG, 75 CLOSE, 77 MID, 47 FAR. For the control set we chose CA/PA pairs in which the distances between the CA and the PA pairs was equivalent to the distances of the AA pairs of the relevant set. Due to the high discrepancy between the size of the true AA set and the control CA/PA pairs (specifically in FAR and MID) for each group we randomly chose a control set which was 3-fold the size of the target set, accounting for ∼1% of the total number of sequences in the control group. Overall we included 231 MID and 141 FAR. Since the total number of available CLOSE CA/PA pairs in our data was 382, to keep consistent with the 3-fold ratio we randomly chose 225 events. For the NAGNAG control set we extracted from the human genome all NAGNAG acceptors for which there was no evident for AS. These were split in two groups: 177 NAGNAG motifs in which the distal site is a CA and the proximal is a competitive PA (NAGNAG-distal); and 397 NAGNAG motifs in which the distal site is a PA and the proximal is CA (NAGNAG-proximal). The genomic coordinates (UCSC, hg17) and sequences of all AA and CA/PA pairs can be found in Supplementary Data file 1 and 2, respectively.
Features analyzed
Splice site parameters
Here we concentrated only on the first nt preceding the invariant AG, ignoring neighboring nts. For simplicity, the first nucleotide in the proximal splice site (Np) and the distal splice site (Nd) were scored 1, 2, 3, 4 for G, A, T, C, respectively. The relative strength between the two N's was calculated as the ratio between the scores. To ignore directionality the higher or equal value was always taken as the numerator (Np/Nd). In addition, we provided an overall score for the tandem acceptor reflecting the strength of both sites (STRE). This parameter included values ranging from 1 to 9 for AAG-[N]n-AAG, AAG-[N]n-TAG, AAG-[N]n-CAG, TAG-[N]n-AAG, TAG-[N]n-TAG, CAG-[N]n-AAG, CAG-[N]n-TAG and CAG-[N]n-CAG, respectively, N accounts for any nt and n is the number of nts between the splice site pairs.
Polypyrimidine tract (PPT)
The region between the distal splice site and 100 nt upstream of the proximal or pseudo splice site was screened for the existence of PPTs. The screening process involved four major steps:
Detecting the seed PPT: Starting from the distal (or constitutive) splice site a seed PPT was defined as the first (upstream) stretch of nts of size 12 including a minimum of 75% pyrimidines. The threshold of 75% was chosen based on a preliminary analysis we have applied on a set of experimentally validated intronic PPTs extracted from the ASD database (http://www.ebi.ac.uk/asd/).
PPT expansion: The PPT seed was expanded both upstream and downstream adding 1 nt at each iteration. The PPT was extended only if the pyrimidine concentration increased.
Detecting additional PPTs: After defining a PPT, the region from the 5′ side of the PPT to the distal splice site was masked. Additional PPTs were detected applying step 1 and 2 on the remaining sequence.
Ranking of PPTs: The PPTs found within the 100 nt upstream of the proximal site were further ranked according to their length. The two largest PPTs were denominated ‘PPT1’ and ‘PPT2’, respectively.
For each PPT, the following characteristics were computed: Percent of pyrimidine after extension (defined also as the PPT score), PPT length, the distance of the PPT to the proximal site (PPT∼P) and to the distal site (PPT∼D). In addition, we computed a series of binary features describing whether the PPT is placed upstream, downstream or overlapping the proximal (or pseudo) splice site. The position of PPT relative to the splice site was defined as described below:
PPT-p: Only one PPT was found and was placed upstream of the proximal site or else when two PPTs were placed upstream of the proximal site
PPT-[p]: Only one PPT was found and overlapped the proximal site.
p-PPT: Only one PPT was found and was downstream of the proximal site or else when two PPTs were placed downstream of the proximal site.
PPT-p-PPT: The proximal site was placed between two PPTs.
PPT-PPT[p]: The PPT overlapped the proximal site and was followed by a second PPT.
PPT[p]-PPT: The PPT overlapped the proximal site and was preceded by a second PPT.
Branch point (BP)
Branch points (BP) were predicted according to the method developed by Kol et al. (11) with minor modifications. Putative BPs were scanned for in a region of 15 nt upstream and 15 nt downstream of the 5′ boundary of the predicted PPT. BPs were scored using a PSSM for mammalian BPs (31) requiring an ‘A’ at position 6 of the PSSM, which represents the invariant adenine of the BP. In addition, the distance to the proximal (BP∼P) and distal (BP∼D) splice sites were computed.
Intronic conservation
Human–mouse (hg17/mm7) pairwise alignments were extracted and conservation scores were calculated based on the number of conserved base pairs in five overlapping windows of length 10 nt (IC1–5). Matching positions (M) were scored 0.1, mismatching positions (m) were scored 0. Gaps (g) in the human strands were skipped, thus the actual sliding window size was 10 + g. Gaps in the mouse strand were treated as mismatches. The conservation score was calculated as
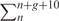 where n is the position between 0 and −30 nt upstream of the HAG. In addition the average conservation in an upstream region of 100 nt (IC100) was calculated. In cases were the alignments for 100 nt were not available (∼10% of the data) the average conservation was computed for the available alignment, which was always ≥30 nt. The sequence alignments of the upstream regions of all AA and CA/PA pairs are given in Supplementary Data file 3.
where n is the position between 0 and −30 nt upstream of the HAG. In addition the average conservation in an upstream region of 100 nt (IC100) was calculated. In cases were the alignments for 100 nt were not available (∼10% of the data) the average conservation was computed for the available alignment, which was always ≥30 nt. The sequence alignments of the upstream regions of all AA and CA/PA pairs are given in Supplementary Data file 3.
GC content
The frequency of G and C was computed for each sequence at the region of 100 nt upstream of the proximal or the pseudo splice site.
ESE and ESS density
The frequencies of the regulatory element candidates (ESEs and ESSs) were computed for the 100 nt upstream of the proximal or pseudo splice site. The list of ESEs were extracted from the ESE-RESCUE database (http://genes.mit.edu/burgelab/rescue-ese/). The two sets of ESSs (hex2 and hex3) were downloaded from the FAS-ESS server http://genes.mit.edu/fas-ess/. The occurrence of all regulatory sequences within a given list (i.e. ESE, ESS-hex 2, ESS-hex3) was calculated per each sequence in the subgroup (e.g. CLOSE AA). The average frequency is reported for each subset.
Occurrence of pseudo splice sites
The occurrence of NAG triplets in the region of 100 nt upstream of the proximal or the pseudo splice site was computed. The occurrence was calculated separately for each of the four different triplets (AAG, CAG, GAG and TAG) as well as for all HAG's (AAG, CAG and TAG) as a group.
Statistical analysis
We performed an F-test and a Student's t-test (assuming equal variance) on the FAR, MID, CLOSE, NAGNAG-proximal and NAGNAG-distal datasets using the R Stats package (http://stat.ethz.ch/R-manual/R-patched/library/stats/html/).
In the different groups we analyzed the following features: Np, Nd, IC100, PPT score, PPT length, PPT∼D, PPT∼P, BS score, BS∼D, BS∼P, ESE/ESS density, HAG and GC (as described above). The significance of the F- and t-tests were determined using the Westfall–Young method for i-value adjustment (32). Briefly, we re-sampled the set of AA and CA/PA pairs and calculate the F- or t-test P-value. The process was carried 1000 times for each test and the minima of the new P-value was retained and compared to the original one, namely the P-value of the AA versus CA/PA set without re-sampling. If the latter P-value was smaller than the minima of the Westfall–Young procedure and <0.05 the result of the test was considered significant. To estimate the sample size we have calculated the power of the t-test (1−β) using the R package, only tests which yielded a power ≥0.9 were further considered significant.
SVM training and testing
SVM is a machine-learning algorithm used to detect and exploit complex patterns in data. The SVM is a kernel-based method applying linear classification techniques to non-linear classification problems. It has been widely used to explore biological problems (33) including alternative splicing (23,25). In this study, we used the gist-train-svm software http://bioinformatics.ubc.ca/gist/ with a linear kernel. Input data was normalized by rescaling the columns to values between −1 and 1. All tests were conducted by applying a ‘leave one out’ cross-validation (jackknife) procedure. The following feature sets were used for training the FAR, MID and CLOSE classifiers: PPT parameters (PPT score, PPT length, PPT∼D, PPT∼P, UP, DN, OVLP), splice site parameters (Nd, Np, Nd/Np, STRE), Intronic conservations (IC100, IC1–5), frequency of pseudo splice sites (AAG, CAG, GAG, TAG, HAG) and GC content. The UP, DN and OVLP parameters refer to the relative position of the PPT, whether it is placed upstream (UP), downstream (DN) or overlapping (OVLP) the proximal (or pseudo) splice site. For the NAGNAG classifiers the same feature sets were used with the exception of the PPT∼P. The later was exempted since in NAGNAG motifs the PPT∼P is equivalent to the PPT∼D.
The SVM performance was evaluated by the ROC (receive operating characteristics) analysis which plots the true positive rate (TPR) versus True negative rate (TNR) for different cutoffs. The AUC (area under curve) was reported for each test. In addition, we calculated the total accuracy (TA), sensitivity (SN), specificity (SP) and the Matthews correlation coefficient (MCC).
 |
ΔAUC calculations
The ΔAUC for each parameter was defined as the difference between the AUC value obtained with the full feature vector and the AUC value reached when a specific feature set was eliminated. The parameter sets included: splice site (splice site parameter), IC (intronic conservation), GC (GC content), PPT (PPT parameters), AG (occurrence of pseudo splice sites) and CIS (occurrence of cis-regulatory elements).
RESULTS
Dataset assembly
In an attempt to better understand the mechanism of AA selection, we have analyzed evolutionarily conserved AA pairs in distances ranging from 3 to 100 nt. These include all events in which both the proximal (upstream) 3′SS and a distal (downstream) 3′SS are evidently involved in splicing in both the human and mouse genomes, based on the existence of EST and mRNA transcripts in both species (1). The number of conserved AS events represent a lower bound of the AS events in humans, nevertheless, they are expected to be biologically significant (34). It is generally accepted that two putative splicing acceptors which are located in close proximity are highly competitive (6,7,9). To test the dependency between the location of the putative splice site and the splice site selection we divided the data into four separate sets according to the distance between the splice site pairs. The groups were designated: FAR, MID, CLOSE and NAGNAG. The FAR, MID and CLOSE groups were composed of AA pairs separated by 40–100, 13–39, 4–12 nt, respectively. The NAGNAG group included only AA pairs placed in tandem. While in the CLOSE and NAGNAG datasets, we expect both the AA to be placed downstream of the BP, which is generally found between 18 and 40 nt upstream of the splice site (6,7,9,11), the FAR dataset included sequences in which the first splice site is expected to be upstream of the BP. However, as reported in Gooding et al. (9), in some cases the BP could be located upstream of the proximal site also in the FAR group. The borderline cases were grouped together in the MID dataset. In addition, we compiled a series of control sets containing constitutive acceptors (CA) separated from an upstream potential competitor or pseudo acceptor (PA) by an AG-depleted stretch of variable lengths. The sequences for the control set were deliberately selected so the distribution of the distances between the CA and the PA in the control sets will be equivalent to that of the corresponding alternative dataset.
The unique features of AAs
In order to study the AA pairs and compare them to 3′SS which are constitutively chosen, we have analyzed a series of intronic properties calculated for each group. Among the features we included were splice site strength, intronic evolutionary conservation (excluding the splice sites), length and score of the PPT and its position relative to the splice sites, BP score and distance to the splice sites, GC content, ESE/ESS density and the occurrence of other AGs dinucleotides (see Methods section). The latter were previously found to affect the recognition of the splice sites when they occur in upstream intronic regions (7,8). As in the majority of cases [excluding the class of distal BPs (9)], the splicing regulatory elements are close to the 3′SS, we restricted the analysis to 100 nt upstream of the proximal site.
The above properties were calculated for all sequences in each AA subset and compared to the corresponding subset of CA/PA pairs. In the case of the NAGNAG group, comparisons were conducted against two independent sets of CA/PA pairs: NAGNAG motifs in which the distal site is constitutively spliced and the proximal is a competitive pseudo acceptor (NAGNAG-distal); and the reverse case where the proximal site is constitutively spliced and the distal NAG serves as a pseudo acceptor (NAGNAG-proximal). Although competitive sites are generally placed upstream of the splice site (6,7), in the unique case of NAGNAG, we chose to test the two control sets since, in tandem acceptors, both the distal and proximal sites were previously suggested to contribute to the competition. Furthermore, these NAGNAG motifs are of special interest as they are widely distributed throughout the human genome (10,14).
For each of the properties analyzed, we have carried out a statistical analysis applied to all datasets pairs (AA versus CA/PA). A summary of the statistical analysis is given in Table 1 (detailed results are given in Table 1S). Interestingly, in each subgroup (defined by the splice site distance) we found a different set of features that deviated between the AA and CA/PA pairs. For example, splice site features were discriminative only in the NAGNAG group, both when comparing it to the NAGNAG-proximal and to the NAGNAG-distal (Table 1SA and B). This is in agreement with previous studies which observed correlation between the splice site strength and the splicing pattern at the NAGNAG motif (10,13). In addition, consistent with our previous results (10), the intronic conservation in the 100 nt upstream of the proximal splice site was significantly higher in alternative NAGNAGs compared to the NAGNAG-proximal group (P-value for F-test = 4.E−04). Interestingly, the intron conservation did not appear to differ significantly when comparing alternative NAGNAGs to the NAGNAG-distal group. The latter groups both demonstrate a high intronic conservation which may suggest similar regulatory constraints (10). Furthermore we observed a significant difference in the GC content between alternative NAGNAGs and the NAGNAG-distal group. Surprisingly, the high GC content in the NAGNAG-distal group was higher than the average GC content found generally upstream of constitutive acceptor sites (Table 1S).
Table 1.
P-values for Student's; t- and F-tests comparing alternative acceptors against constitutive/pseudo acceptors based for the following features: distal splice sites (Dist SS), proximal splice sites (Prox SS), average intronic conservation in 100 nt upstream of the proximal splice site (IC100), PPT length, PPT score, distance of PPT to the distal site (PPT∼D) and the proximal site (PPT∼P), ESE/ESS density, pseudo HAG sites and GC content
| Feature | NAGNAG-P | NAGNAG-D | CLOSE | MID | FAR | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| t P-val | F P-val | t P-val | F P-val | t P-val | F P-val | t P-val | F P-val | t P-val | F P-val | |
| Dist SS | 2.E−16 | 0.215 | 1.E−09 | 2.E−16 | 0.371 | 0.183 | 0.321 | 0.005 | 0.003 | 0.049 |
| Prox SS | 2.E−06 | 1.E−07 | 0.236 | 0.026 | 0.730 | 0.241 | 0.083 | 0.169 | 0.063 | 0.260 |
| IC100 | 4.E−04 | 0.144 | 0.007 | 0.037 | 0.013 | 0.435 | 2.E−10 | 0.373 | 3.E−06 | 0.465 |
| PPT Length | 0.226 | 0.344 | 0.996 | 0.030 | 0.823 | 0.004 | 0.001 | 2.E−14 | 0.061 | 2.E−10 |
| PPT Score | 0.525 | 0.748 | 0.211 | 0.199 | 0.012 | 6.E−13 | 4.E−08 | 8.E−07 | 3.E−07 | 3.E−10 |
| PPT∼D | 0.054 | 0.233 | 0.901 | 0.516 | 5.E−09 | 0.002 | 0.854 | 0.038 | 1.E−05 | 0.161 |
| PPT∼P | na | na | na | na | 1.E−06 | 0.007 | 0.986 | 0.055 | 0.023 | 0.001 |
| BP score | 0.525 | 0.748 | 0.2388 | 0.2338 | 0.289 | 0.539 | 0.4418 | 0.239 | 0.4564 | 0.550 |
| BP∼D | 0.054 | 0.233 | 0.6724 | 0.9282 | 8.88E−07 | 0.017 | 0.002 | 0.154 | 1.30E−05 | 0.021 |
| BP∼P | na | na | na | na | 2.90E−04 | 0.031 | 1.22E−04 | 0.143 | 0.005 | 2.85E−04 |
| ESE | 0.042 | 0.020 | 0.850 | 0.812 | 0.708 | 0.618 | 0.031 | 0.331 | 0.042 | 0.020 |
| ESS.hex2 | 0.873 | 0.252 | 0.582 | 0.948 | 0.721 | 0.186 | 0.064 | 0.157 | 0.176 | 0.135 |
| ESS.hex3 | 0.350 | 0.023 | 0.710 | 0.779 | 0.985 | 0.170 | 0.606 | 0.112 | 0.324 | 0.428 |
| HAG | 0.888 | 0.561 | 0.601 | 0.023 | 0.023 | 0.723 | 2.E−04 | 0.490 | 1.E−04 | 0.868 |
| GC | 0.265 | 0.512 | 1.E−04 | 0.714 | 2.E−04 | 0.711 | 0.044 | 0.999 | 0.149 | 0.312 |
Results are shown for the different datasets: FAR, MID, CLOSE, NAGNAG-proximal and NAGNAG-distal. Significant values (based on Westfall–Young correction) are indicated in bold.
Nevertheless, in the CLOSE group in which the splice sites are very close to each other but not adjacent (Table 1SC), the features which were significantly different between AA and CA/PA pairs were the distance of the PPT and BP to the splice sites and the PPT score (which was statistically significant in the CLOSE group when applying the F test). Generally, in AA pairs the PPT and BP appeared closer both to the proximal and distal sites and the PPT displayed a wider variance of scores. As in the NAGNAG-distal group, in the CLOSE group we also observed a relatively lower GC content upstream of the AA pairs. In contrast to the NAGNAG group, in the CLOSE group we did not observe significant differences either in the splice site composition or in intronic conservation between AA and CA/PA pairs. In the MID group we observed weaker PPTs (i.e. lower PPT score) in AA compared to CA/PA pairs. In addition, the intronic conservation levels were higher upstream of AA pairs and the AG dinucleotides were slightly underrepresented (Table 1S D). Among the most discriminating features in the FAR group were the score and the relative position of the PPT and BP. These were found to be weaker in AA pairs and farther from the distal splice site. In addition, the intronic conservation was significantly higher in AA pairs and the occurrence of intronic AGs was underrepresented.
Overall, our results suggest that only when the two splice sites are placed in tandem (NAGNAG) the splice site composition, namely the identity of the nucleotide preceding the conserved AG, appeared to be discriminative. In contrast, when the distance between splice sites is larger, differences in the PPT composition and the relative location of the PPT and BP to the splice sites seem to play an important role. Consistently, the level of intronic conservation appeared to be discriminative in the MID and FAR groups in which the AA distance is >12 nt (Table 1S, Figure 1). However, it is important to note that, in the unique group of the NAGNAG acceptors, the intronic conservation was also found to be statistically significant (though to a lesser extent) only when comparing tandem acceptors to the proximal NAGNAG group and not in comparison to the distal group. Likewise, we observed that the intronic evolutionary conservation in the CLOSE group is relatively high (though not statistically significant) for AA pairs when compared to the average intronic conservation upstream of randomly chosen constitutive splice sites (Figure 1A). A relative high intronic conservation was also detected in the CA/PA pairs only in the close region adjacent to the pseudo splice site (Figure 1). The high intronic conservation levels found generally upstream of alternative splice site are consistent with other studies suggesting the existence of regulatory elements involved in splice site selection (29). Nevertheless, the relative high intronic conservation close to the splice site in CA/PA pairs, specifically in the CLOSE group, could be due to the presence of regulatory elements which may be involved in controlling constitutive splicing when potential competitors are present. It is important to note that in our dataset the pseudo splice sites (PAs) themselves are not evolutionarily conserved and thus the higher intronic conservation observed cannot be related to an overall high conservation of the site or an alignment artifact.
Figure 1.

Human–mouse evolutionary conservation shown for the CLOSE (A) and FAR (B) groups. Conservation was calculated for the 30 nt upstream of the proximal (or pseudo) splice site in overlapping windows of length 10. Gray circles account for alternative acceptor (AA) pairs, black triangles for constitutive/pseudo acceptor (CA/PA) pairs and the gray crosses for a set of 1000 randomly selected constitutive acceptors (CA). For the CA set, the conservation was calculated upstream of the constitutive splice site.
PPT analysis
The PPT is a key feature in splicing regulation. Previously, it has been shown that both the composition and the distance of the PPT can influence splice site selection (19). The PPT is commonly identified by splicing factors such as the splicing repressor PTB and the splicing enhancer U2AF65. Recent reports demonstrate that both factors can compete with each other for binding the PPT (35). Although these proteins are considered basic splicing factors, it has been suggested that they also play an important role in the regulation of alternative 3′ splice sites (36). As described in the pervious section, we have conducted a comprehensive analysis of the PPT in AA and CA/PA pairs in the different subsets. Figure 2A and B illustrate the relationship between the distances of the PPT to the distal splice site and the distance between the proximal splice site to the distal splice site in AA pairs and in CA/PA pairs, respectively (in CA/PA pairs CA is equivalent to distal and PA to proximal). Each dot represents the relationship in one sequence. As shown, when the distance between the proximal and distal splice sites is 3 (in the NAGNAG subset) in both alternative and constitutive splice sites a PPT is found anywhere between 0 and 90 nt upstream of the splice sites. However, when the splice sites are not in tandem, in AA pairs the predicted PPT is found close to the distal splice site only when the splice sites are relatively close to each other, separated by <40 nt (Figure 2A). This is most probably related to the dependency between proximal site and the PPT in the AA pairs. As demonstrated by the dots lying along the diagonal in Figure 2A, in the majority of AA pairs the predicted PPT falls in close proximity to the proximal splice site. In the CA/PA pairs (Figure 2B), although we do observe a large proportion of PPTs located in proximity to the distal site we could not detect any clear relationship between the location of the PPT and the PA. To ensure that these results are not due to the smaller sample size of the AA pairs, we have randomly selected from the full set of all constitutive events 1000 sets of equal size to the AA group. For each set we calculated the number of cases in which the PPT was adjacent to the distal site (<5 nt apart) in CA/PA pairs separated by ≤40 and >40 nt and compared it to the distribution in the AA set. We have applied a series of Fisher-exact tests comparing each random set to the AA set and all cases showed a significant difference between the groups (using the Bonferroni correction P < 5*10−5). These results confirmed that the dependency between the ‘distal site-PPT’ distance and the ‘distal-proximal’ distance is restricted to the AA set.
Figure 2.

PPT distribution. (A) The distance between the most downstream nt of the PPT and position −1 (or N site) at the distal NAG site is plotted against the number of nts between position −1 of the proximal and the distal splice site. (B) A control set in which the PPT-to-constitutive splice sites distance is plotted against the constitutive-to-pseudo splice site distance. The diagonal indicate positions for which the PPT is adjacent to the proximal (or pseudo) splice site.
We have further conducted a detailed analysis of the proximity between the largest PPT and the proximal splice site. Though the analysis was conducted on all subgroups, we concentrated specifically on the FAR group in which the distance between the splice site allows the detection of full-size PPTs. The analysis has revealed a surprisingly high number of AA pairs in which the PPT was found completely overlapping the proximal splice site (∼57%). This phenomenon was not found in the set CA/PA pairs, where we found only 8% of cases in which the predicted PPT overlapped the PA site (Table S2). In contrast, in ∼65% of the constitutive events the PPT was predicted downstream of the pseudo splice site (close to the constitutive splice site) while in only ∼13% of the AA pairs the PPT was found downstream of the proximal site. In both groups, we observe a similar proportion of events in which the PPT was found relatively far, upstream of the proximal (or pseudo) splice site. Important to mention is that in most cases in which the PPT overlaps the splice sites, these were embedded within the PPT usually in the 5′ half but not at the edge (the frequency of events showing the position of the splice site relative to the PPT is given in Figure 1S).
Previous studies have suggested that efficient repression by PTB depends on the existence of two binding sites which can mediate the formation of a stem-loop structure by protein–protein interactions between PTB monomers, known as the ‘looping out’ model (37). This mechanism was originally suggested in order to explain the regulation of alternative exons; however, BPs where also proposed to be looped out and avoided by the splicing machinery (38). In addition, a recent computational study reported the existence of two PPTs flanking the upstream splice site when alternative 3′SS are separated by ≥8 nt (39). To further identify features which could be used to differentiate between alternative and constitutive splice acceptors at a genomic level, we searched for the existence of an additional polypyrimidine stretch in the region of 100 nt upstream of the splice site (both surrounding AA and CA/PA pairs). The definition used to automatically assign the second PPT is described in detail in the Methods section. Generally, the assignment of PPTs was done based on their relative size, PPT1 being the longest stretch. Overall, we did not observe a clear difference between the alternative and constitutive splice sites when simply considering the length of PPT2 (P = 0.060) or its relative strength (P = 0.596). It is important to note that in ∼20% of the AA CA/PA pairs we were not able to find an additional PPT, while in 19% of AA and 27% of CA/PA pairs we found both PPTs either upstream or downstream of the splice site (Table S3).
Subsequently, we evaluated the relative position of both PPTs in all AA and CA/PA pairs, concentrating on the FAR group. We found that in 53% of the AA pairs, one PPT was found overlapping the splice site and the other one was found upstream of it [PPT-PPT(p)]. In contrast, this pattern accounted for only ∼6% of all CA/PA pairs (Figure 3). These observations reinforce that the high occurrence of overlap between the proximal splice site and the PPT is not restricted to close splice site pairs, in which the proximal site by default falls within the PPT due to space restriction as in the CLOSE and MID groups (Figure 2S). These results are also consistent with previous observations by Dou et al. (39). In addition, in most AA pairs analyzed in our study, the PPT that was found to overlap the splice site was the larger one among the two PPTs (Table 4S). Furthermore, we observed that ∼54% of the pseudo splice sites in CA/PA pairs were found to be flanked (but do not overlap) by two PPTs (PPT-p-PPT). This pattern was observed only in ∼17% of the AA pairs (Figure 3). Generally, in AA pairs the PPTs were always found upstream of the proximal splice site or overlapping the splice site, but never downstream of the proximal site (Figures 3 and 2S). The fact that PPTs were not detected between AA pairs could be due to the coding potential of this region. Nevertheless, it could suggest that a downstream PPT alone is not capable of regulating an upstream splice site.
Figure 3.

Position of PPTs relative to the proximal splice site in the FAR group. The bars indicate the percent of observations in the data. I–III are cases in which only one PPT was found upstream (I), overlapping (II) or downstream (II) the proximal splice site. IV–VI are cases in which two PPTs were observed (IV) flanking the proximal site, (V) one PPT overlapping the splice site and the second one upstream and (VI) one PPT overlapping the splice site and the second one downstream of the proximal splice site. The gray bars represent alternative acceptor pairs and the black bars represent constitutive/pseudo acceptor pairs in which the pseudo splice site mimics the proximal site. The number of occurrences is shown in brackets.
Combining features for splice site classification
To test whether the combination of features described above can better separate AA from CA/PA pairs, we have built a SVM classifier for each of the datasets: NAGNAG, CLOSE, MID and FAR. SVM is a supervised machine-learning algorithm which is trained to separate between two sets of data. It has been previously applied to automatically identify alternative exons based on both exonic and intronic properties, including evolutionary conservation, length of PPT and splice sites composition (23). In addition, a recent study has applied SVM to differentiate between alternative and constitutive 3′/5′ splice sites based on parameters such as PPT, splice site composition and frame preservation (25). Here we applied the SVM algorithm to distinguish alternative versus constitutive 3′ss events in each subset independently. The feature set was composed of intronic features calculated in the previous section including the splice site composition, PPT properties; intronic conservation, pseudo splice site occurrence and GC content (see the Methods section for details). Since the position of the predicted BP relative to splice site was found to be highly correlated with the relative position of the PPT (Pearson correlation ∼0.9), we did not include this parameter in the SVM feature set. Additionally, we have not included the ESE and ESS density as parameters for SVM as they were not found to be statistically significant in any of the subsets. To estimate the performance of our method, we have performed a ‘hold one out’ cross-validation test (also known as the ‘jackknife’ test). For each test, we plotted a receiving operating characteristics (ROC) plot and calculated the area under the curve (AUC), the sensitivity, specificity, total accuracy and The Matthews correlation coefficient (Table 2). As shown, the best SVM performance was achieved for the AA FAR versus CA/PA FAR classifier, for which the AUC was 0.94, followed by the MID (AUC = 0.91), the CLOSE (AUC = 0.80) and lastly the NAGNAG-P (AUC = 0.78) (Figure 4 and Table 2). As shown in the NAGNAG-D classifier, we encountered a notable decrease in SVM performance (AUC = 0.69). These results are in accordance with our previous reports showing similarity in genomic properties between alternative spliced NAGNAG motifs and NAGNAG motif in which the distal site is chosen constitutively (10). Overall in agreement with the recent study by Xia et al. (25) we observe that the performance of the SVM increases as a function of the distance between the splice site pairs. Different from Xia et al. (25), in the current study we also obtained a considerably high performance for the classification of splice site pairs which are in close proximity, with sensitivity varying between 60 and 85% in the different subgroups (Table 2). The difference in SVM performance obtained for the close sites in the different studies is probably due to the unique features which were selected for the study.
Table 2.
SVM performance
| FP | FN | TP | TN | SN | SP | TA | MCC | AUC | |
|---|---|---|---|---|---|---|---|---|---|
| FAR | 12 | 7 | 40 | 129 | 85.106 | 91.489 | 89.894 | 0.741 | 0.936 |
| MID | 37 | 14 | 63 | 194 | 81.818 | 83.983 | 83.442 | 0.608 | 0.913 |
| CLOSE | 51 | 22 | 53 | 174 | 70.667 | 77.333 | 75.667 | 0.437 | 0.802 |
| NAGNAG-P | 121 | 47 | 150 | 276 | 76.142 | 69.521 | 71.717 | 0.432 | 0.785 |
| NAGNAG-D | 53 | 75 | 122 | 124 | 61.929 | 70.056 | 65.775 | 0.32 | 0.698 |
The table displays the number of false positives (FP), true positives (TP), false negatives (FN), true negatives (TN), sensitivity (SN), specificity (SP), total accuracy (TA) as well as The Matthews correlation coefficient (MCC) and the AUC value for the different datasets.
Figure 4.

ROC plot summarizing the SVM results: False positive rate is plotted against the true positive rate for alternative acceptors versus constitutive/pseudo acceptor pairs in the FAR (black line), MID (red dashed), CLOSE (green line), NAGNAG-proximal (blue dots) and NAGNAG-distal (black dots) groups.
Feature selection
In order to estimate the contribution of the different parameters to the learning process, we have performed a simple (backward) feature selection procedure whereby we exclude one feature set in each SVM run and evaluate the change in the performance. A single feature set was defined as a set of all parameters that have a common property and are highly interdependent; for example, intron conservation was calculated for five overlapping windows, the average conservation values for each window separately and the average over all windows together were considered as single features in the vector while a set of all these features together was considered as a feature set, named ‘intron conservation’. For each set we computed a ΔAUC, which is the difference between the overall AUC value obtained with the full vector and the AUC value reached when the specific set was eliminated.
As shown in Figure 5, the features which were found to contribute mostly to SVM performance in the FAR and MID groups were the intronic conservation, (especially in the MID group) and the PPT features, including both the PPT length and the relative distance of the PPT to the splice site. Interestingly, the effect of removing either the PPT or the intronic conservation feature sets from the FAR group was very similar, suggesting that they are both important in the latter group. In contrast, the removal of the conservation set from the MID group had a remarkable effect compared to the removal of the PPT set, suggesting that in this subset the contribution of each of the parameters to the learning process is different. This result could be due to the fact that the MID group includes the borderline sequences and may represent a mixed distribution. In the CLOSE group, the most significant change in the AUC value arose when the PPT features were eliminated. In addition, we observed a lower reduction in SVM performance when eliminating each of the other features from the CLOSE classifier. This indicates that in the CLOSE group (different from the FAR and MID group) the learning process is mostly ruled by a unique feature set. In the NAGNAG-proximal group, the most notable features were the splice sites followed by the intronic conservation. This is in agreement with the statistical analysis results of the current study and previous observations (10,13). Overall, the feature selection test was consistent with the statistical analysis. Nevertheless, the latter results reinforce that the differences between AS and CS do not rely on unique parameters, but rather a combination of several sequence features.
Figure 5.

ΔAUC values for the different features sets are plotted for the FAR (black), MID (red), CLOSE (green) and NAGNAG-proximal (blue) groups. The feature sets are splice sites (SS), intronic conservation (CON), polypyrimidine tract (PPT), pseudo splice sites (PSE) and GC content (GC).
DISCUSSION
In an attempt to understand the regulation of alternative splice site selection, we have conducted a comprehensive analysis of alternative acceptor (AA) pairs separated by a range of distances. In order to concentrate on functional AA, we have restricted our study to alternative splicing events conserved between humans and mice (1). Applying both classical statistics and machine-learning approaches, we demonstrate that a combination of splicing canonical elements found in the introns show major variations between alternative and constitutive acceptors. Most importantly, we find that the ensemble of properties which distinguish between alternative and constitutive splicing strongly depends on the distance between the spice sites.
In agreement with previous work, the splice site composition was found to contribute mostly to SVM performance when splice sites are placed in tandem, as in the NAGNAG motif (10,13,40). In addition, we found considerable contribution of the intronic evolutionary conservation levels flanking the NAGNAG motif in comparison to the NAGNAG-proximal control set, but not compared to the NAGNAG-distal set. This is consistent with the high conservation levels that were previously observed both upstream of alterative spliced NAGNAGs and upstream of NAGNAG motifs in which the distal splice site is constitutively chosen (10). Furthermore, in this study we observed that when the splice sites are placed nearby (4–12 nt), but not in tandem, there is a relatively high evolutionary conservation level both upstream of alternative splice sites and PAs. In both cases, the conservation was higher than the background conservation level found upstream of constitutive splice sites. The similarity in the intronic properties flanking AA and CA/PA pairs when the splice sites are relatively close was reinforced by the relative low contribution of the intronic conservation feature to the SVM performance in the CLOSE group. Nevertheless, when the distance between the splice sites increased (MID, FAR), we found that elimination of the intronic conservation features strongly affected the SVM performance. It is known that two AG sites placed in close proximity are highly competitive (6,7,9); hence, the relatively high intronic conservation levels observed upstream AA and CA/PA pairs could be indicative of regulatory elements important to avoid the selection of alternative (or pseudo) AG sites that by default would be preferred by the splicing machinery (6,7). These results are consistent with recent work by Wang et al., which observed high levels of regulatory elements in the exonic regions between competitive splice sites (29).
In accordance with a recent report (10,25), here we have also observed that the most important feature to discriminate between alternative and constitutive acceptors was the PPT. PPT-related features were statistically significant in the CLOSE, MID and FAR groups and were found to play a significant role when combined with other features during the learning process. The fact that removing the PPT had a lesser effect on SVM performance in the MID and FAR compared to the CLOSE group could be related to compensation by other features such as the intronic evolutionary conservation. A striking observation in the current study was the high frequency of PPTs overlapping the proximal splice site, which was predominately observed in AA pairs. This observation coincides with previous studies describing the existence of two PPTs upstream AA pairs separated by ≥8 nt. Different from AAs, pseudo acceptors were found to be mostly located between, but not overlapping two PPTs. This could indicate a mechanism by which the two PTB-binding sites mediate looping-out of the pseudo splice sites. Although the looping out mechanism was originally suggested to explain alternative splice selection (37,38) our data imply that it could also be involved in avoiding the selection of pseudo splice sites. The high occurrence of AA pairs in which the proximal splice site is located inside the PPT suggests an important contribution of the PPT to the regulation of proximal splice site selection. This is supported by previous studies describing the competition between the splice factor U2AF and PTB for binding to the PPT (35) and the involvement of both factors in the regulation of alternative 3′ss (36). Moreover, it was observed that most of the disease related de novo 3′splice sites are found within the PPT (17), indicating that the overlap between the PPT and the 3′SS enhances the likelihood of an AG site to be chosen. Further experimental analysis will be needed to uncover the effect of the overlap between the splice site and the PPT on alternative 3′ss selection.
In summary, this study supplies further evidence of the involvement of basal splicing elements in the regulation of alternative splicing. Overall, our results suggest that differences may exist in the regulation of splice site recognition depending on the distance to the neighboring splice site candidates. Generally our findings, which are based on a bioinformatics analysis of only human–mouse conserved AA are in agreement with several experimental studies which have demonstrated that the proximity between AG pairs can affect splice site selection (6–8).
Availability
The package 3pred including the program for predicting AA vs CA/PA can be downloaded from: http://biology.technion.ac.il/facultywebsites/Yael/database.html
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We would like to thank Manny Ares for valuable comments on the manuscript. Thanks to Omer Haber for computer assistance. This work was supported by the ‘Mallat family fund’ and by the Israeli Science Foundation Grant number 923/05. Funding to pay the Open Access publication charges for this article was provided by ISF grant number 923/05.
Conflict of interest statement. None declared.
REFERENCES
- 1.Sugnet CW, Kent WJ, Ares M, Jr, Haussler D. Transcriptome and genome conservation of alternative splicing events in humans and mice. Pac. Symp. Biocomput. 2004;2004:66–77. doi: 10.1142/9789812704856_0007. [DOI] [PubMed] [Google Scholar]
- 2.Black DL. Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 2003;72:291–336. doi: 10.1146/annurev.biochem.72.121801.161720. [DOI] [PubMed] [Google Scholar]
- 3.Shapiro MB, Senapathy P. RNA splice junctions of different classes of eukaryotes: sequence statistics and functional implications in gene expression. Nucleic Acids Res. 1987;15:7155–7174. doi: 10.1093/nar/15.17.7155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yeo G, Burge CB. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 2004;11:377–394. doi: 10.1089/1066527041410418. [DOI] [PubMed] [Google Scholar]
- 5.Kay PH, Ziman MR. Alternate Pax7 paired box transcripts which include a trinucleotide or a hexanucleotide are generated by use of alternate 3′ intronic splice sites which are not utilized in the ancestral homologue. Gene. 1999;230:55–60. doi: 10.1016/s0378-1119(99)00049-9. [DOI] [PubMed] [Google Scholar]
- 6.Smith CW, Chu TT, Nadal-Ginard B. Scanning and competition between AGs are involved in 3′ splice site selection in mammalian introns. Mol. Cell. Biol. 1993;13:4939–4952. doi: 10.1128/mcb.13.8.4939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chua K, Reed R. An upstream AG determines whether a downstream AG is selected during catalytic step II of splicing. Mol. Cell. Biol. 2001;21:1509–1514. doi: 10.1128/MCB.21.5.1509-1514.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lev-Maor G, Sorek R, Shomron N, Ast G. The birth of an alternatively spliced exon: 3′ splice-site selection in Alu exons. Science. 2003;300:1288–1291. doi: 10.1126/science.1082588. [DOI] [PubMed] [Google Scholar]
- 9.Gooding C, Clark F, Wollerton MC, Grellscheid SN, Groom H, Smith CW. A class of human exons with predicted distant branch points revealed by analysis of AG dinucleotide exclusion zones. Genome Biol. 2006;7:R1. doi: 10.1186/gb-2006-7-1-r1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Akerman M, Mandel-Gutfreund Y. Alternative splicing regulation at tandem 3′ splice sites. Nucleic Acids Res. 2006;34:23–31. doi: 10.1093/nar/gkj408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kol G, Lev-Maor G, Ast G. Human-mouse comparative analysis reveals that branch-site plasticity contributes to splicing regulation. Hum. Mol. Genet. 2005;14:1559–1568. doi: 10.1093/hmg/ddi164. [DOI] [PubMed] [Google Scholar]
- 12.Zhuang Y, Weiner AM. The conserved dinucleotide AG of the 3′ splice site may be recognized twice during in vitro splicing of mammalian mRNA precursors. Gene. 1990;90:263–269. doi: 10.1016/0378-1119(90)90189-x. [DOI] [PubMed] [Google Scholar]
- 13.Chern TM, van Nimwegen E, Kai C, Kawai J, Carninci P, Hayashizaki Y, Zavolan M. A simple physical model predicts small exon length variations. PLoS Genet. 2006;2:e45. doi: 10.1371/journal.pgen.0020045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hiller M, Huse K, Szafranski K, Jahn N, Hampe J, Schreiber S, Backofen R, Platzer M. Widespread occurrence of alternative splicing at NAGNAG acceptors contributes to proteome plasticity. Nat. Genet. 2004;36:1255–1257. doi: 10.1038/ng1469. [DOI] [PubMed] [Google Scholar]
- 15.Wu S, Romfo CM, Nilsen TW, Green MR. Functional recognition of the 3′ splice site AG by the splicing factor U2AF35. Nature. 1999;402:832–835. doi: 10.1038/45590. [DOI] [PubMed] [Google Scholar]
- 16.Gaur RK, Beigelman L, Haeberli P, Maniatis T. Role of adenine functional groups in the recognition of the 3′-splice-site AG during the second step of pre-mRNA splicing. Proc. Natl Acad. Sci. USA. 2000;97:115–120. doi: 10.1073/pnas.97.1.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kralovicova J, Christensen MB, Vorechovsky I. Biased exon/intron distribution of cryptic and de novo 3′ splice sites. Nucleic Acids Res. 2005;33:4882–4898. doi: 10.1093/nar/gki811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chua K, Reed R. The RNA splicing factor hSlu7 is required for correct 3′ splice-site choice. Nature. 1999;402:207–210. doi: 10.1038/46086. [DOI] [PubMed] [Google Scholar]
- 19.Coolidge CJ, Seely RJ, Patton JG. Functional analysis of the polypyrimidine tract in pre-mRNA splicing. Nucleic Acids Res. 1997;25:888–896. doi: 10.1093/nar/25.4.888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Singh R, Valcarcel J, Green MR. Distinct binding specificities and functions of higher eukaryotic polypyrimidine tract-binding proteins. Science. 1995;268:1173–1176. doi: 10.1126/science.7761834. [DOI] [PubMed] [Google Scholar]
- 21.Pollard AJ, Krainer AR, Robson SC, Europe-Finner GN. Alternative splicing of the adenylyl cyclase stimulatory G-protein G alpha(s) is regulated by SF2/ASF and heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1) and involves the use of an unusual TG 3′-splice Site. J. Biol. Chem. 2002;277:15241–15251. doi: 10.1074/jbc.M109046200. [DOI] [PubMed] [Google Scholar]
- 22.Baek D, Green P. Sequence conservation, relative isoform frequencies, and nonsense-mediated decay in evolutionarily conserved alternative splicing. Proc. Natl Acad. Sci. USA. 2005;102:12813–12818. doi: 10.1073/pnas.0506139102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dror G, Sorek R, Shamir R. Accurate identification of alternatively spliced exons using support vector machine. Bioinformatics. 2005;21:897–901. doi: 10.1093/bioinformatics/bti132. [DOI] [PubMed] [Google Scholar]
- 24.Wang M, Marin A. Characterization and prediction of alternative splice sites. Gene. 2006;366:219–227. doi: 10.1016/j.gene.2005.07.015. [DOI] [PubMed] [Google Scholar]
- 25.Xia H, Bi J, Li Y. Identification of alternative 5′/3′ splice sites based on the mechanism of splice site competition. Nucleic Acids Res. 2006;34:6305–6313. doi: 10.1093/nar/gkl900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sorek R, Ast G. Intronic sequences flanking alternatively spliced exons are conserved between human and mouse. Genome Res. 2003;13:1631–1637. doi: 10.1101/gr.1208803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sugnet CW, Srinivasan K, Clark TA, O’Brien G, Cline MS, Wang H, Williams A, Kulp D, Blume JE, et al. Unusual intron conservation near tissue-regulated exons found by splicing microarrays. PLoS Comput. Biol. 2006;2:e4. doi: 10.1371/journal.pcbi.0020004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang J, Smith PJ, Krainer AR, Zhang MQ. Distribution of SR protein exonic splicing enhancer motifs in human protein-coding genes. Nucleic Acids Res. 2005;33:5053–5062. doi: 10.1093/nar/gki810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Z, Xiao X, Van Nostrand E, Burge CB. General and specific functions of exonic splicing silencers in splicing control. Mol. Cell. 2006;23:61–70. doi: 10.1016/j.molcel.2006.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aebi M, Hornig H, Padgett RA, Reiser J, Weissmann C. Sequence requirements for splicing of higher eukaryotic nuclear pre-mRNA. Cell. 1986;47:555–565. doi: 10.1016/0092-8674(86)90620-3. [DOI] [PubMed] [Google Scholar]
- 31.Lim LP, Burge CB. A computational analysis of sequence features involved in recognition of short introns. Proc. Natl Acad. Sci. USA. 2001;98:11193–11198. doi: 10.1073/pnas.201407298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Westfall PH, Young SS. On adjusting p-values for multiplicity. Biometrics. 1993;49:941–944. [Google Scholar]
- 33.Byvatov E, Schneider G. Support vector machine applications in bioinformatics. Appl. Bioinformatics. 2003;2:67–77. [PubMed] [Google Scholar]
- 34.Sorek R, Shamir R, Ast G. How prevalent is functional alternative splicing in the human genome? Trends Genet. 2004;20:68–71. doi: 10.1016/j.tig.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 35.Sauliere J, Sureau A, Expert-Bezancon A, Marie J. The polypyrimidine tract binding protein (PTB) represses splicing of exon 6B from the beta-tropomyosin pre-mRNA by directly interfering with the binding of the U2AF65 subunit. Mol. Cell. Biol. 2006;26:8755–8769. doi: 10.1128/MCB.00893-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin CH, Patton JG. Regulation of alternative 3′ splice site selection by constitutive splicing factors. RNA. 1995;1:234–245. [PMC free article] [PubMed] [Google Scholar]
- 37.Amir-Ahmady B, Boutz PL, Markovtsov V, Phillips ML, Black DL. Exon repression by polypyrimidine tract binding protein. RNA. 2005;11:699–716. doi: 10.1261/rna.2250405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oberstrass FC, Auweter SD, Erat M, Hargous Y, Henning A, Wenter P, Reymond L, Amir-Ahmady B, Pitsch S, et al. Structure of PTB bound to RNA: specific binding and implications for splicing regulation. Science. 2005;309:2054–2057. doi: 10.1126/science.1114066. [DOI] [PubMed] [Google Scholar]
- 39.Dou Y, Fox-Walsh KL, Baldi PF, Hertel KJ. Genomic splice-site analysis reveals frequent alternative splicing close to the dominant splice site. RNA. 2006;12:2047–2056. doi: 10.1261/rna.151106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hiller M, Huse K, Szafranski K, Jahn N, Hampe J, Schreiber S, Backofen R, Platzer M. Single-nucleotide polymorphisms in NAGNAG acceptors are highly predictive for variations of alternative splicing. Am. J. Hum. Genet. 2006;78:291–302. doi: 10.1086/500151. [DOI] [PMC free article] [PubMed] [Google Scholar]