


Abstract
T4 DNA ligase is used in standard cyclization assays to trap double-stranded DNA (dsDNA) in low-probability, cyclic or highly bent conformations. The cyclization probability, deduced from the relative yield of cyclized product, can be used in conjunction with statistical mechanical models to extract the bending stiffness of dsDNA. By inserting the base analog 2-aminopurine (2-AP) at designated positions in 89 bp and 94 bp dsDNA fragments, we find that T4 DNA ligase can have a previously unknown effect. Specifically, we observe that addition of T4 ligase to dsDNA in proportions comparable to what is used in the cyclization assay leads to a significant increase in fluorescence from 2-AP. This effect is believed to originate from stabilization of local base-pair opening by formation of transient DNA-ligase complexes. Non-specific binding of T4 ligase to dsDNA is also confirmed using fluorescence correlation spectroscopy (FCS) experiments, which reveal a systematic reduction of dsDNA diffusivity in the presence of ligase. ATP competes with regular DNA for non-covalent binding to the T4 ligase and is found to significantly reduce DNA-ligase complexation. For short dsDNA fragments, however, the population of DNA-ligase complexes at typical ATP concentrations used in DNA cyclization studies is determined to be large enough to dominate the cyclization reaction.
INTRODUCTION
In eukaryotic cells, genomic sequences are repetitively and compactly packaged into nucleosomes where they are in close association with histone proteins. In this setting, histone proteins and DNA form tightly knit complexes that sterically occlude DNA from interacting with other proteins (1). The stability of the nucleosome controls nearly all DNA-templated biological process, and can be partly modulated using intrinsic regulation mechanisms that rely upon the ability of DNA sequences to create well-ordered, reproducible nucleosome structures (2,3). In other words, the nucleosome stability, and consequently protein access, is likely encoded in the DNA sequence itself through sequence-dependent DNA bending flexibility, which is inversely related to the energetic cost of forming the nucleosome particles.
A variety of experimental tools have been developed to investigate DNA bending flexibility (4–6), including comparative gel electrophoresis, crystallography, electron microscopy, transient electric birefringence and DNA cyclization, as well as more recent single molecule manipulation techniques (7–9). DNA cyclization has distinguished itself from most other approaches by its simplicity, robust theoretical foundation, high sensitivity and accuracy, and the fact that it can be used to provide a simple indication of the local chain stiffness (10–13). In a typical DNA cyclization experiment, DNA is designed with ligatable single-stranded ends. T4 DNA ligase is added to initiate the cyclization reaction. The ratio of equilibrium constants for ligatable unimoleuclar and bimolecular forms are measured under precise conditions to determine the so-called Jacobson-Stockmayer factor, J-factor. The experimentally obtained J are then fitted to an analytical or numerical model, e.g. the continuous worm-like chain model (WLC) (14–19), to obtain the bending flexibility of the particular sequence used.
Recent cyclization data reported by Cloutier and Widom (20,21), have produced much excitement in the literature because they indicate that short (<100 bp) double stranded DNAs (dsDNA) repeats are dramatically more flexible than anticipated by any current model for DNA bending. Marko and co-workers performed a follow-up theoretical study to explain Cloutier and Widom's experimental results. These authors found that the unusually high cyclization efficiencies reported can be reproduced by a kinkable worm like chain (KWLC) model for the DNA analyte (22–24), though the fundamental origin of kinks in the analytes used in the experiments remains unknown. An even more recent study by Vologodskii and co-workers (25) has challenged the enhanced DNA flexibility reported by Cloutier and Widom's experiments on different grounds. These authors contend that the exceptionally high cyclization efficiency reported is correlated with the higher than normal T4 ligase concentration used in the measurements.
Yuan and co-workers (26) reported a Fluorescence Resonance Energy Transfer (FRET)-based technique to measure the probability distribution of short dsDNA fragments with and without base pair mismatches. These authors observed that the presence of local unpaired bases in the dsDNA fragment leads to dramatic increases in the probability of highly bent, kinked states implying that local base unpairing can produce the kinked states postulated by Marko and co-workers.
Here we utilize fluorescence measurements to test a key assumption about T4 DNA ligase used in cyclization measurements. Specifically, in cyclization measurements T4 ligase is used to perform a single task. Namely, to efficiently trap low-probability, highly bent DNA conformations in which the 3′ hydroxyl end of one dsDNA molecule approaches the 5′ phosphorylated end of the same molecule. The reports by Vologodskii et al. and by Yuan et al. motivate a perhaps obvious question: Does T4 ligase always restrain itself to the assigned task? To answer this question, DNA molecules containing base analog, which can be used as indicators of the base pair stacking status, are used to evaluate the impact of ligase on DNA conformation under the conditions used for cyclization studies reported in references (20,21,25).
MATERIALS AND METHODS
DNA samples
DNA oligos with 2-AP inserted at designated positions were custom synthesized by Integrated DNA technologies (Coraville, IA). All oligos used in the study were purified by the manufacturer using high performance liquid chromatography (HPLC); oligo sequences are detailed in the Supplementary Data section, 1a. Unless otherwise specified, the 5′ and 3′ ends of the oligos are comprised of hydroxyl groups. dsDNA were produced by the standard annealing procedure (26). For samples containing 2-AP, the ratio of the oligo containing 2-AP to the complementary strand is maintained at 3:4 to minimize the relative amount of 2-AP in the non-helical states. The composition of dsDNA is summarized in the Supplementary Data, Section 1b. All dsDNA fragments contain mutually complementary 4 nt overhangs at both ends.
Fluorescence measurements
Fluorescence excitation and emission spectra were collected using a fluorophotometer (PTI inc) with a 76 mW source power, 5 nm slit width, and integration time of 0.5 s. Quartz fluorometer cells with 3 mm path length (Starna Cells Inc., CA) were used for all measurements. All spectra reported were corrected in real-time for lamp fluctuations. The fluorescence measurement buffer contains 50 mM Tris-HCl, 10 mM MgCl2, 10 mM DTT and 25 μg/ml BSA. The buffer conditions used here are similar to those employed in the standard cyclization assay, except for the absence of ATP (21,25). To evaluate the effect of T4 DNA ligase on 2-AP fluorescence, solutions containing DNA and T4 DNA ligase (New England Biolabs, MA) at various concentrations were made-up and mixed using a gentle vortexing procedure; solutions were stabilized at room temperature for at least 30 min prior to the measurements. The concentration of T4 DNA ligase is provided by the manufacturer in New England Biolab (NEB) units. The equivalent molar concentration can be quantified using Coomassie Plus Assay (Pierce Biotech, IL). Based on this procedure, the NEB units can be converted to standard molar concentrations using the relation, 1 unit/ml = 0.02 nM (±25%). It should be noted that the original NEB ligase contains 200 μg/ml BSA, the measured ligase concentration must therefore be corrected by subtracting the BSA contribution.
Characteristic diffusion times of DNA molecules in the presence of T4 ligase at various concentrations were determined using a home-built fluorescence correlation spectroscopy (FCS) apparatus (27–29). In a typical FCS measurement, fluctuations in the fluorescence intensity are quantified by temporally autocorrelating the intensity signal. Samples containing a fixed concentration of DNA (25 nM) end-labeled with fluorescein were excited with a 488 nm laser, such that only fluorescently labeled DNA molecules or their complexes are visible under the microscope. The recorded autocorrelation function G(τ) was analyzed using well-known procedures (27,29). If the sample contains only one fluorescent diffusive species, G(τ) can be written as:
| (1) |
where τD is the diffusion time, N is the number of fluorescent molecules within focal volume, and ω is the ratio of the axial/radial dimensions of the observation volume. The value of ω was characterized using Alexa 488 dye before the measurements, and is taken to be a constant for the subsequent analysis.
When the ligase and DNA coexists in solution, DNA-ligase complexes contribute to G(τ) as a second diffusive species. The autocorrelation function can therefore be written as,
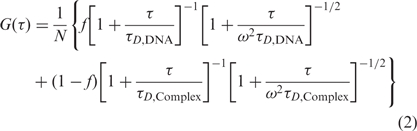 |
(2) |
where f is the fraction of the total labeled DNA complexed with ligase.
The characteristic diffusion time for DNA (τD,DNA) can be easily determined from FCS control experiments in which no ligase is added. In the presence of ligase, the autocorrelation function is analyzed with a ‘two-diffusive-species’ model (Equation 2), in which the diffusion time of DNA (τD,DNA) is fixed at the value obtained from the control experiment.
To probe the effect of ligase concentration on DNA diffusion, DNA solutions in buffer containing ligase concentrations ranging from 500 units/ml to 4000 units/ml were employed in the FCS experiments. It is possible that changes in the ligase concentration in the medium can in of itself alter the medium viscosity, and hence the apparent diffusivity of DNA. To quantify this effect, control experiments were performed in which the diffusivity of labeled dsDNA was determined in media containing BSA (MW 67 kDa) at concentrations equivalent to those used for the T4 ligase (MW 55.3 kDa) studies. These measurements reveal no change in the characteristic diffusion time of DNA upon addition of BSA, indicating that the ligase concentrations used in the study are low-enough that trivial changes to the medium viscosity produced by the ligase itself can be ignored in analyzing DNA diffusion.
RESULTS AND DISCUSSION
2-AP as an indicator of DNA base pair flip-out
The nucleotide 2-AP is a fluorescent analog of guanosine and adenosine and has been used as a site-specific probe of nucleic acid structure and dynamics (30,31). Fluorescence emission from 2-AP can be readily excited at a wavelength of 320 nm, far away from any DNA absorption, or absorption due to DNA-protein complexes. This feature allows selective excitation of fluorescence from the 2-AP base even in the presence of protein residues. Significantly, the fluorescence emission from a 2-AP nucleotide is highly quenched in the stacked state, and its quantum yield increases approximately 10-fold in the un-stacked state (32,33). Local conformational changes, especially stacking status changes, can therefore be assessed from the fluorescence of 2-AP, i.e. the 2-AP nucleotide manifests enhanced fluorescence intensity when assuming an extra helical conformation (see inset of Figure 1) (32–37). By selectively exciting the sample at 320 nm, the fluorescence intensity increase can be used to report the population of spontaneously ‘flipped-out’ bases.
Figure 1.

Fluorescence emission spectra for E89-23 and E89-16. Inset: emission spectra for 89 bp DNA containing 2-AP inserted in single stranded (oligo) and double stranded (E89-23) DNA fragments. The dsDNA concentration is 1 μM.
To demonstrate the sensitivity of these measurements, dsDNA containing 2-AP inserted in different sequence contexts were synthesized. In the E89-23 sample (see Supplementary Data 1a), 2-AP is inserted in an AT rich region, while for E89-16 2-AP is located within a random genomic sequence context. The fluorescence emission spectra collected when the two DNA samples, at a concentration of 1.0 μM DNA in buffer, are excited at 320 nm are reported in Figure 1. The background emission was independently measured using the neat buffer (i.e. in the absence of DNA), and subtracted from the sample spectra. It is apparent from the figure that dsDNA containing 2-AP in the AT rich region (E89-23) exhibits substantially higher fluorescence than the sample where 2-AP is inserted in the random sequence context (E89-16). Increasing the complementary oligo concentration, which does not contain the 2-AP insertion, has virtually no effect on the fluorescence intensity, indicating that the global thermal stability of the as formed dsDNA does not contribute to the observed fluorescence increase. Figure 1 then simultaneously shows that even in the absence of ligase some 2-AP bases are quite exposed and the effect is dramatically larger for ‘softer’ A-T rich sequence domains.
The difference of 2-AP fluorescence within different sequence contexts can originate from basic differences in the energy transfer mechanism between stacked bases [E89-23: (AApT); E89-16: (GApT)], (36–38), as well as from differences in the rate of spontaneous base un-pairings, the so-called base flip-out rate. The relatively low base pairing and stacking energies within the AT rich region [the stacking energy difference between E89-23 and E89-16 is estimated to be around 0.2 ∼ 0.3 kcal/mol for regular AA and GA dinucleotides, (39)], have been argued to make it easier for a single nucleotide to bypass the energetic barrier, and spontaneously un-pair with its complement (39–41). It has been demonstrated through extensive studies, that the fluorescence of 2-AP can be used as a sensitive reporter of the enhanced base pair un-pairing that would result from such events (31–34).
T4 DNA ligase and its effect on DNA conformation
Local base pair flip-out or local defects in short dsDNA fragments can contribute significantly to the measured J-factors deduced from cyclization measurements. Here we wish to determine what influence, if any, other species used in these measurements might have on such defects, and thereby the apparent bending stiffness reported by the measurements. The ATP dependent ligation reaction involves three principal steps: (i) Activation of the enzyme through formation of a covalent protein-AMP intermediate, accompanied by release of PPi. (ii) Transfer of the nucleotide to a phosphorylated 5′-end of the nick or the sticky end to produce an inverted (5′)-(5′) phyrophosphate bridge structure. (iii) Catalysis of the transesterification reaction resulting in the joining of the nick and release of free AMP (42,43). Three components are essential for the successful and timely completion of a ligation reaction, namely, the 5′ phosphorylated end and the presence of ATP and ligase. By controlling the relative abundance of these three elements, the effect of ligase on the DNA conformation at different stages of the reaction can be clarified. For these experiments, the same two 89 bp dsDNA, containing 2-AP as in the previous experiments is used. The 5′ ends of the dsDNA are unphosphorylated to prevent ligation, so the molecular size remains unchanged during the measurement. Fluorescence measurements performed using the buffer containing an equal concentration of ligase are used to quantify any background effects, which are subtracted from the raw spectra to isolate the effect of T4 ligase concentration on DNA fluorescence.
Figure 2a illustrates the effect of increasing the amount of T4 DNA ligase on the fluorescence emission intensity of E89-23. DNA concentration is maintained constant, while the ligase concentration is gradually increased. Background effects are subtracted from all spectra using the procedure outlined in the last section. The spectra shown are averages from at least two runs. It is apparent from the data that as the ligase concentration is increased the fluorescence of 2-AP manifests a corresponding increase. To confirm our findings, excitation spectra are independently measured on the same DNA samples containing varying concentration of ligase. The DNA solution without ligase is treated as background during these measurements, and the final spectra are obtained by subtracting the respective background spectra from the measured sample excitation spectra. The excitation spectra collected at a fixed emission wavelength of 370 nm, should therefore solely reflect any changes to 2-AP fluorescence induced by ligase. Figure 2b summarizes the main results. It is apparent from this figure that with increased ligase concentration, the protein excitation peak at 280 nm increases, as expected. However, the secondary excitation peak centered at around 310 nm, which originates from direct excitation of 2-AP also changes from insignificant to significant with increasing ligase concentration. This effect has been confirmed using a larger DNA fragment, 94 bp (E94-25), indicating that the observation is quite general. Control experiments using 2-AP inserts in single-stranded DNA indicate that T4 ligase does not induce any changes in 2-AP fluorescence in single stranded DNA. This last observation is significant because it eliminates the trivial possibility that the presence of ligase might change some bulk property of the medium (e.g. its dielectric constant) and thereby alter the quantum yield of 2-AP.
Figure 2.

(a) Fluorescence emission spectra for E89-23 at various ligase concentrations. A fixed excitation wavelength (320 nm) is used throughout. Emission spectra for DNA-free buffer solutions containing equal amounts of ligase are taken as background and subtracted from the measured fluorescence spectra. (b) Excitation spectra of E89-23 at various ligase concentrations. Fluorescence emission is collected at a fixed wavelength of 370 nm, and buffer solutions containing equal amounts of DNA is taken as the background and subtracted from the measured spectra.
These observations can be interpreted either in terms of a higher base pair flip-out rate induced by the ligase or in terms of an increase in the population of dsDNA containing extra helical 2-AP nucleotides induced by the presence of DNA ligase. Either mechanism can in principle provide a physical source for the ‘kinked’ states postulated by Marko and co-workers (22–24), and as such are tempting candidates to explain cyclization data revealing enhanced dsDNA flexibility. It must be kept in mind, however, that the DNA concentration employed in our measurements is 100–1000-fold larger than those normally used in cyclization experiments, and that the ligase concentration is substantially higher than the 100 units/ml value recommended by Du et al. as a standard example for studies of very short DNA. Implications of both effects (higher DNA concentration and higher than recommended ligase concentration) will be addressed in detail in subsequent sections of the article.
The relative enhancement in base pair flip-out, or equivalently the increase in the population of DNA containing local defects, can be directly related to the increase in the fluorescence emission intensity (Figure 3). Since the quantum yield of 2-AP in its non-stacking status is almost 10 times higher than in the stacked status, the relative amount of DNA with ligase induced 2-AP flip-out, i.e. base pair flip-out, can be estimated to be around one-tenth of the fluorescence emission intensity increase, i.e. the fractional increase referenced to the zero-ligase control. The observed ligase-induced fluorescence enhancement evidently does not exhibit a significant length dependence, which implies that it originates from the general destabilization of bases along the DNA backbone. From the slope of the fitted line in Figure 3, it is also possible to estimate that the enzyme binds to the 2-AP site or 2-AP containing region with Kd ≈ 50 μM. The apparent Kd for free state ATP, which is structurally very similar to the 2-AP nucleotide, in forming the non-convalent ligase-ATP complex is reported to be below 0.15 μM.44 The nearly three orders difference in Kd means that the inserted 2-AP nucleotide is well incorporated inside the helix.
Figure 3.

Enhancement of 2-AP fluorescence emission as a function of ligase concentration; [DNA] = 1 μM.
Results from other experiments using a dsDNA sample in which the 5′ end of the dsDNA is phosphorylated (E89-23p), but in the absence of ATP to prevent further ligation are presented in Figure 4. The results for E89-23p show similar increases in fluorescence intensity from dsDNA (E89-23p) as the ligase concentration is gradually increased. A noteworthy feature of all of these experiments is that as [Ligase] increases, the enhancement in 2-AP fluorescence seen in the presence of ligase becomes larger.
Figure 4.

Fluorescence emission spectra of E89-23p at various ligase concentrations, fluorescence are excited at 320 nm. Emission spectra for buffer solutions containing equal amounts of ligase are taken as background and subtracted from the measured spectra.
It is well known that to initiate the ligation reaction, ATP must be present in the reaction buffer as an essential cofactor. Specifically, ATP is known to be essential for triggering the first step of the ligation reaction by forming a transient AMP-ligase complex that can bind non-specifically to the DNA target. Also, ATP has been documented as a fluorescence quencher of 2-AP. However, for the low ATP concentrations used in this study, the characteristic emission of the 2-AP is anticipated to be nearly unaffected in the presence of ATP (38). Fluorescence measurements performed by simply adding ATP to dsDNA with zero ligase concentration, in fact reveal no measurable effect of ATP on the characteristic fluorescence excitation or emission spectra.
Addition of ATP to dsDNA solutions in buffers containing T4 ligase produces a very different effect. This effect is illustrated in Figure 5 where the effect of ATP concentration on ligase-enhanced 2-AP fluorescence is shown. To avoid the potential complication of molecular size change due to the ligation, E89-23 which has hydroxyl groups on both 5′ and 3′ ends are used for these measurements. The fluorescence background is obtained using buffer solutions containing the same concentration of DNA and ATP and subtracted from the sample spectra. It is apparent from Figure 5 that the addition of ATP to E89-23 solutions containing ligase gradually reverses the effect of the ligase on 2-AP fluorescence.
Figure 5.

Emission spectra for E89-23 at various ATP concentrations, Fluorescence are excited at 320 nm. Emission spectra for buffer solutions containing equal amounts of DNA and ATP are taken as background and subtracted from the measured spectra.
The crystal structure of DNA ligases have been studied by several groups (45–47). These studies reveal highly modular structures, all with preserved nucleotide-binding domains or an oligomer-binding (OB) fold. This DNA binding domain is generally positively charged and is thus capable of stabilizing the dsDNA molecules containing transiently unpaired bases by binding non-specifically to the DNA. The ATP binding domain is generally postulated to be close to the DNA binding domain (45). The diminishing ‘ligase-enhanced’ fluorescence observed after ATP addition suggests that ATP competes with DNA for binding to the ligase. Two plausible mechanisms could explain the observed phenomena. One is that after ATP binds to the ligase, it triggers a conformational change of the protein and thus precludes the ligase from acting as a stabilizing agent for spontaneously unpaired bases in the dsDNA fragments. The other is that ATP binding enhanced the specific binding of ligase towards its end and thus makes the internal binding on DNA backbone less favorable.
Effect of T4 DNA ligase on diffusivity of DNA
From the emission spectra of 2-AP incorporated inside DNA, it already seems clear that T4 DNA ligase is capable of enhancing the 2-AP base flip out, although 2-AP is generally well incorporated inside the double helix and does not affect the conformation of B-type DNA. It nonetheless remains unclear whether the existence of this transient DNA-ligase complex is, not only demonstrated but also, somehow triggered by the 2-AP incorporation. To answer this question, we performed independent FCS measurements to determine what effect, if any, T4 DNA ligase has on the transport properties of the dsDNA fragments without the 2-AP insertion. If, as argued above, T4 ligase enhances base-pair flip out by non-specific binding to dsDNA, formation of the postulated DNA-ligase complex should be reflected as a reduction in the translation diffusivity of the dsDNA fragments measured in the presence of ligase. This effect should be above and beyond any trivial reduction of DNA diffusivity produced by the ligase-induced enhancement of the bulk viscosity of the medium.
FCS measurements were performed at 25°C using a fluorescently labeled 89 bp non-phosphorylated DNA, without 2-AP insertion. The inset in Figure 6 illustrates the effect of ligase concentration on the characteristic diffusion time deduced from the FCS data by fitting the fluorescence correlation function for a single diffusing species. It is evident from the figure that the average diffusion time of the DNA is, significantly increased by the addition of ligase. Furthermore, as the concentration of ligase is increased, the diffusion time at first increases quite strongly but then manifests a weaker but steady increase with ligase concentration, over nearly four decades of concentration. The apparent ligase-induced slowing down of DNA diffusion is consistent, at least qualitatively, with formation of the DNA-ligase complexes hypothesized on the basis of the 2-AP fluorescence data. We emphasize here that the DNA specimen used for FCS measurements do not contain 2-AP, demonstrating that the insertion of 2-AP in the previous fluorescence experiments does not create preferential binding site for the ligase. Also, essentially identical results are obtained in similar experiments using 89 bp DNA sample without 2-AP insertion or 4 nt dangling ends, which indicates that T4 DNA ligase does not preferentially bind to the dangling end of the DNA.
Figure 6.

Relative ratio of DNA-ligase complex to DNA as a function of ligase concentration determined by analyzing the autocorrelation function using two diffusing species. Inset: characteristic diffusion time of DNA, obtained by analyzing the autocorrelation function using a single diffusive species model, as a function of ligase concentration.
More careful scrutiny of the fluorescence correlation function indicates that addition of ligase does not uniformly slow down diffusion of the DNA. Specifically, by fitting the correlation function data to a model for two diffusing species, the ligase is found instead to produce a second slow-diffusing DNA (only DNA is fluorescently labeled) component with a slower characteristic time τD;slow, that increases as the ligase concentration is increased. Significantly, even in buffers with high ligase concentration, the two species model indicates that there is always a significant population of DNA that remains effectively unaffected by the ligase (i.e. τD;fast ≈ τD;DNA, irrespective of [Ligase]).
Considering the low concentration of DNA used for the measurements the relative abundance of the slower, presumably ligase-complexed, species can be estimated as the relative ratio of slow diffusion (f) species. Figure 6 shows the effect of ligase concentration on f calculated in this manner. The slope of the straight-line fits to the data is inversely related to
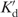 , the dissociation constant of the DNA-ligase complex. At the lowest ligase concentration (i.e. in the range where DNA bending studies are performed using cyclization) we find
≈ 1.5 μM, while at higher ligase concentrations we obtain
≈ 15 μM. These values are, respectively, 30-times and 3-times smaller than the Kd value calculated previously using the 2-AP fluorescence data. Taken at face value, they suggest that the DNA-ligase complexes are most stable at the lowest ligase concentrations. These conclusions obviously overlook the complicating influence of multiple ligase molecules binding to a single DNA fragment. Specifically, Kd as calculated previously, reflects the dissociation constant when ligase is bound ‘specifically’ to the 2-AP site or the 2-AP incorporated regions. To take into account the possibility of multiple binding sites on a single substrate, relative abundance data are conventionally analyzed using the Hill Function, p = [Ligase]n/(K + [Ligase]n).The solid line in Figure 6, is obtained by fitting this functional form to the experimental data. The fit is evidently not as good as the dual straight line fit, particularly at low [Ligase]. We nonetheless proceed to extract values of n ≈ 0.3 ± 0.1 and K ≈ (5 ± 16) × 109 from the Hill function fit to the data. A Hill coefficient, n, less than unity implies that the binding event is negatively cooperative, while the value of K has less physical meaning both due to its large uncertainty and the lack of a direct correlation with the binding constant for the reaction. Based on both analyses, we therefore conclude that ligase has a tendency to bind non-specifically to the DNA and to form a transiently stable complex with a dissociation constant estimated to be 2 – 25 μM, depending on the ligase concentration.
, the dissociation constant of the DNA-ligase complex. At the lowest ligase concentration (i.e. in the range where DNA bending studies are performed using cyclization) we find
≈ 1.5 μM, while at higher ligase concentrations we obtain
≈ 15 μM. These values are, respectively, 30-times and 3-times smaller than the Kd value calculated previously using the 2-AP fluorescence data. Taken at face value, they suggest that the DNA-ligase complexes are most stable at the lowest ligase concentrations. These conclusions obviously overlook the complicating influence of multiple ligase molecules binding to a single DNA fragment. Specifically, Kd as calculated previously, reflects the dissociation constant when ligase is bound ‘specifically’ to the 2-AP site or the 2-AP incorporated regions. To take into account the possibility of multiple binding sites on a single substrate, relative abundance data are conventionally analyzed using the Hill Function, p = [Ligase]n/(K + [Ligase]n).The solid line in Figure 6, is obtained by fitting this functional form to the experimental data. The fit is evidently not as good as the dual straight line fit, particularly at low [Ligase]. We nonetheless proceed to extract values of n ≈ 0.3 ± 0.1 and K ≈ (5 ± 16) × 109 from the Hill function fit to the data. A Hill coefficient, n, less than unity implies that the binding event is negatively cooperative, while the value of K has less physical meaning both due to its large uncertainty and the lack of a direct correlation with the binding constant for the reaction. Based on both analyses, we therefore conclude that ligase has a tendency to bind non-specifically to the DNA and to form a transiently stable complex with a dissociation constant estimated to be 2 – 25 μM, depending on the ligase concentration.
It must be noted that the FCS experiments are carried out under conditions with higher ligase concentration as well as with a reaction buffer without added ATP as compared with typical cyclization reaction. The higher ligase concentration is essential for resolving the slow diffusive species, and at lowest ligase concentration being explored, less than 1% of DNA forms complex by binding to the ligase. The effect of ATP is also examined in the FCS measurement. With 1 mM ATP present in the reaction buffer, the binding of the ligase to the DNA molecules is effectively suppressed, exhibiting constant diffusion times independent of the added ligase concentration as indicated in the inset of Figure 6.
Implications for DNA cyclization reactions
The findings reported in the last section suggest that by stabilizing transiently unpaired bases, T4 ligase can dramatically lower the bending stiffness of DNA. This effect is analogous to that produced local defects, e.g. base pair mismatches, gaps and bulges, on the bending stiffness of DNA (26,48–49). However, we have already reported that ATP can suppress such non-specific binding of ligase to the DNA backbone, suggesting that the effect can be reversed by employing a sufficiently large ATP concentration in cyclization measurements. To further evaluate this possibility, we now employ the thermodynamic constants obtained in the last section to determine the effect of ATP on the population of DNA containing defects. We begin by setting the ligase concentration to be around 100–400 units/ml (2–8 nM), as commonly used in the cyclization assay. At zero ATP concentration, 10−4–10−3 (
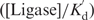 ) DNA contains local defects induced and/or stabilized by ligase binding. As the ATP concentration increases, the relative percentage decreases. For example, with 1 mM ATP, as in a typical ligation buffer [Kd,ATP = 0.15 μM, (39–40)], only 10−8–10−7
) DNA contains local defects induced and/or stabilized by ligase binding. As the ATP concentration increases, the relative percentage decreases. For example, with 1 mM ATP, as in a typical ligation buffer [Kd,ATP = 0.15 μM, (39–40)], only 10−8–10−7
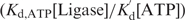 DNA contains the induced defects. This is a very small population of the DNA molecules, and its effect generally negligible when the length of DNA is much larger than its persistence length. However, as the length of DNA decreases, the population of cyclizable species reduces dramatically and finally approaches and becomes even smaller than this value. For example for the 89 bp DNA used in reference (15) the J-factor predicted by the WLC model, with a persistence length of 50 nm, is 10−15 M, which is equivalent to 10−12–10−11 of the total DNA molecules. Since DNA molecules containing local defects stabilized by ligase are expected to have a much higher tendency to be cyclized, the contribution to the J-factor measured in the cyclization assay is clearly very important even at the ATP concentrations currently employed.
DNA contains the induced defects. This is a very small population of the DNA molecules, and its effect generally negligible when the length of DNA is much larger than its persistence length. However, as the length of DNA decreases, the population of cyclizable species reduces dramatically and finally approaches and becomes even smaller than this value. For example for the 89 bp DNA used in reference (15) the J-factor predicted by the WLC model, with a persistence length of 50 nm, is 10−15 M, which is equivalent to 10−12–10−11 of the total DNA molecules. Since DNA molecules containing local defects stabilized by ligase are expected to have a much higher tendency to be cyclized, the contribution to the J-factor measured in the cyclization assay is clearly very important even at the ATP concentrations currently employed.
To provide accurate measurements of DNA stiffness using cyclization, our results in fact indicate that for a fixed concentration of ligase, the relative ratio of ATP and DNA must be carefully tuned to prevent non-specific binding of ligase to DNA. However, since ATP is generally consumed during the ligation reaction, reaction time and substrate concentration should also be taken into consideration while selecting the optimal ATP concentration. To illustrate, consider the cyclization measurements conducted on 105 bp DNA by Du and co-workers (25). These authors managed to recover the cyclization efficiency predicted by WLC model for 106 bp DNA, using 25 units/ml ligase, 0.025 nM DNA and 1 mM ATP (25). Under these conditions, the relative abundance of DNA-ligase complexes is calculated to be 10−10–10−9, compared with ∼10−10 cyclizable DNA relative to all DNA molecules present. It is then possible to estimate that either an ATP concentration of over 10 M or ligase concentration <10−2 units/ml is needed to fully suppress the contribution of ligase induced DNA destabilization to the measured J-factor.
Our observations therefore appear to provide a plausible answer to the longstanding controversy as to the variability of bending stiffness values for short DNA fragments reported by various groups (20,21,25). Specifically, our results indicate that the presence of large amounts of ligase in cyclization experiments might well create rather than simply trap highly bent DNA conformations; implying that the cyclization assay may be inherently not suitable for extracting bending stiffness of very short DNA fragments.
CONCLUSIONS
Using the base analog 2-AP, whose fluorescence is sensitive to its microenvironment, the local conformational change of DNA molecule under standard conditions of the cyclization assay is tested. Existence of T4 DNA ligase enhanced 2-AP fluorescence indicates that ligase is capable of inducing DNA conformation change. The non-specific interaction between DNA and ligase increases the subpopulation of DNA molecules containing local defects, i.e. extra-helical nucleotides, which can lower the apparent stiffness of DNA determined from cyclization data. The presence of ATP can reduce this ‘ligase enhanced’ fluorescence by competitively binding to the functional domain of ligase and thus suppress the DNA conformational change due to the non-specific DNA ligase contact.
In performing a meaningful cyclization assay, the ligase and ATP concentration must be selected with extra caution. Besides satisfying the kinetic assumption underlying the J analysis (25), the ligase induced DNA conformational change must be suppressed efficiently. Performing the reaction in an ATP rich medium seems to be a promising resolution to this problem. But as DNA size reduces, sufficiently high ligase concentration is essential for efficient completion of ligation reaction and the preference of DNA with local defects in the cyclization reaction can shift the binding competition between ATP and DNA towards the DNA side. All of these factors might contribute to the complexity in analyzing the cyclization data of short DNA strands and a measurement of DNA bending stiffness without external agent could be required in resolving this issue.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We are grateful to the National Science Foundation (grant DMR0551185) for supporting this study. Funding to pay the Open Access publication charges for this article was provided by National Science Foundation (Grant DMR 0551185).
Conflict of interest statement: None declared.
REFERENCES
- 1.Richmond TJ, Davey CA. The structure of DNA in the nuclesome core. Nature. 2003;423:145–150. doi: 10.1038/nature01595. [DOI] [PubMed] [Google Scholar]
- 2.Thastrom A, Lowary PT, Widlund HR, Cao H, Kubista M, Widom J. Sequence motifs and free energies of slected natural and non-natural nucleosome positioning DNA sequence. J. Mol. Biol. 1999;288:213–229. doi: 10.1006/jmbi.1999.2686. [DOI] [PubMed] [Google Scholar]
- 3.Lowary PT, Widom J. New DNA sequence rules fro high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol. 1999;276:19–42. doi: 10.1006/jmbi.1997.1494. [DOI] [PubMed] [Google Scholar]
- 4.Bloomfield VA, Crothers DM, Tinoco I. Nucleic Acids: Structures, Properties, and Functions. Sauslito, California: University Science Books; 2000. [Google Scholar]
- 5.Hagerman PJ. Investigation of the flexibility of DNA using transient electric birefringence. Biopolymers. 1981;7:1503–1535. doi: 10.1002/bip.1981.360200710. [DOI] [PubMed] [Google Scholar]
- 6.Hagerman PJ. Flexibility of DNA. Ann. Rev. Biophys. Biophys. Chem. 1988;17:265–286. doi: 10.1146/annurev.bb.17.060188.001405. [DOI] [PubMed] [Google Scholar]
- 7.Bustamante C, Marko JF, Siggia ED, Smith S. Entropic elasticity of lambda-phage DNA. Science. 1994;265:1599–1600. doi: 10.1126/science.8079175. [DOI] [PubMed] [Google Scholar]
- 8.Wang MD, Yin H, Landick R, Gelles J, Block S. Stretching DNA with optical tweezer. Biophysical J. 1997;72:1335–1346. doi: 10.1016/S0006-3495(97)78780-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bouchiat C, Wang MD, Allemand JF, Strick T, Block SM, Croquette V. Estimating the persistence length of a worm-like chain molecule from force-extension measurements. Biophysical J. 1999;76:409–413. doi: 10.1016/s0006-3495(99)77207-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crothers DM, Drak J, Kahn JD, Levene SD. DNA bending, flexibility and helical repeat by cyclization kinetics. Methods Enzymol. 1992;212:3–29. doi: 10.1016/0076-6879(92)12003-9. [DOI] [PubMed] [Google Scholar]
- 11.Shore D, Langowski J, Baldwin RL. DNA flexibility studied by covalent closure of short fragments into circles. PNAS. 1981;78:4833–4837. doi: 10.1073/pnas.78.8.4833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shimada J, Yamakawa H. Ring-closure probabilities for twisted wormlike chains-application to DNA. Macromolecules. 1984;17:689–698. [Google Scholar]
- 13.Zhang Y, Crothers DM. Statistical mechanics of sequence-dependent circular DNA and its application for DNA cyclization. Biophysical J. 2003;84:136–153. doi: 10.1016/S0006-3495(03)74838-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benham CJ. Elastic model of supercoiling. PNAS. 1977;74:2397–2401. doi: 10.1073/pnas.74.6.2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hao MH, Olson WK. Global equilibrium-configurations of supercoiled DNA. Macromolecules. 1989;22:3292–3303. [Google Scholar]
- 16.Bauer WR, Lund RA, White JH. Twist and writhe of a DNA loop containing intrinsic bends. PNAS. 1993;90:833–837. doi: 10.1073/pnas.90.3.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Balaeff A, Mahadevan L, Schulten K. Elastic rod model of a DNA loop in the LAC operon. Phys. Rev. Lett. 1999;83:4900–4903. [Google Scholar]
- 18.Yang Y, Tobias I, Olson WK. Finite element analysis of DNA supercoiling. J. Chem. Phys. 1993;98:1673–1686. [Google Scholar]
- 19.Yang Y, Westcotts TP, Pedersen SC, Tobias I, Olson WK. Effects of localized bending on DNA supercoiling. Trends Biochem. Sci. 1995;20:313–319. doi: 10.1016/s0968-0004(00)89058-1. [DOI] [PubMed] [Google Scholar]
- 20.Cloutier TE, Widom J. Spontaneous sharp bending of double-stranded DNA. Mol. Cell. 2004;14:355–362. doi: 10.1016/s1097-2765(04)00210-2. [DOI] [PubMed] [Google Scholar]
- 21.Cloutier TE, Widom J. DNA twisting flexibility and the formation of sharply looped protein-DNA complex. PNAS. 2005;102:3645–3650. doi: 10.1073/pnas.0409059102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yan J, Marko JF. Localized single-stranded bubble mechanism for cyclization of short double helix DNA. Phys. Rev. Lett. 2004;93:108108. doi: 10.1103/PhysRevLett.93.108108. [DOI] [PubMed] [Google Scholar]
- 23.Wiggins PA, Phillips R, Nelson PC. Exact theory of kinkable elastic polymers. Phys. Rev. E. 2005;71:021909. doi: 10.1103/PhysRevE.71.021909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yan J, Kawamura R, Marko JF. Statistics of loop formation along double helix DNAs. Phys. Rev. E. 2005;71:061905. doi: 10.1103/PhysRevE.71.061905. [DOI] [PubMed] [Google Scholar]
- 25.Du Q, Smith C, Shiffeldrim N, Vologodskaia M, Vologodskii A. Cyclization of short DNA fragments and bending fluctuations of the double helix. PNAS. 2005;102:5397–5402. doi: 10.1073/pnas.0500983102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yuan C, Rhoades E, Lou XW, Archer LA. Spontaneous sharp bending of DNA: Role of melting bubbles. Nucleic Acids Res. 2006;34:4554–4560. doi: 10.1093/nar/gkl394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rhoades E, Ramlall TF, Webb WW, Eliezer D. Quantification of α-Synuclein binding to lipid vesicles using fluorescence correlation spectroscopy. Biophys. J. 2006;90:4692–4700. doi: 10.1529/biophysj.105.079251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park HY, Qiu X, Rhoades E, Korlach J, Kwok LW, Zipfel WR, Webb WW, Pollack L. Achieving uniform mixing in microfluidic device: hydrodynamic focusing prior to mixing. Analytical Chem. 2006;78:4465–4473. doi: 10.1021/ac060572n. [DOI] [PubMed] [Google Scholar]
- 29.Magde D, Elson E, Webb WW. Thermodynamic fluctuation in a reacting system measurement by fluorescence correlation spectroscopy. Phys. Rev. Lett. 1972;29:705–708. [Google Scholar]
- 30.Ward DC, Reich E, Stryer L. Fluorescence studies of nucleotides and polynucleotides. J. Biol. Chem. 1969;244:1228–1237. [PubMed] [Google Scholar]
- 31.Millar DP. Fluorescence studies of DNA and RNA structure and dynamics. Curr. Opin. in Struct. Biol. 1996;6:322–326. doi: 10.1016/s0959-440x(96)80050-9. [DOI] [PubMed] [Google Scholar]
- 32.Su TJ, Tock MR, Egelhaaf SU, Poon WCK, Dryden DTF. DNA bending by M. EcoKI methyltransferase is coupled to nucleotide flipping. Nucleic Acids Res. 2005;10:3235–3244. doi: 10.1093/nar/gki618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu C, Martin CT. Fluorescence characterization of the transcription bubble in elongation complexes of T7 RNA polymerase. J. Mol. Biol. 2001;308:465–475. doi: 10.1006/jmbi.2001.4601. [DOI] [PubMed] [Google Scholar]
- 34.Holz B, Klimasauskas S, Serva S, Weinhold E. 2-Aminopurine as a fluorescent probe for DNA base flipping by methyltransferases. Nucleic Acids Res. 1998;26:1076–1083. doi: 10.1093/nar/26.4.1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stivers JT. 2-Aminopurine fluorescence studies of base stacking interactions at a basic site in DNA: a metal-ion and base sequence effect. Nucleic Acids Res. 1998;26:3837–3844. doi: 10.1093/nar/26.16.3837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rai P, Cole TD, Thompson E, Millar DP, Linn S. Steady-state and time-resolved fluorescence studies indicate an unusual conformation of 2-aminopurine within ATAT and TATA duplex DNA sequences. Nucleic Acids Res. 2003;31:2323–2332. doi: 10.1093/nar/gkg339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jean JM, Hall KB. 2-Aminopurine fluorescence quenching and lifetimes: role of base stacking. PNAS. 2001;98:37–41. doi: 10.1073/pnas.011442198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rachofsky EL, Osman R, Ross JBA. Probing structure and dynamics of DNA with 2-aminopurine: effects of local environment. Biochemisty. 2001;40:946–956. doi: 10.1021/bi001664o. [DOI] [PubMed] [Google Scholar]
- 39.SantaLucia J, Hicks D. The thermodynamics of DNA structural motifs. Annu. Rev. Biomol. Struct. 2004;33:415–420. doi: 10.1146/annurev.biophys.32.110601.141800. [DOI] [PubMed] [Google Scholar]
- 40.Friedman RA, Honig B. A free energy analysis of nucleic acid base stacking in aqueous solution. Biophysical J. 1995;69:1528–1533. doi: 10.1016/S0006-3495(95)80023-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yahovchuk P, Protozanova E, Frank-Kamenetskii MD. Base-stacking and base-pairing contributions into thermal stability of the DNA double helix. Nucleic Acids Res. 2006;34:564–574. doi: 10.1093/nar/gkj454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lehman IR. DNA ligase: structure, mechanism, and function. Science. 1974;186:790–797. doi: 10.1126/science.186.4166.790. [DOI] [PubMed] [Google Scholar]
- 43.Rossi R, Montecucco A, Ciarrocchi G, Biamonti G. Functional characterization of the T4 DNA ligase: a new insight into the mechanism of action. Nucleic Acids Res. 1997;25:2106–2113. doi: 10.1093/nar/25.11.2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cherpepanov AV, de Vries S. Binding of Nucleotides by T4 DNA ligase and T4 RNA ligase: optical absorbance and fluorescence studies. Biophys. J. 2002;81:3545–3559. doi: 10.1016/S0006-3495(01)75985-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Subramanya HS, Doherty AJ, Ashford SR, Wigley DB. Crystal structure of an ATP-dependent DNA ligase from bacteriophage T7. Cell. 1996;85:607–615. doi: 10.1016/s0092-8674(00)81260-x. [DOI] [PubMed] [Google Scholar]
- 46.Singleton MR, Hakansson K, Timson DJ, Wigley DB. Structure of the adenylation domain of an NAD (+)-dependent DNA ligase. Structure. 1999;7:35–42. doi: 10.1016/s0969-2126(99)80007-0. [DOI] [PubMed] [Google Scholar]
- 47.Lee JY, Chang C, Song HK, Moon J, Yang JK, Kim HK, Kwon ST, Suh SW. Crystal structure of NAD (+)-dependent DNA ligase: modular architecture and functional implications. EMBO J. 2000;19:1119–1129. doi: 10.1093/emboj/19.5.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schallhorn KA, Freedman KO, Moore JM, Lin J, Ke PC. Single-molecule DNA flexibility in the presence of base-pair mismatch. App. Phys. Lett. 2005;87:033901. [Google Scholar]
- 49.Kahn JD, Yun E, Crothers DM. Detection of localized DNA flexibility. Nature. 1994;368:163–166. doi: 10.1038/368163a0. [DOI] [PubMed] [Google Scholar]