

Abstract
Individual protein binding sites on DNA can be measured in bits of information. This information is related to the free energy of binding by the second law of thermodynamics, but binding kinetics appear to be inaccessible from sequence information since the relative contributions of the on- and off-rates to the binding constant, and hence the free energy, are unknown. However, the on-rate could be independent of the sequence since a protein is likely to bind once it is near a site. To test this, we used surface plasmon resonance and electromobility shift assays to determine the kinetics for binding of the Fis protein to a range of naturally occurring binding sites. We observed that the logarithm of the off-rate is indeed proportional to the individual information of the binding sites, as predicted. However, the on-rate is also related to the information, but to a lesser degree. We suggest that the on-rate is mostly determined by DNA bending, which in turn is determined by the sequence information. Finally, we observed a break in the binding curve around zero bits of information. The break is expected from information theory because it represents the coding demarcation between specific and nonspecific binding.
INTRODUCTION
Transcription factors bind to a variety of sequences with different affinities (1). The amount of sequence variability within a set of binding sites is limited by physical requirements for binding, as well as the ability for the site to be distinguished from non-sites in the genome (2). A range of affinities allows for a subtle regulation of transcription. In the case of activators, higher affinity sites will presumably be bound longer than lower affinity sites, and have a greater probability of stabilizing the initiation complex, which in turn has a greater probability of transcribing a gene. Therefore, the affinity of the protein for a site is a direct indicator of the degree that that site will affect the gene expression.
Being able to predict binding affinities for different DNA targets is useful in characterizing genetic regulatory pathways. To do this, we use an information theory-based weight matrix to quantify protein binding to individual sequences (3).
Information theory was developed by Claude Shannon in the late 1940s to describe the movement of information in communications (4). When applied to biological systems it has proven to be useful (2,5–7). Based on the frequency of each base at each position in a set of aligned binding sites, we can determine the strength of an individual site in bits of information. This strength is called the individual information, Ri (rate of individual information transfer, bits per site) for a site (3). Advantages of this approach are discussed in Materials and Methods.
It has been shown that the protein–DNA dissociation constant, KD, varies with DNA sequences, and can be approximated by different weight matrix approaches (8–12). The information in a binding site should be related to the binding energy (13). Binding energy, in turn, is proportional to the logarithm of the ratio of the association (kon) and dissociation (koff) rate constants of binding. Since the on-rate depends on diffusion of the protein to the DNA binding site, we expected that the on-rate would be independent of the binding sequence. This suggests that the information of binding sites (Ri) should be linearly related to the logarithm of the off-rate. Others have reported differences in binding rate constants as a function of sequence (14–16), but they did not report any relationship between the rate constants and affinity predictions. No one has shown how information theory predictions of individual binding sites are related to binding and dissociation kinetics.
To address this issue, we used surface plasmon resonance (SPR) technology (17–19) and electrophoretic mobility shift assays (EMSA) to measure the binding kinetics for 13 Fis binding sites ranging in predicted site strength, based on our information theory approach. Fis is a pleiotropic homodimeric DNA binding protein involved in site-specific recombination, chromosomal compaction and transcriptional regulation (6,20,21). Because many genomic sites have been experimentally identified, a reliable Fis model could be constructed and verified (6,22), making it a good protein for this analysis.
MATERIALS AND METHODS
Constructing the Fis model
The Fis binding site model was built using the standard Delila programs (23,24) (Figure 1), and was originally presented in (6). Individual information analysis (3,25) of Fis binding sites was computed using a weight matrix from the equation:
| 1 |
where f (b, l) is the frequency of each base b at each position l for all positions in an aligned set of binding sites. e(n) is a sample correction value where n is 120, the number of Fis binding sites and their complements that make up our frequency matrix. To determine the strength of a site (Ri), a DNA sequence is compared to the Riw (b,l) weight matrix and the information contribution of each base is summed across the site. There are several advantages to our approach. First, our models are composed of only experimentally verified binding sites, and do not require a training set of unproven ‘non-sites’ like many neural-networks or HMMs (26–29). Second, our method has no arbitrary parameters, and the theory predicts that all sites with greater than zero bits of information have a negative ΔG of binding (3). Third, the units of measurement, bits, allow direct comparison between different molecular systems. Fourth, the average Ri for all binding sites that define an Riw (b,l) is Rsequence, or the total information content [the area under a sequence logo (2)]. The information content is a measure of the sequence conservation and it is determined by the evolution of the sites in the genome (30).
Figure 1.

Sequence logo for the Fis protein (6). The heights of letters in each stack are proportional to the frequency of each base at that position. The height of the stack is the information content for that position (23). The total conservation, summed for all positions in the range −7 to +7 is Rsequence= 7.18 ± 0.23 bits per site (2), which is also the average of the individual information of all of the sites (3). The sine wave above the logo represents the 10.6 bp helical twist of B-form DNA (24). The positions presumably bound by the D helices from the major groove at ± 7 are marked with squares, the pyrimidine/purine steps that kink the DNA at ± 4 and ± 3 are marked with filled triangles, and the A/T bases that allow bending into the minor groove are marked with open triangles.
We used a Fis model ranging from −7 to +7 throughout this article. This assumes that positions outside this region do not affect binding and it is consistent with known footprinting data (6). The small amount of information observed in positions −10 to −8 and +8 to +10 (Figure 1) may correspond to overlapping adjacent Fis sites (22).
Individual information analysis was done using the program scan and sequence walkers were generated using lister (3,31) (Figure 2).
Figure 2.

Sequences used for analysis. Information analysis for individual sequences was computed (3) and displayed using sequence walkers (31). Those positions that are favored according to the Riw(b,l) weight matrix (contribute positive information) are represented by bases above the x-axis, whereas those bases that are not favored (contribute negative information) are below the x-axis. The height of each base is its information contribution to the site strength. The sum of all base heights is Ri for the sequence, and this is given on the right of the sequence walker. These sequences correspond to those in Table 1. The sequences are sorted by their strength in bits and the saturation of a colored rectangle behind each walker is proportional to that strength. As in Figure 1, the sine wave above the walker represents the 10.6 bp helical twist of B-form DNA (24).
Oligo construction
Thirteen oligos of varying information content were synthesized to measure binding kinetics. Ten of these contain naturally occurring Fis binding sites, where binding has been experimentally verified (32–38). These sites are presented in Table 1 and Figure 2. We chose these oligos to cover a spectrum of strengths from 4.9 to 12.7 bits, as assessed by our information theory approach. The three remaining oligos do not contain characterized binding sites, but have been engineered by us to test binding at additional site strengths.
Table 1.
Kinetics as determined by SPR
| Oligo name | Ri (bits) | Number of experiments | Stability (s−1) | Reference |
|---|---|---|---|---|
| anti-con | −30.6 | 6 | 2.21 × 10−1 ± 4.08 × 10−3 | This work |
| cin-336 | 4.9 | 2 | 1.24 × 10−1 ± 2.48 × 10−3 | (33) |
| hin-1096 | 5.4 | 2 | 7.39 × 10−2 ± 6.07 × 10−4 | (32) |
| lacP-560 | 6.6 | 4 | 1.67 × 10−2 ± 6.49 × 10−5 | (34) |
| ndhII-188 | 8.2 | 7 | 7.37 × 10−3 ± 1.10 × 10−4 | (35) |
| comp-ndhII-188 | 8.2 | 2 | 6.24 × 10−3 ± 4.19 × 10−5 | (35) |
| fis-333 | 10.1 | 1 | 3.45 × 10−3 ± 3.21 × 10−5 | (37) |
| tgt-1824 | 10.2 | 1 | 2.62 × 10−2 ± 1.21 × 10−4 | (36) |
| hin-180 | 10.4 | 2 | 7.83 × 10−3 ± 4.80 × 10−5 | (32) |
| thrU-87 | 10.9 | 1 | 4.06 × 10−3 ± 6.54 × 10−5 | (38) |
| ndhI-137 | 12.7 | 1 | 2.90 × 10−3 ± 5.80 × 10−5 | (35) |
| mut-con | 12.8 | 6 | 8.65 × 10−4 ± 4.29 × 10−6 | This work |
| con | 14.9 | 1 | 9.40 × 10−4 ± 1.39 × 10−5 | This work |
‘Oligo’ is the name of the synthetic DNA hairpin as defined in this article or the name of the adjacent gene and base coordinate of the site in a GenBank entry (6). The sequences are given in Figure 2 and Supplementary Data Figure 1. Ri is the individual information for that site. ‘Number of experiments’ is the number of measurements made with each oligo. ‘Stability’ is the apparent koff that we measure using SPR and analyzed with Scrubber. ‘Reference’ is the reference describing the binding of Fis to that sequence.
The first engineered oligo is the Fis consensus of 5′-ATTGGTTAAATTTTAACCAAT-3′ over the range −10 to +10, containing three extra natural bases on each end (Figure 1), which is presumably the highest strength site (14.9 bits), and it does not occur in the Escherichia coli genome (named con in Table 1, Figure 2). The second oligo is a slight modification of this consensus, where we mutated the T at position +1 to a G to decrease the strength of the site to 12.8 bits (named mut-con in Table 1, Figure 2). The third oligo is the Fis anti-consensus of 5′-CGGCTGACCCCGGGTCAGCCG-3′, which is made up of the least favorable base at each position (named anti-con in Table 1, Figure 2). The kinetics of binding to this sequence are presumably those of nonspecific interactions of Fis with DNA.
All sequences were inserted into the same hairpin construct: 5′-GCTATCGCG-[Sequence]-ACGATCGCGC-GAA-GCGCGATCGT-[Complement of Sequence]-CGCGA-3′, where there is a 5′ 4 bp overhang of GCTA to allow for future modification, and a 3 bp loop of GAA in the center. This construct has been shown to form tight hairpins (6,39). All oligos were synthesized carrying a 5′-biotin tag (Synthegen, LLC) to allow immobilization of the oligos onto NeutrAvidin (NA)-coated sensor chips (B1 chips, Biacore Inc.). To test whether the orientation of a sequence in the hairpin affects binding, we inverted the ndhII-188 sequence in the hairpin to create comp-ndhII-188.
SPR analysis
NeutrAvidin was purchased from Pierce. EDTA, SDS, NaCl and HEPES (pH 7.4) were purchased from Invitrogen. Potassium glutamate was purchased from Sigma-Aldrich. Tris-HCl (pH 7.5) was purchased from Quality Biological, Inc. Binding experiments were performed on Biacore 2000 and Biacore 3000 instruments (Biacore Inc.). NeutrAvidin was diluted to a final concentration of 25 g/ml in 10 mM sodium acetate, pH 4.5. An immobilization wizard within the Biacore control software was used to immobilize no more than 4000 RU of NA. One RU, or resonance unit, corresponds to a change in the angle of the intensity minimum by 0.0001 ○ as detected by the Biacore. The oligos were diluted to a final concentration of 1 mg/ml in immobilization buffer (10 mM Tris-HCl pH 7.5, 300 mM NaCl, 1 mM EDTA). To prepare double-stranded DNA, the oligos were heated to 95°C for 5 min, snap cooled on ice for 5 min, and incubated at room temperature for 15 min. The sample was then diluted 750-fold in immobilization buffer and injected manually over the surface until between 100 and 150 RUs were captured on the B1 sensor chip.
Purified Fis protein (22) was serially diluted in 1×running buffer (10 mM HEPES pH = 7.4, 350 mM potassium glutamate (40), 3.4 mM EDTA, 0.01% BSA) to concentrations ranging from 100 nM for the high affinity oligos to 1000 nM for the low affinity oligos and injected at 25°C at a flow rate of 100 μl/min for 90 s. All oligos reached a stochastic steady state of Fis binding. Dissociation times were typically 90–360 s depending upon the stability of the complex. Disruption of any complex that remained bound after dissociation was achieved using two 50 μl injections of regeneration solution (0.1% SDS, 3.4 mM EDTA) followed by one EXTRACLEAN command, a running buffer wash to eliminate carry-over into the next experiment. At the beginning of each cycle, the needle was pre-dipped in running buffer before an injection of 100 μl running buffer. Similarly, each cycle was ended by an injection of 100 μl running buffer and an EXTRACLEAN command. Typically, every concentration of protein was injected twice from separate vials. In order to subtract any background noise from each data set, all samples were also run over a sensor chip surface of NA without oligo and injections of running buffer were performed for every experiment (‘double referencing’) (41). Data were fit to a single exponential decay model using both of the programs Scrubber 1.10 (42) and Biaevaluation 3.1 (Biacore, Inc).
Fis competition electrophoretic mobility shift assay (EMSA)
Using EMSA, we found that nonspecific binding occurred with the long oligos used in the SPR experiments. Therefore we used hairpin oligos containing a Fis site (−7 to +7) with no additional bases, a loop (5′-GCGAAGC-3′) and the complementary sequence of the Fis site for EMSA. (See Supplementary Data Figure 1 for the sequences used.)
Competition EMSA between conF37, a 5′ 6-FAM labeled oligo 5′-GGTTAAATTTTAACC-GCGAAGC-GGTTAAAATTTAACC-3′ (Integrated DNA Technologies) containing the consensus Fis binding site, and unlabeled oligos containing naturally occurring and mutated Fis binding sites, was used to determine the KD of the sites. When a potassium glutamate-containing buffer was used for EMSA, Fis–DNA complexes smear on a gel, therefore we used the following buffer. Binding reactions were carried out in 10 μl of solution, containing 7.7 mM Bis Tris Propane-HCl, 10 mM NaCl, 0.5% glycerol, 10 mM MgCl2, 1 mM DTT, 800 nM Fis, 40 nM labeled conF37 oligo and 1.0, 1.5 or 2.0 μM competitors for 5 min at room temperature, followed by 2.2% agarose gel electrophoresis in 5 mM sodium borate pH = 8.5 (43) for 20 min at 5 V/cm and the gel was scanned by a FMBIO II fluorescent scanner (Hitachi) with 505 nm emission filter (Figure 5). (See Supplementary Data for how the data were analyzed.)
Figure 5.

Competition electrophoretic mobility shift assay with three different concentrations of oligos containing different Fis binding sites. (See Supplementary Data Figure 1 for the sequences.) For each concentration, the top band is Fis bound to the consensus 5′ 6-FAM labeled oligo and the bottom band is unbound labeled oligo (see Materials and Methods). The competitor concentrations shown are approximately: 1.0 μM low, 1.5 μM medium, 2.0 μM high; the exact values for each competitor are given in Supplementary Data. Lanes 1 to 13: competitor oligos 1 to 13; Lane 14: no competitor.
RESULTS AND DISCUSSION
The Fis sequence logo is consistent with models of the Fis/DNA complex (Figure 1) (6,44,22). Sequence conservation at positions ± 7 above 1 bit suggests that Fis binds two major grooves on the same face of the DNA (24). However, the distance between the D helices which bind these two major contacts is less than 10.6 bases, one helical twist of B-form DNA, suggesting that the DNA must bend to enable positions ± 7 to contact the D helices (24). Indeed, Fis bends DNA (44). The relatively low information content of 7.18 ± 0.23 bits over the range ± 7 bases, suggests that Fis is a fairly prolific binder (2, 30). This is consistent with the observed high concentration of Fis in response to nutrient upshifts (as many as 50 000 dimers per cell) (37). Finally, DNA methylation and DNase I hypersensitivity results are consistent with positions of significant sequence conservation (6). The correspondence between the physical and biochemical characterization of Fis binding with the sequence conservation supports the information-theory based Fis binding model.
We chose ten naturally occurring Fis binding sites and three synthetic sites for kinetic analysis. These sites covered a spectrum of strengths and are reported in Figure 2. The terms anti-consensus (anti-con) and consensus (con) refer to the weakest and strongest possible sites based on our model respectively (3).
In order to measure the binding kinetics of these oligos, we used SPR technology. Protein can be flowed over a mat of DNA tethered to a thin gold surface. As the protein associates and dissociates, the change in density on the surface can be monitored, and kon and koff can be determined (17,45). The SPR plots appeared to have one-stage binding, suggesting a simple association–dissociation mechanism (Figure 3).
Figure 3.

Sensogram of Fis bound to different DNA sequences. All curves were normalized so that saturation of the chip is set to 1. At time zero, Fis was washed onto the SPR chip. At time 90 s, Fis was washed off the chip. The stability measurements reported in Table 1 were determined from the curve after 90 s.
All data obtained for the Fis dimer (22.4 kDa) on the Biacore machine were transport limited (46). That is, the kinetics of binding that are inferred from these experiments are not only a measurement of binding, but also a measure of the delivery of Fis to the chip surface. However, we were able to measure an apparent koff or ‘stability’ which is the rate of dissociation of Fis from the surface. Although this is not the true koff, because of the transport limitation, it is proportional since the rate of transport (
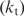 ) is constant for all measurements. Additionally, surface effects such as nonspecific interactions of Fis with the chip surface could affect the SPR measure so that it does not entirely represent in vivo or in-solution conditions, but as with the rate of transport, such effects should also be constant for all measurements.
) is constant for all measurements. Additionally, surface effects such as nonspecific interactions of Fis with the chip surface could affect the SPR measure so that it does not entirely represent in vivo or in-solution conditions, but as with the rate of transport, such effects should also be constant for all measurements.
The stability kinetics measurement is strongly correlated to the individual information of the sites, with r2 = 0.84 (Figure 4). These values are presented in Table 1. The complexes of Fis with oligos ndhII and comp-ndhII had similar stabilities (7.4 × 10−3 and 6.2 × 10−3 s−1 respectively) suggesting that orientation within the hairpin had little affect on the stability measurement. The dissociation of the protein from the anti-con oligo is faster than the dissociation from the weakest observed natural binder cin-336, 0.22 s−1 versus 0.12 s−1. This is presumably related to the energy difference between the weakest possible specific binding and nonspecific binding for Fis. The stability of the protein with the consensus and mutated consensus is very high, 9.4 × 10−4 and 8.7 × 10−4 s−1, respectively.
Figure 4.

Binding site information is correlated to stability. For each sequence described in Figure 2 and Table 1, we plotted the stability versus the information Ri. Scrubber and Biaevaluation are two implementations of curve fitting by a single exponential decay describing the dissociation. Both were used to evaluate all of the data and slight differences were observed from small deviations in the start and stop points chosen for analysis. We plot each measurement independently. Although the anti-con oligo is presumably nonspecific at −30.6 bits, we plotted it as having 0 bits of information. All points at zero bits are for the anti-con oligo. The regression line (excluding the anti-con) is shown as a red line (r2 = −0.84). 99% confidence limits for the regression are shown with blue lines. The equation for the regression line is log2(Stability) = −0.70 × Individual information −0.84.
The logic of our experiment is based on a series of simple relations:
- Information is related to energy by a version of the Second Law of thermodynamics (13). The relationship is generally proportional (TDS in preparation) so we expect that the individual information should relate to the binding energy:
This is supported by experiments in a number of systems (8,9,47).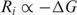
2 - The binding energy is related to the binding constant:
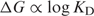
3 - The binding constant is a function of the on and off rates:
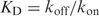
4 Once a protein is at a binding site, it will frequently bind irrespective of how strong the binding is, so the on-rate, kon should be roughly constant and this is observed in various other genetic systems (14–16, 48).
- Combining the above
so the more information a binding site has, the larger the number of contacts it can make with the protein (49) and correspondingly the more difficult it becomes for thermal noise to separate the two once they are bound together. The off-rate is strongly dependent on the detailed binding contacts since all of these have to be broken to release the protein.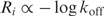
5
Although our Biacore experiments gave the relationship of Equation (5) (Figure 4), they did not give us kon values. To investigate kon, we performed competitive EMSA experiments to determine KDs (Figure 5). The results show a linear relationship between
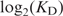 and Ri:
and Ri:
| 6 |
with r2 = −0.73 (Supplementary Data). The experiment was repeated and similar results were obtained (data not shown).
Since koffs and KDs were measured by different techniques, the relative ratios between the sites should be correct but they may differ from the absolute values by an unknown multiplicative factor. On the log scale, this is in the additive constant. Using the KDs measured by EMSA and the koffs measured in the Biacore experiments, we calculated kon according to Equation (4) for each DNA. Unexpectedly, we observed that kon is related to the information.
By using linear regression of
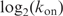 against Ri and
against Ri and
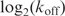 against Ri,
against Ri,
| 7 |
and
| 8 |
we found that 49% of the variance of
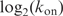 and 78% of the variance of
is explained by the variance of Ri (Supplementary Data). Thus most of the off-rate is explained by the information in the sequence. In addition, a good portion of the on-rate is explained by the sequence, implying that another factor—we suggest sequence bendability—may be involved in the initial binding.
and 78% of the variance of
is explained by the variance of Ri (Supplementary Data). Thus most of the off-rate is explained by the information in the sequence. In addition, a good portion of the on-rate is explained by the sequence, implying that another factor—we suggest sequence bendability—may be involved in the initial binding.
Are the evolved binding targets of Fis the result of the physical properties of DNA? It is possible that the bases that are specifically contacted have been adapted through natural selection to facilitate binding through bending. If this is true, then there should be a correlation between kon and koff. Indeed, kon and koff increase together with a positive correlation
| 9 |
and 85% of the
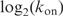 variance is explained by
, suggesting that some of the positions are important for both binding and bending (Supplementary Data Figure 2).
variance is explained by
, suggesting that some of the positions are important for both binding and bending (Supplementary Data Figure 2).
This proposal is consistent with our previous observations on the sequence logo of Fis (22). We found that patterns of bases in the Fis sites can be explained in two distinct ways. In Figure 1, the outer bases at ± 7, mostly G and C, are consistent with direct binding by Fis into the major groove but these contacts are too close to allow the D helices of Fis to fit into the major groove unless the DNA is also bent. Positions ± 4 and ± 3 contain pyrimidines and purines (respectively, on the 5′↣ 3′ strand) which could be contacted directly through the major groove or which could provide a bendable step. Likewise positions −2 to +2 contain A or T which is also consistent with either direct minor groove contacts or with bending into the minor groove. Since the central positions from −4 to +4 do not appear to be contacted in our 3D model (22), binding of Fis may first involve specific contacts followed by bending that perhaps releases those contacts. This implies that the binding rate requires DNA sequence-dependent bending. If so, kon is controlled by the degree of flexibility of the DNA and that, in turn, is controlled by the DNA sequence. However, if Fis makes direct contacts to the central bases while bound (despite our modeling) then DNA sequence should determine the strength of binding, and this is indeed observed. We are led to suggest that both bending and direct contacts are involved in both of the on- and off-stages of Fis binding. Similar experiments relating the information content of binding sites for other proteins that do not bend DNA as strongly as Fis may reveal further insights into the binding process.
The experiments described here suggest that Ri is mostly dependent upon the logarithm of koff. It has previously been shown that the average Ri for all sites is Rsequence, the sequence conservation of a set of binding sites (3). Therefore, the results imply that the sequence conservation (the amount of variability among a set of binding sites) for a protein is directly related to the binding kinetics of that protein to its targets. A stronger binding protein that covers the same length of DNA will have a less variable site. Another aspect is that Rsequence evolves to match the information needed to find the sites in the genome, Rfrequency, which is a function of the size of the genome and number of sites (2,30). As a protein evolves to bind a greater number of targets, the average specific binding energy of that protein to its targets would decrease by increased koff.
Our experiment provides preliminary data supporting a distinction between two approaches to understanding the DNA recognition process. In Figure 4, no data points were obtained between the anti-consensus at −30.6 bits and 0 bits, however the lowest positive Fis site, at 4.9 bits has a
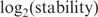 around −3 and the anti-consensus is around −2 so the curve is linear with a negative slope to near zero bits and then presumably is essentially flat from there to −30.6 bits. As shown in Figure 6, a similar result occurs with a plot of binding energy (
around −3 and the anti-consensus is around −2 so the curve is linear with a negative slope to near zero bits and then presumably is essentially flat from there to −30.6 bits. As shown in Figure 6, a similar result occurs with a plot of binding energy (
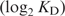 ) versus information. We suggest that this apparent break at zero bits is a manifestation of the Second Law of Thermodynamics and the channel capacity. That is, the Second Law predicts that sites with positive information should have negative ΔG values and those with negative information should not bind because they have positive ΔG values (13). Shannon's channel capacity theorem predicts threshold effects in coded systems where there is a sharp boundary between recognized and unrecognized signals (50). The break in the curve therefore provides support for a coding interpretation of the binding interaction between Fis and DNA. This is in contrast with thermodynamic theories of binding, which generate a scale starting at the consensus, and which do not predict a specific boundary (8).
) versus information. We suggest that this apparent break at zero bits is a manifestation of the Second Law of Thermodynamics and the channel capacity. That is, the Second Law predicts that sites with positive information should have negative ΔG values and those with negative information should not bind because they have positive ΔG values (13). Shannon's channel capacity theorem predicts threshold effects in coded systems where there is a sharp boundary between recognized and unrecognized signals (50). The break in the curve therefore provides support for a coding interpretation of the binding interaction between Fis and DNA. This is in contrast with thermodynamic theories of binding, which generate a scale starting at the consensus, and which do not predict a specific boundary (8).
Figure 6.

Binding energy is linearly related to binding site information for positive information binding sites but apparently flat for sites with negative information. The curve appears to break near zero bits. The average KD values were normalized so that the Hin-180 sequence has the published value of Hin-D, 2× 10-9 M (51). Excluding the anti-consensus at −30.6 bits, the regression line is given in Equation (6) with r2 = −0.73.
The individual information appears to be well correlated to the kinetics of binding. This not only gives greater confidence in our previous information theory based models, but also shows that it is a reliable approach to characterize genetic systems in silico. Furthermore, the relationship between information and energy is subtle (13), and this correlation helps ground the information theory approach into thermodynamics.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We would like to thank Zehua Chen for testing the oligos, Alan Moses, Greg Bowman, Matt Fivash, Jack Kirsch and Danielle Needle for useful discussions. This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract N01-CO-12400. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the U.S. Government. This Research was supported [in part] by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. Funding to pay the Open Access publication charges for this article was provided by NCI.
Conflict of interest statement. None declared.
REFERENCES
- 1.Record MT, Jr, Ha JH, Fisher MA. Analysis of equilibrium and kinetic measurements to determine thermodynamic origins of stability and specificity and mechanism of formation of site-specific complexes between proteins and helical DNA. Methods Enzymol. 1991;208:291–343. doi: 10.1016/0076-6879(91)08018-d. [DOI] [PubMed] [Google Scholar]
- 2.Schneider TD, Stormo GD, Gold L, Ehrenfeucht A. Information content of binding sites on nucleotide sequences. J. Mol. Biol. 1986;188:415–431. doi: 10.1016/0022-2836(86)90165-8. http://www.ccrnp.ncifcrf.gov/~toms/paper/schneider1986/ [DOI] [PubMed] [Google Scholar]
- 3.Schneider TD. Information content of individual genetic sequences. J. Theor. Biol. 1997;189:427–441. doi: 10.1006/jtbi.1997.0540. http://www.ccrnp.ncifcrf.gov/~toms/paper/ri/ [DOI] [PubMed] [Google Scholar]
- 4.Shannon CE. A mathematical theory of communication. Bell System Tech. J. 1948;27:379–423. 623–656. http://cm.bell-labs.com/cm/ms/what/shannonday/paper.html. [Google Scholar]
- 5.Shultzaberger RK, Bucheimer RE, Rudd KE, Schneider TD. Anatomy of Escherichia coli ribosome binding sites. J. Mol. Biol. 2001;313:215–228. doi: 10.1006/jmbi.2001.5040. http://www.ccrnp.ncifcrf.gov/~toms/paper/flexrbs/ [DOI] [PubMed] [Google Scholar]
- 6.Hengen PN, Bartram SL, Stewart LE, Schneider TD. Information analysis of Fis binding sites. Nucleic Acids Res. 1997;25:4994–5002. doi: 10.1093/nar/25.24.4994. http://www.ccrnp.ncifcrf.gov/~toms/paper/fisinfo/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rogan PK, Faux BM, Schneider TD. Information analysis of human splice site mutations. Hum. Mutat. 1998;12:153–171. doi: 10.1002/(SICI)1098-1004(1998)12:3<153::AID-HUMU3>3.0.CO;2-I. Erratum in: Hum Mutat 1999;13(1):82. http://www.ccrnp.ncifcrf.gov/~toms/paper/rfs/ [DOI] [PubMed] [Google Scholar]
- 8.Berg OG, von Hippel PH. Selection of DNA binding sites by regulatory proteins, statistical-mechanical theory and application to operators and promoters. J. Mol. Biol. 1987;193:723–750. doi: 10.1016/0022-2836(87)90354-8. [DOI] [PubMed] [Google Scholar]
- 9.Barrick D, Villanueba K, Childs J, Kalil R, Schneider TD, Lawrence CE, Gold L, Stormo GD. Quantitative analysis of ribosome binding sites in E. coli. Nucleic Acids Res. 1994;22:1287–1295. doi: 10.1093/nar/22.7.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roulet E, Bucher P, Schneider R, Wingender E, Dusserre Y, Werner T, Mermod N. Experimental analysis and computer prediction of CTF/NFI transcription factor DNA binding sites. J. Mol. Biol. 2000;297:833–848. doi: 10.1006/jmbi.2000.3614. [DOI] [PubMed] [Google Scholar]
- 11.Liu X, Clarke ND. Rationalization of gene regulation by a eukaryotic transcription factor: calculation of regulatory region occupancy from predicted binding affinities. J. Mol. Biol. 2002;323:1–8. doi: 10.1016/s0022-2836(02)00894-x. [DOI] [PubMed] [Google Scholar]
- 12.Udalova IA, Mott R, Field D, Kwiatkowski D. Quantitative prediction of NF-κB DNA-protein interactions. Proc. Natl Acad. Sci. USA. 2002;99:8167–8172. doi: 10.1073/pnas.102674699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schneider TD. Theory of molecular machines. II. Energy dissipation from molecular machines. J. Theor. Biol. 1991;148:125–137. doi: 10.1016/s0022-5193(05)80467-9. http://www.ccrnp.ncifcrf.gov/~toms/paper/edmm/ [DOI] [PubMed] [Google Scholar]
- 14.Kim JG, Takeda Y, Matthews BW, Anderson WF. Kinetic studies on Cro repressor-operator DNA interaction. J. Mol. Biol. 1987;196:149–158. doi: 10.1016/0022-2836(87)90517-1. [DOI] [PubMed] [Google Scholar]
- 15.Schaufler LE, Klevit RE. Mechanism of DNA binding by the ADR1 zinc finger transcription factor as determined by SPR. J. Mol. Biol. 2003;329:931–939. doi: 10.1016/s0022-2836(03)00550-3. [DOI] [PubMed] [Google Scholar]
- 16.Linnell J, Mott R, Field S, Kwiatkowski DP, Ragoussis J, Udalova IA. Quantitative high-throughput analysis of transcription factor binding specificities. Nucleic Acids Res. 2004;32:e44. doi: 10.1093/nar/gnh042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fisher RJ, Fivash M, Casas-Finet J, Erickson JW, Kondoh A, Bladen SV, Fisher C, Watson DK, Papas T. Real-time DNA binding measurements of the ETS1 recombinant oncoproteins reveal significant kinetic differences between the p42 and p51 isoforms. Protein Sci. 1994;3:257–266. doi: 10.1002/pro.5560030210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fisher RJ, Fivash MJ, Stephen AG, Hagan NA, Shenoy SR, Medaglia MV, Smith LR, Worthy KM, Simpson JT, et al. Complex interactions of HIV-1 nucleocapsid protein with oligonucleotides. Nucleic Acids Res. 2006;34:472–484. doi: 10.1093/nar/gkj442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rich RL, Myszka DG. Survey of the year 2005 commercial optical biosensor literature. J. Mol. Recognit. 2006;19:478–534. doi: 10.1002/jmr.808. [DOI] [PubMed] [Google Scholar]
- 20.Travers A, Schneider R, Muskhelishvili G. DNA supercoiling and transcription in Escherichia coli: The FIS connection. Biochimie. 2001;83:213–217. doi: 10.1016/s0300-9084(00)01217-7. [DOI] [PubMed] [Google Scholar]
- 21.Ussery D, Larsen TS, Wilkes KT, Friis C, Worning P, Krogh A, Brunak S. Genome organisation and chromatin structure in Escherichia coli. Biochimie. 2001;83:201–212. doi: 10.1016/s0300-9084(00)01225-6. [DOI] [PubMed] [Google Scholar]
- 22.Hengen PN, Lyakhov IG, Stewart LE, Schneider TD. Molecular flip-flops formed by overlapping Fis sites. Nucleic Acids Res. 2003;31:6663–6673. doi: 10.1093/nar/gkg877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schneider TD, Stephens RM. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 1990;18:6097–6100. doi: 10.1093/nar/18.20.6097. http://www.ccrnp.ncifcrf.gov/~toms/paper/logopaper/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schneider TD. Reading of DNA sequence logos: Prediction of major groove binding by information theory. Methods Enzymol. 1996;274:445–455. doi: 10.1016/s0076-6879(96)74036-3. http://www.ccrnp.ncifcrf.gov/~toms/paper/oxyr/ [DOI] [PubMed] [Google Scholar]
- 25.Schneider TD, Rogan PK. 1999. Computational analysis of nucleic acid information defines binding sites. United States Patent 5867402. [Google Scholar]
- 26.Stormo GD, Schneider TD, Gold L, Ehrenfeucht A. Use of the ‘Perceptron’ algorithm to distinguish translational initiation sites in E. coli. Nucleic Acids Res. 1982;10:2997–3011. doi: 10.1093/nar/10.9.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lukashin AV, Anshelevich VV, Amirikyan BR, Gragerov AI, Frank-Kamenetskii MD. Neural network models for promoter recognition. J. Biomol. Struct. Dyn. 1989;6:1123–1133. doi: 10.1080/07391102.1989.10506540. [DOI] [PubMed] [Google Scholar]
- 28.Weller K, Recknagel RD. Promoter strength prediction based on occurrence frequencies of consensus patterns. J. Theor. Biol. 1994;171:355–359. doi: 10.1006/jtbi.1994.1239. [DOI] [PubMed] [Google Scholar]
- 29.GuhaThakurta D, Stormo GD. Identifying target sites for cooperatively binding factors. Bioinformatics. 2001;17:608–621. doi: 10.1093/bioinformatics/17.7.608. [DOI] [PubMed] [Google Scholar]
- 30.Schneider TD. Evolution of biological information. Nucleic Acids Res. 2000;28:2794–2799. doi: 10.1093/nar/28.14.2794. http://www.ccrnp.ncifcrf.gov/~toms/paper/ev/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schneider TD. Sequence walkers: a graphical method to display how binding proteins interact with DNA or RNA sequences. Nucleic Acids Res. 1997;25:4408–4415. doi: 10.1093/nar/25.21.4408. Erratum: NAR 1998, 26(4):1135. http://www.ccrnp.ncifcrf.gov/~toms/paper/walker/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Glasgow AC, Bruist MF, Simon MI. DNA-binding properties of the Hin recombinase. J. Biol. Chem. 1989;264:10072–10082. [PubMed] [Google Scholar]
- 33.Finkel SE, Johnson RC. The Fis protein: it's not just for DNA inversion anymore (erratum) Mol. Microbiol. 1992;6:1023. doi: 10.1111/j.1365-2958.1992.tb02193.x. [DOI] [PubMed] [Google Scholar]
- 34.Pan CQ, Johnson RC, Sigman DS. Identification of new Fis binding sites by DNA scission with Fis-1,10-phenanthroline-copper(I) chimeras. Biochemistry. 1996;35:4326–4333. doi: 10.1021/bi952040z. [DOI] [PubMed] [Google Scholar]
- 35.Green J, Anjum MF, Guest JR. The ndh-binding protein (Nbp) regulates the ndh gene of Escherichia coli in response to growth phase and is identical to Fis. Mol. Microbiol. 1996;19:1043–1055. doi: 10.1111/j.1365-2958.1996.tb02545.x. [DOI] [PubMed] [Google Scholar]
- 36.Slany RK, Kersten H. The promoter of the tgt/sec operon in Escherichia coli is preceded by an upstream activation sequence that contains a high affinity FIS binding site. Nucleic Acids Res. 1992;20:4193–4198. doi: 10.1093/nar/20.16.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ball CA, Osuna R, Ferguson KC, Johnson RC. Dramatic changes in Fis levels upon nutrient upshift in Escherichia coli. J. Bacteriol. 1992;174:8043–8056. doi: 10.1128/jb.174.24.8043-8056.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bosch L, Nilsson L, Vijgenboom E, Verbeek H. FIS-dependent trans-activation of tRNA and rRNA operons of Escherichia coli. Biochim. Biophys. Acta. 1990;1050:293–301. doi: 10.1016/0167-4781(90)90184-4. [DOI] [PubMed] [Google Scholar]
- 39.Lyakhov IG, Hengen PN, Rubens D, Schneider TD. The P1 phage replication protein RepA contacts an otherwise inaccessible thymine N3 proton by DNA distortion or base flipping. Nucleic Acids Res. 2001;29:4892–4900. doi: 10.1093/nar/29.23.4892. http://www.ccrnp.ncifcrf.gov/~toms/paper/repan3/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Merickel SK, Sanders ER, Vazquez-Ibar JL, Johnson RC. Subunit exchange and the role of dimer flexibility in DNA binding by the Fis protein. Biochemistry. 2002;41:5788–5798. doi: 10.1021/bi020019+. [DOI] [PubMed] [Google Scholar]
- 41.Myszka DG. Improving biosensor analysis. J. Mol. Recognit. 1999;12:279–284. doi: 10.1002/(SICI)1099-1352(199909/10)12:5<279::AID-JMR473>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 42.Myszka DG, Morton TA. CLAMP: a biosensor kinetic data analysis program. Trends Biochem. Sci. 1998;23:149–150. doi: 10.1016/s0968-0004(98)01183-9. [DOI] [PubMed] [Google Scholar]
- 43.Brody JR, Kern SE. Sodium boric acid: a Tris-free, cooler conductive medium for DNA electrophoresis. Biotechniques. 2004;36:214–216. doi: 10.2144/04362BM02. [DOI] [PubMed] [Google Scholar]
- 44.Yuan HS, Finkel SE, Feng J-A, Kaczor-Grzeskowiak M, Johnson RC, Dickerson RE. The molecular structure of wild-type and a mutant Fis protein: relationship between mutational changes and recombinational enhancer function or DNA binding. Proc. Natl Acad. Sci. USA. 1991;88:9558–9562. doi: 10.1073/pnas.88.21.9558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Myszka DG, Jonsen MD, Graves BJ. Equilibrium analysis of high affinity interactions using BIACORE. Anal. Biochem. 1998;265:326–330. doi: 10.1006/abio.1998.2937. [DOI] [PubMed] [Google Scholar]
- 46.Karlsson R. Affinity analysis of non-steady-state data obtained under mass transport limited conditions using BIAcore technology. J. Mol. Recognit. 1999;12:285–292. doi: 10.1002/(SICI)1099-1352(199909/10)12:5<285::AID-JMR469>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 47.Berg OG, vonHippel PH. Selection of DNA binding sites by regulatory proteins. Trends Biochem. Sci. 1988;13:207–211. doi: 10.1016/0968-0004(88)90085-0. [DOI] [PubMed] [Google Scholar]
- 48.Das N, Valjavec-Gratian M, Basuray AN, Fekete RA, Papp PP, Paulsson J, Chattoraj DK. Multiple homeostatic mechanisms in the control of P1 plasmid replication. Proc. Natl Acad. Sci. USA. 2005;102:2856–2861. doi: 10.1073/pnas.0409790102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mirny LA, Gelfand MS. Structural analysis of conserved base pairs in protein-DNA complexes. Nucleic Acids Res. 2002;30:1704–1711. doi: 10.1093/nar/30.7.1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shannon CE. Communication in the presence of noise. Proc. IRE. 1949;37:10–21. [Google Scholar]
- 51.Pan CQ, Finkel SE, Cramton SE, Feng JA, Sigman DS, Johnson RC. Variable structures of Fis-DNA complexes determined by flanking DNA-protein contacts. J. Mol. Biol. 1996;264:675–695. doi: 10.1006/jmbi.1996.0669. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.