

Abstract
Insufficient expression of factor VIII (fVIII) is a major hurdle in the development of successful nucleic acid treatments for hemophilia. However, we recently showed that under myeloablative and reduced-intensity total body irradiation (TBI) conditioning, transplantation of hematopoietic stem cells (HSCs) transduced with recombinant retroviruses containing B domain–deleted porcine fVIII (BDDpfVIII) sequences provides curative fVIII levels in a hemophilia A mouse model. In the current study, we tested BDDpfVIII activity after nonmyeloablative conditioning with busulfan, cyclophosphamide, or fludarabine and immunosuppressive agents CTLA4-Ig + anti-CD40L or anti-(murine)thymocyte serum (ATS). ATS is similar in action to anti-(human)thymocyte globulin (ATG), which is used clinically with busulfan in bone marrow transplantations to increase donor cell engraftment. Mice conditioned with busulfan + ATS and that received a transplant of BDDpfVIII-transduced stem-cell antigen 1-positive cells exhibited moderate levels of donor cell chimerism (between 20% and 60%) and achieved sustained fVIII levels more than 1 U/mL. Similar results were observed in mice preimmunized with human fVIII and conditioned with 5 Gy TBI + ATS or busulfan + ATS. These data demonstrate that it is possible to achieve sufficient fVIII expression after transplantation of BDDpfVIII-transduced HSCs following low-toxicity pretransplantation conditioning with targeted immunosuppression, potentially even in the context of preexisting inhibitors.
Introduction
Hemophilia A is an X-linked recessive genetic disorder leading to a deficiency of functional clotting factor VIII (fVIII). One in 5000 males is affected by hemophilia A, which in its severe form (accounting for approximately 50% of all cases) is life threatening. Clinically, the disease is characterized by spontaneous or traumatic bleeding, usually into joints or soft tissues but also into critical closed spaces such as the cranial cavity or retroperitoneal space. Spontaneous bleeds are most frequently observed in severe hemophiliacs, defined as those with less than 0.01 U/mL fVIII activity. Currently, the treatment of choice is prophylactic administration of recombinant fVIII protein with the goal of maintaining fVIII levels above 0.01 U/mL. Recombinant fVIII therapy for hemophilia A, while thought to be safer than plasma derived therapy, can be extremely costly with the average patient using up to $100 000 of fVIII product per year. Therapy can also be complicated by infection or thrombosis of the central venous port typically required for repeated intravenous access in pediatric patients. In addition, up to 30% of severe hemophiliacs develop antibodies against fVIII, which often represents a neoantigen to their immune system, significantly increasing the complexity and cost of their care.1
In order to surmount the ongoing risk and cost of recombinant fVIII therapy, gene therapy has been proposed as a potential cure for hemophilia A. As a monogenic disorder requiring only modest improvements in circulating protein levels to afford significant clinical improvement, hemophilia A seems to be an ideal candidate for gene therapy. Despite promising data in murine and canine studies, clinical trials thus far have been hampered by meager levels of circulating fVIII and have had little clinical impact (for review see High2). All trials to date used human fVIII (hfVIII) transgenes. However, Doering et al recently demonstrated that B domain–deleted porcine fVIII (BDDpfVIII) is expressed at 10- to 14-fold greater levels than hfVIII in vitro and that the increased expression is due to enhanced secretion.3 Consistent with these results, we showed that mesenchymal stem cells transduced with recombinant retroviruses containing BDDpfVIII sequences express fVIII more efficiently than those transduced with recombinant retroviruses containing hfVIII sequences.4 In addition, genetic modification and transplantation of hematopoietic stem cells (HSCs), which offer a means to introduce the BDDpfVIII transgene into cells that are long-lived and undergo self-renewal and differentiation,5 under myeloablative and reduced-intensity pretransplantation conditioning resulted in curative fVIII levels. FVIII expression was sustained for greater than 1 year posttransplantation in a mouse model of hemophilia A, thus overcoming the barrier of low fVIII expression.4 Alternatively, Moayeri et al achieved sustained levels of a bioengineered hfVIII under myeloablative conditions by transplanting high doses of genetically modified bone marrow cells.6 Since prior studies have shown that in vivo transduction can lead to a host immune response and elimination of transduced cells,7 these successes indicate that transduction of cells ex vivo and transplantation of the genetically modified cells under myelosuppressive conditioning may be necessary.
In the current study, we aimed to develop and test clinically relevant hematopoietic stem cell transplantation (HSCT) regimens using (1) low multiplicity of infection (moi) transduction, (2) nonmyeloablative conditioning, (3) transplantation of low cell numbers, and (4) transient immunosuppression to prevent inhibitor formation. Hemophilia A mice received a transplant of MSCV-BDDpfVIII-transduced stem cell antigen-1-positive (sca-1+) cells following conditioning with low-dose total body irradiation (TBI) or various chemotherapy regimens including busulfan (BU), cyclophosphamide (CY), and fludarabine (FLU). We show that following nonmyeloablative transplantation of BDDpfVIII-modified HSCs into hemophilia A mice it is possible to achieve fVIII levels similar to that observed in healthy mice and humans.
Materials and methods
This study was approved by Emory's IACUC.
Materials
DNA and RNA extraction kits were purchased from Qiagen (Valencia, CA). DNA extraction from peripheral blood was performed using the Puregene DNA purification system from Gentra (Minneapolis, MN). Polymerase chain reaction (PCR) reagents were purchased from Applied Biosystems (Foster City, CA). All antibodies for flow cytometry were purchased from BD Pharmingen (San Diego, CA). Recombinant cytokines were purchased from R&D Systems (Minneapolis, MN). COATEST SP fVIII assay kits were purchased from DiaPharma (West Chester, OH). Cell culture media and supplements were purchased from Invitrogen (Carlsbad, CA). Magnetic separation columns were purchased from Miltenyi Biotec (Auburn, CA). Pooled normal human plasma (FACT) and human fVIII-deficient plasma were purchased from George King Biomedical (Overland Park, KA). Recombinant hfVIII (ADVATE; Baxter) was generously provided by Hemophilia of Georgia (Atlanta, GA) and Amy Dunn (Aflac Cancer Center and Blood Disorder Services, Atlanta, GA). Activated partial thromboplastin reagent was purchased from Organon Technika (Durham, NC); BU, from PDL BioPharma (Fremont, CA); CY, from Sigma Aldrich (St Louis, MO); and FLU, from Gensia Sicor Pharmaceuticals (Irvine, CA). Anti-CD40 ligand and CTLA4-Ig were a kind gift from Dr Christian Larsen (Emory University, Atlanta, GA). Antithymocyte serum was purchased from Intercell Technologies (Jupiter, FL). Anti-BDDpfVIII and anti-hfVIII monoclonal antibodies were a kind gift of Dr Pete Lollar (Aflac Cancer Center and Blood Disorder Services). Exon 16-disrupted hemophilia A mice and enhanced green fluorescence protein (eGFP) transgenic mice have been previously described.8,9 EGFP transgenic mice were used as donors in the HSC transplantation experiments. All mice that underwent transplantation were between 8 to 12 weeks old. TBI was performed using a Gammacell 40 Exactor (Nordion, Ottawa, Canada). Complete blood counts were performed on a Heska CBC-Diff Veterinary Hematology System (Heska, Fribourg, Switzerland).
Production of MSCV-fVIII vectors and viral concentrates
MSCV-BDDpfVIII viral supernatant was produced as previously described.4 Briefly, BD RetroPack PT67 viral producer cells (BD Biosciences, San Jose, CA) were transfected transiently with the MSCV-BDD porcine fVIII plasmid. Virus-containing medium was collected daily and placed onto BD EcoPack2 cells (BD Biosciences). Conditioned medium was collected daily, 0.45-μm filtered, and stored at − 80°C until use. Virus was concentrated overnight by centrifugation at 9000g at 4°C, resuspended in 1/40th the original volume, and stored at − 80°C in 1-mL aliquots. Viral titer determination has been previously described.4 Stable MSCV-BDD porcine fVIII ecotropic retroviral producer cell lines were established that routinely produced viral stocks containing 105 to 106 TUNIH3T3/mL following concentration.
Isolation and transduction of murine stem cell antigen-1+ cells
Sca-1+ cell isolation was performed as previously described using positive immunomagnetic bead selection.4 Prior to transduction, cells were stimulated for 2 days in serum-free media containing murine stem cell factor (100 ng/mL), murine interleukin-3 (20 ng/mL), human interleukin-11 (100 ng/mL), and human Flt-3 ligand (100 ng/mL). One million cells per well of a 6-well plate were transduced consecutively on days 3 and 4 with 1 to 2 × 106 TU each day.
Transplantation of sca-1+ cells
Transduced cells were transplanted intravenously into conditioned recipient mice via tail vein injection on day 0. CY was administered intraperitoneally at 100 mg/kg per day on days − 3 and − 2 relative to transplantation. BU was given intraperitoneally either as 17.5 mg/kg per day on days − 3 and − 2 (35 mg/kg total) or as a single dose of 20 mg/kg on day − 2. FLU was administered intraperitoneally at 30 mg/kg per day on days − 3, − 2, and − 1. CTLA4-Ig and anti-CD40L were given as intraperitoneal injections at 500 μg/dose on days 0 and + 2. ATS was administered intraperitoneally on days − 1 and 0 at 30 mg/kg except for the 3 Gy TBI + ATS mice that received ATS only on day 0. When given on day 0, CTLA4-IG, anti-CD40L, and ATS were given 2 to 4 hours prior to transplantation. Total donor cell (eGFP+) engraftment and fVIII expression were analyzed by flow cytometry using a BD LSR-II. Gene-marking analysis was performed using real-time PCR as described for viral titering with the following modifications. The oligonucleotide sequences were as follows: porcine fVIII forward, 5′-TCAGTCTACTGGCACGTGATTGGA-3′ and reverse, 5′-AGCAGTGAGGAAAGTTAG TGGCGA-3′. Final primer concentrations were 250 nM.
FVIII analyses
FVIII activity was measured using the activated partial thromboplastin one-stage coagulation assay and chromogenic fVIII activity assay as described previously.10 Total anti-fVIII IgG levels were determined by enzyme-linked immunosorbent assay (ELISA) as described previously.11,12 FVIII inhibitory activity was determined using a modified Bethesda assay13 in which experimental plasma samples were incubated with human fVIII-deficient plasma reconstituted with recombinant BDD porcine fVIII for 2 hours and assayed for residual fVIII activity.14 Intracellular staining for flow cytometry was performed using the Cytofix/Cytoperm kit according to the manufacturer's suggestions (BD Biosciences) with a biotinylated mouse anti-BDDpfVIII primary antibody.
Statistical analysis
All values are expressed as mean plus or minus sample standard deviation. Student t test was used for comparisons between groups unless otherwise noted.
Results
Toxicities of nonmyeloablative and reduced-intensity regimens in hemophilia A mice
To translate genetically modified HSCT to clinical applications for the treatment of hemophilia A, it is necessary to achieve sustained levels of fVIII following transplantation under conditioning regimens with an acceptable risk-benefit ratio. In the current study, we explored the myelosuppressive and immunosuppressive properties of several clinically relevant chemotherapeutic and immunosuppressive agents in a murine model of hemophilia A (Table 1). We estimated that 10% to 50% engraftment of donor cells would be necessary to allow for engraftment of sufficient genetically modified cells, yet we anticipated the possibility of anti-BDDpfVIII inhibitory antibody formation under conditions that are less stringent than those previously demonstrated to be successful (ie, 11 and 5.5 Gy TBI). Therefore, we also tested the effectiveness of coadministrating mild immunosuppression.
Table 1.
Conditioning agents, doses, and dosing schedules for reduced-intensity BDDpfVIII transduced HSCTs
| Conditioning therapy | Day relative to transplantation | Total dose(s) |
|---|---|---|
| TBI | 0 | 2 Gy |
| TBI and CY | 0/−3, −2 | 2 Gy, 200 mg/kg |
| CY | −3, −2 | 200 mg/kg |
| CY and FLU | −3, −2/−3, −2, −1 | 200 mg/kg, 90 mg/kg |
| BU | −3, −2 | 35 mg/kg* |
| BU and CY | −3, −2 | 35 mg/kg, 200 mg/kg |
BU was administered at 35 mg/kg for toxicity and transplantation studies unless otherwise noted.
Complete blood counts (cbcs) were performed following each of the conditioning regimens listed in Table 1 as a means to gauge both the immunosuppressive and myelosuppressive properties (Figure 1A,B). Little effect was observed on peripheral leukocyte counts with BU, and while we did observe the characteristic rise and subsequent fall in platelet counts, no suppression was observed in the peripheral blood T-cell compartment (Figure 1C,D).15 Although several conditioning regimens resulted in similar levels of myelosuppression compared with 5.5 Gy TBI, engraftment of donor cells was not achieved under these conditions (Figure 1E). For these engraftment studies, 3 × 105 transduced sca-1+ cells were transplanted. This dose was chosen following testing of the engraftment levels obtained using various numbers of sca-1+ cells, where we found that 5 × 104 to 2 × 105 cells resulted in variable engraftment in animals conditioned with 5.5 Gy TBI prior to transplantation (data not shown). However, transplantation of 3 × 105 sca-1+ cells resulted in reproducible engraftment of more than 50% donor cells (data not shown),4 indicating this cell dose is near the minimum needed to achieve engraftment of sufficient genetically modified cells under stringent (ie, 5.5 Gy TBI) pretransplantation conditioning.
Figure 1.

Toxicity of nonmyeloablative conditioning regimens. Mice were conditioned with chemotherapeutic agents or TBI as described in “Materials and methods.” (A) White blood cell counts in mice conditioned with 2 Gy TBI (▵), 2 Gy TBI + 200 mg/kg CY (□), 200 mg/kg CY (◇), 90 mg/kg FLU + 200 mg/kg CY (▿), 20 mg/kg BU (●), and 35 mg/kg BU (♦). TBI (5.5 Gy, ○) is shown for comparison. BU + CY groups were omitted for clarity as they closely resembled the CY-only group. (B) Mean number of days to recovery of granulocytes to more than 500 per mm3. (C) Platelet counts for BU- and BU + CY-treated mice. (D) CD3+ T-cell counts (solid line) as well as the percentage of CD4+ and CD8+ subpopulations of CD3+ cells were followed by flow cytometry after conditioning with 35 mg/kg BU. (E) Percent donor chimerism measured by flow cytometry of eGFP+ donor cells in the peripheral blood following HSCT.
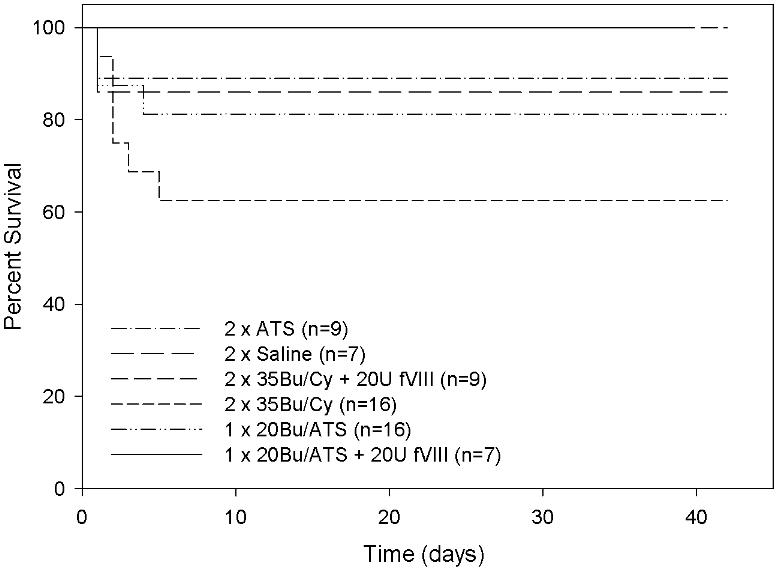
While hemophilia A mice have a normal life expectancy with careful handling, we observed injection-associated toxicity following intraperitoneal administration of chemotherapeutic agents. In particular, the BU + CY-treated mice showed elevated morbidity and mortality that was not observed in saline-injected animals (Figure S1, available on the Blood website; see the Supplemental Materials link at the top of the online article). Based on necropsy findings of intraperitoneal hemorrhage and previously normal cbcs indicating lack of drug toxicity, we conclude that multiple intraperitoneal injections of some chemotherapy agents in this hemophilia A murine model that is prone to bleeding does lead to hemorrhage and even death in some animals. This conclusion is supported by the observation that concomitant administration of recombinant hfVIII completely prevents conditioning-related mortality.
FVIII expression and inhibitor formation following BU + CY conditioning
Following transplantation of 3 × 105 sca-1+ cells, no engraftment was observed using BU + CY as conditioning agents, even though these regimens showed similar toxicity profiles compared with 5.5 Gy and others have observed donor engraftment using higher cell doses.16 Despite the observation that animals conditioned with BU or BU + CY had no detectable engraftment of donor cells, plasma fVIII activity was observed on day +7 after transplantation, suggesting that transplanted progenitor and/or short-term repopulating cells are capable of fVIII production under these conditions. Mean fVIII activity levels were 0.05 plus or minus 0.06 U/mL (5% of normal human fVIII levels) and 0.85 plus or minus 0.68 U/mL (85% of normal human fVIII levels) on day 7 with BU and BU + CY conditioning, respectively. But, plasma fVIII activity was no longer detectable on and subsequent to day + 14 after transplantation (Figure 2A).
Figure 2.

Transient fVIII activity and subsequent inhibitor formation following BU + CY conditioning and HSCT/ BDDpfVIII gene therapy. Mice were conditioned with 35 mg/kg BU and 200 mg/kg CY on days − 3 and − 2, then received a transplant of 3 × 105 BDDpfVIII-transduced sca-1+ cells on day 0. (A) FVIII activity levels on days + 7 and + 14 as measured by chromogenic fVIII assay. Dashed line indicates lower limit of detection. Mice were assayed biweekly after day + 14 and no fVIII activity was detected in any mice. (B) Anti-BDDpfVIII inhibitory antibody titers as determined by the Bethesda assay. Solid line indicates upper limit of detection.
Having shown that these mice were exposed to BDDpfVIII produced from genetically modified cells during the first week following transplantation, we next determined if they mounted an immune response against BDDpfVIII. Circulating anti-BDDpfVIII inhibitory antibodies were detected on day + 14 following transplantation (Figure 2B). Compared with the BU-only group, Bethesda titers were lower (< 10 Bethesda units) and more transient in the BU + CY-treated animals. When rechallenged with 20 U BDDpfVIII at 6 months after transplantation, the BU + CY mice exhibited an anamnestic response equal to that of the BU-only-treated mice, as mean inhibitor titers were 103 Bethesda units one month after challenge in the BU + CY group versus 111 Bethesda units in the BU-only group.
Costimulation blockade prevents inhibitory antibody formation
Based on the anti-BDDpfVIII immune response observed in the BU and BU + CY studies, we hypothesized that further immunosuppression is likely needed at the time of transplantation of BDDpfVIII-transduced sca-1+ cells to prevent inhibitory antibody formation, and while the inclusion of CY in the conditioning regimen did decrease overall inhibitor formation, it did not prevent an immune response to BDDpfVIII. Others have demonstrated that anti-fVIII antibody formation following naked plasmid delivery can be blocked using transient immunosuppression,17 and that mixed hematopoietic chimerism has been achieved following low-dose BU combined with costimulation blockade.18 We hypothesized that similar findings might be observed for HSC gene transfer and, therefore, tested a short course of costimulation blockade in addition to myelosuppressive conditioning. Animals received CTLA4-Ig and anti-CD40L at 500 μg per dose on days 0 and +2 following conditioning with BU at 35 mg/kg. Low-dose costimulation blockade (2 doses versus ≥ 4 doses typically used by others17) had no effect on peripheral T-cell counts in hemophilia A mice (Figure 3A), and compared with the BU-only group, these mice engrafted at significantly higher levels (Figure 3B versus Figure 1E) (P = .001). Mean engraftment was 35.7% plus or minus 22.4% in the peripheral blood, 34.4% plus or minus 22.8% in the bone marrow, and 27.6% plus or minus 17.9% in the spleen, as determined by flow cytometry at 6 months after transplantation. Sustained therapeutic levels (> 1% of the normal human level or ∼ 0.01 U/mL) of fVIII were observed in 7 of 8 animals (range, 0.02-0.79 U/mL) (Figure 3B). Mean proviral copy number in these mice was 0.03 plus or minus 0.02 (range, 0-0.07) in the peripheral blood and 0.15 plus or minus 0.20 (range, 0.01-0.51) in the bone marrow at the time of death (Figure 3C).
Figure 3.

Toxicity, fVIII activity, and donor cell engraftment following BU conditioning with costimulation blockade. Mice were conditioned with 35 mg/kg BU on days − 3 and − 2 and then received a transplant of 3 × 105 BDDpfVIII-transduced sca-1+ cells on day 0. CTLA4-Ig and anti-CD40L were administered on days 0 and + 2 relative to transplantation. (A) The percentages of B and T cells over time were determined by flow cytometry after administration of BU + costimulation blockade. (B) eGFP+ donor-cell chimerism in the peripheral blood versus fVIII activity at 2 weeks, 8 weeks, and 24 weeks after transplantation. (C) Percent eGFP+ donor-cell chimerism versus mean proviral copy number in the peripheral blood versus fVIII activity at the time of death, 6 months after transplantation.
In vivo T-cell depletion facilitates increased fVIII activity following HSCT gene therapy
Increased engraftment and fVIII activity following transplantation using costimulation blockade led us to explore further T-cell depletion regimens as part of the HSCT gene therapy protocol (Table 2). Antithymocyte globulin (ATG) has been used widely in clinical bone marrow transplantation (BMT) to prevent and to treat graft-versus-host disease.19 Additionally, ATG is increasingly being used as an adjuvant to nonmyeloablative conditioning regimens as a means of boosting donor cell engraftment. We show that antithymocyte serum (ATS), which is a polyclonal rabbit antimouse thymocyte product, effectively depletes peripheral blood T cells in vivo with mean recovery of 42 days after transplantation (Figure 4A).20
Table 2.
Summary of the results from reduced-intensity conditioning regimens with and without immunosuppression
| Conditioning agent(s) | Sca-1+ cell dose | Immunosuppression | Percent chimerism | FVIII activity, U/mL |
|---|---|---|---|---|
| BU, 35 mg/kg | 300 000 | none | 0 | 0 |
| BU, 35 mg/kg and CY, 200 mg/kg | 300 000 | none | 0 | 0 |
| BU, 35 mg/kg | 300 000 | Anti-CD40L + CTLA4Ig | 29.6 ± 18.1 | 0.12 ± 0.19 |
| BU, 20 mg/kg | 300 000 | ATS | 7.6 ± 9.6 | 0.01 ± 0.01 |
| BU, 20 mg/kg | 1 000 000 | ATS | 27.2 ± 18.6 | 2.66 ± 1.96 |
| BU, 35 mg/kg | 300 000 | ATS | 41.1 ± 25.5 | 4.04 ± 1.66 |
| BU, 35 mg/kg | 1 000 000 | ATS | 18.5 ± 13.6 | 3.44 ± 2.07 |
| TBI, 3 Gy | 300 000 | none | 0 | 0 |
| TBI, 3 Gy | 300 000 | ATS | 17.4 ± 7.3 | 2.05 ± 0.87 |
| TBI, 3 Gy | 1 000 000 | ATS | 53.8 ± 15.2 | 3.73 ± 1.08 |
Data shown are from 14 to 16 weeks after transplantation.
Figure 4.

BU + ATS conditioning facilitates sustained, therapeutic levels of fVIII. Mice were conditioned with BU on day − 3 and − 2, treated with 30 mg/kg ATS on days − 1 and 0, and then received a transplant of BDDpfVIII-transduced sca-1+ cells on day 0. (A) In vivo T-cell depletion following 2 intraperitoneal doses of ATS with representative flow cytometry analysis on days − 2 and + 3. (B) Mean fVIII plasma activity levels for mice treated with 20 mg/kg BU + ATS, 3 × 105 sca-1+ cells (●), 20 mg/kg BU + ATS, 106 sca-1+ cells (▴), 35 mg/kg BU + ATS, 3 × 105 sca-1+ cells (●), and 20 mg/kg BU + ATS, 106 sca-1+ cells (■). (C-E) Percentage eGFP+ donor-cell chimerism in the peripheral blood versus fVIII activity for mice conditioned with (C) 35 mg/kg BU + ATS and that received a transplant of 3 × 105 sca-1+ cells, (D) 20 mg/kg BU + ATS and that received a transplant of 106 sca-1+ cells, and (E) 35 mg/kg BU + ATS and that received a transplant of 106 sca-1+ cells at 14 weeks after transplantation. (F) Mean fVIII activity levels in mice conditioned with 3 Gy TBI ± ATS on day 0.
To compare the use of ATS to the BU + costimulation blockade transplants, a cohort of mice was conditioned with 35 mg/kg BU, treated with ATS on days − 1 and 0, and received a transplant of 3 × 105 BDDpfVIII-transduced sca-1+ cells. At 19 weeks after transplantation, all mice exhibited stable donor-cell chimerism (45.8% ± 34.0%) and therapeutic fVIII activity levels of 3.14 plus or minus 2.83 U/mL (Figure 4B,C). Therefore, ATS facilitated similar engraftment levels (P = .68) with higher fVIII activity (P = .001) compared with costimulation blockade in BU-conditioned mice (Figures 3B and 4B).
We next sought to determine the lower limit of myelosuppression that would allow for sufficient engraftment of genetically modified cells. Adding ATS to the low-dose BU (20 mg/kg) regimen increased engraftment of donor cells above 20 mg/kg BU alone (7.6% ± 9.6% versus 0%, respectively) but did not result in detectable plasma fVIII activity, except for one animal (Figure 4B). In order to determine if engraftment and fVIII activity could be improved simply by transplanting more cells, mice were conditioned with 20 mg/kg BU + ATS and received a transplant of 106 sca-1+ cells. Six of 7 animals engrafted with eGFP+ donor cells and had fVIII activity levels greater than 0.01 U/mL at 14 weeks after transplantation (Figure 4B,D). To determine if fVIII expression and activity could be increased further, a fourth cohort of mice received a transplant of 106 sca-1+ cells under 35 mg/kg BU + ATS conditioning. Mean fVIII activity in these mice was 3.44 plus or minus 2.07 U/mL with mean donor chimerism of 18.7% plus or minus 13.6% at 16 weeks after transplantation, which was not a significant improvement over the 35 mg/kg BU + ATS, 3 × 105 sca-1+ cell dose group (P = .967 and P = .128, respectively, for fVIII activity and donor-cell chimerism) (Figure 4B,E). In addition to measuring fVIII activity by chromogenic assay, hemostatic correction was assessed by a one-stage clotting assay (aPTT) for the 35 mg/kg BU + ATS, 3 × 105 sca-1+ cell, and 20 mg/kg BU + ATS, 106 sca-1+ cell groups. We found a significant correction of clotting times in E16−/− mice that underwent transplantation versus naive E16−/− mice (P < .001) as well as a significant correlation between BDDpfVIII activity levels by chromogenic assay and clot times in these mice (P < .001) (Figure S2). In order to determine if these mice would be tolerant to human fVIII in addition to BDDpfVIII, we administered 10 U hfVIII 5 months after transplantation. No anti-hfVIII or anti-BDDpfVIII ELISA titer was detected 1 week after challenge and no significant change in BDDpfVIII levels was detected.
To determine which cells were responsible for fVIII production, 4 mice were killed from the 35 mg/kg BU + ATS, 3 × 105 cell group and 4 mice from the 20 mg/kg BU + ATS, 106 cell group at 12 weeks after HSCT. Bone marrow and spleen cells were stained for intracellular BDDpfVIII as well as extracellular membrane-bound CD45, B220, CD3, Gr-1, and Mac. FVIII production was observed in eGFP+ donor cells with a higher percentage of fVIII-positive cells in the bone marrow than in the spleen. Within the bone marrow, the Gr-1+/Mac+ population had the highest percentage of fVIII positive cells, at more than 50% (Figure 5). The average proviral copy number for bone marrow and spleen in the 35 mg/kg BU + ATS, 3 × 105 cell group was 0.95 plus or minus 0.49 and in the 20 mg/kg BU + ATS, 1 × 106 cell group the average copy number was 1.44 plus or minus 1.32.
Figure 5.

Intracellular staining for BDDpfVIII in bone marrow and spleen after transplantation with BDDpfVIII-transduced sca-1+ cells. Four mice were killed from both the 35 mg/kg BU + ATS, 3 × 105 cells and the 20 mg/kg BU + ATS, 106 cell groups. Bone marrow and spleen cells were harvested and analyzed by flow cytometry for intracellular BDDpfVIII staining, as well as surface markers for lineage differentiation. Representative data are shown for bone marrow (A-D,L) and spleen (E-H) cells from mice that underwent transplantation. Negative controls are blood from a C57BL/6 mouse (I-K) and an isotype control (L) using a biotinylated mouse anti-hfVIII antibody that is not cross-reactive to porcine or murine fVIII. Panels A-H and L are gated on CD45+ donor cells. The mouse shown was engrafted with 34% and 47% GFP+ donor cells in the bone marrow and spleen, respectively.
We next determined if the positive effects of ATS could be applied to other nonmyeloablative conditioning regimens or if the effect is particular to BU. We used low-dose TBI as it is a reliable and reproducible means of myelosuppression with no variability due to drug metabolism, which is sometimes observed with BU.15 Mice were conditioned with 3 Gy TBI and transplanted with 3 × 105 or 106 BDDpfVIII-transduced sca-1+ cells with or without ATS treatment on the day of transplantation (Figure 4F). The results were similar to those observed with BU conditioning with no observed donor engraftment following conditioning with 3 Gy TBI alone but significant donor engraftment and sustained, curative fVIII levels when ATS was included in the regimen. In the group that received a transplant of 3 × 105 cells, mean engraftment was 16.9% plus or minus 7.1% with mean fVIII activity of 2.38 plus or minus 1.0 U/mL at 20 weeks after HSCT. Donor engraftment was significantly higher in the group that received 106 cells (mean 53.7% ± 12.7%, P = .005), and a significant difference in fVIII activity was observed (mean 4.93 ± 1.08 U/mL, P = .001) (Figure 4F).
In primates, ATG eliminates peripheral blood T cells, while T cells within the lymphatic system are less affected at low ATG doses.21 To determine if ATS acts similarly in hemophilia A mice, a cohort of animals was treated for 2 days with ATS and then blood, lymph nodes, and spleen were collected on day + 5. Similar to observations in nonhuman primate models, T cells were effectively cleared from the peripheral blood, but T cells in the spleen and in the peripheral lymphoid tissues were significantly less affected (P = .09 and P = .68, respectively) (Figure S3).
Sustained fVIII activity in preimmunized mice facilitated by T-cell depletion
Given the success in naive animals, we determined if sustained fVIII activity levels could be achieved in animals preimmunized with hfVIII. We previously showed that lethally irradiated preimmunized mice will engraft and express BDDpfVIII, but under reduced-intensity conditioning (5.5 Gy TBI) some animals failed to engraft with BDDpfVIII-transduced sca-1+ cells.10 In the current study, hemophilia A mice were injected intravenously once every week with recombinant hfVIII for 6 weeks. The mean anti-hfVIII ELISA titer was 408 (range, 61-1260) at the time of transplantation, with 9 of 10 animals demonstrating plasma cross-reactivity to BDDpfVIII (Figure 6A). Three of 10 animals displayed an anti-BDDpfVIII Bethesda titer prior to transplantation. Mice were conditioned with either 5.5 Gy or 35 mg/kg BU, and all mice received ATS on days − 1 and 0. All animals treated with 5.5 Gy + ATS engrafted with donor cells and exhibited sustained fVIII activity at 2.92 plus or minus 2.06 U/mL at19 weeks after transplantation (Figure 6C,D). Of note, the 2 mice with the highest anti-hfVIII ELISA and anti-BDDpfVIII Bethesda titers had the lowest fVIII activity after transplantation (Figure 6B). The 35 mg/kg BU + ATS-treated animals closely resemble animals conditioned with 5.5 Gy TBI without ATS since only 3 of 5 mice showed sustained fVIII activity (1.91 ± 2.13 U/mL), although all of the animals engrafted with eGFP+ cells (Figure 6E,F). Hemostatic correction was assessed by aPTT at 19 weeks and found to correlate with fVIII activity measured by chromogenic assay in these mice as well (Figure S2). In order to determine if preimmunized mice that are making BDDpfVIII are also tolerant to hfVIII, they were challenged with 10 U hfVIII 5 months after transplantation. Anti-hfVIII and anti-BDDpfVIII ELISA titers 1 week following challenge were undetectable in all mice. There was no significant change in plasma fVIII levels in either cohort of animals.
Figure 6.

ATS improves circulating fVIII activity levels following BDDpfVIII-transduced HSCT of preimmunized mice. Hemophilia A mice received 6 weekly intravenous injections of recombinant hfVIII (10 U/injection). One week after the last injection, plasma was collected from each mouse (n = 5 per group) and assayed for anti-hfVIII or anti-BDDpfVIII total Ig (ELISA) and inhibitory activity (Bethesda assay). Panel A shows the anti-BDDpfVIII ELISA versus the anti-hfVIII ELISA for each mouse. Panel B shows BDDpfVIII activity following HSCT gene therapy versus pretransplantation anti-hfVIII ELISA titer for each animal. ELISA titer is defined as the reciprocal of the antibody dilution that leads to a signal 3 times background in 15 minutes. Donor cell engraftment (C) and fVIII activity (D) are shown for each animal conditioned with 5.5 Gy TBI + ATS and that received a transplant of 3 × 105 BDDpfVIII-transduced sca-1+ cells. Panels E and F show the same analysis for mice conditioned with 35 mg/kg BU + ATS and that received a transplant of 3 × 105 BDDpfVIII-transduced sca-1+ cells.
Discussion
Successful HSCT-based gene therapy for hemophilia A depends on the ability to achieve sufficient engraftment of genetically modified cells capable of expressing therapeutic levels of fVIII. In the developed world, hemophilia A is a manageable disease for which quality of life and cost, and less frequently mortality, are the main concerns for patients and physicians.22 In this context, a gene-therapy protocol must offer the patient a superior quality of life at no increased risk beyond that of standard treatment. HSCT gene therapy offers the ability to efficiently introduce the fVIII transgene into stem cells ex vivo, thereby providing for long-term expression while limiting the risk of patient exposure to live virus. The level of engraftment of genetically modified cells is determined by multiple factors including transduction efficiency, total transplanted cell dose, and the intensity of the pretransplantation conditioning. Balance must be achieved between the risks and benefits associated with each of these factors.
The safety of HSCT has improved with the development of reduced-intensity conditioning regimens. With respect to the use of gene-modified HSCT for hemophilia A mice, radiation has been used successfully by us4 and others.6 However, because of toxicity and the potential for secondary malignancy, it is generally assumed that similar radiation-based conditioning regimens will not be an acceptable treatment for the majority of patients with hemophilia A.23 Reduced-intensity conditioning regimens using agents such as BU, FLU, and CY along with the immunosuppressive agent ATG have been tested clinically with the goal of decreasing treatment-related toxicities. These regimens do not completely eradicate host hematopoiesis and, therefore, allow for rapid hematopoietic recovery.
In this study, we found that BU + ATS conditioning and HSCT gene therapy promoted sustained, supraphysiological levels of fVIII. While 12 mg/kg BU is considered to be the limit of nonmyeloablative conditioning in humans, 20 to 35 mg/kg was needed in mice to achieve the same level of engraftment that would be anticipated with 8 to 12 mg/kg BU in humans. As shown, the levels of BU used in this study were minimally myelosuppressive with very little effect observed in peripheral blood counts. Lower than expected donor-cell engraftment may occur since transplantation of eGFP+ C57BL/6 genetically modified donor cells into C57BL/6 hemophilia A mice does not truly replicate the scenario of autologous HSCT, as the expression of both eGFP and fVIII are immunogenic. This factor combined with the 4-day transduction protocol and the transplantation of low sca-1+ cell numbers in the current study can account for the decreased engraftment observed under some reduced-intensity conditioning regimens.
Malignant transformation due to insertional mutagenesis has been proven to be a potential adverse event associated with MSCV-based retroviral gene transfer.24,25 Therefore, another consideration for successfully translating genetically modified HSCT for the treatment of hemophilia A is the necessity of producing sufficient fVIII under conditions of limited proviral insertion per genetically modified cell. In order to decrease the risk of insertional mutagenesis, both the moi and number of genetically modified cells transplanted should be kept to a minimum. We were able to use a relatively low moi (< 5) and perform transplants using a relatively low number of cells (3 × 105). Compared with the transplantation of more than 106 bone marrow cells into a single animal, this experimental design also more accurately reproduces a scenario of autologous transplantation in which the number of stem cells (eg, CD34+ cells) that can be collected from an individual patient prior to selection, transduction, and transplantation is limiting.26 As another safety measure, a platelet-specific promoter was recently used for fVIII gene therapy and was shown to be feasible in a mouse model of hemophilia A.27,28 Our observation that the majority of BDDpfVIII-positive cells in the bone marrow 12 weeks after transplantation were granulocytes may lend some additional insight into the ability of certain lineages of hematopoietic cells to efficiently express this transgene.29 However, as shown by Klug et al,30 the apparent selective expression in granulocytes may be due to LTR inactivation in the T-cell population. So, while further studies are needed to explore the relationship between intracellular staining and fVIII secretion by these cells, the knowledge that a specific cell type efficiently expresses fVIII could lead to a safer retroviral vector using a lineage-specific promoter.
Clinical trials have shown the difficulty of achieving sufficient fVIII expression using several gene transfer strategies, partially because fVIII is an inefficiently produced protein. In addition, Hawley and colleagues observed only modest fVIII levels in mice after transplantation of bone marrow cells genetically modified with a L303E/F309S fVIII double mutant.31 This mutant was previously shown to increase fVIII expression due to enhanced fVIII secretion, which presumably results from more efficient trafficking from the endoplasmic reticulum to the Golgi. We previously showed that BDDpfVIII overcomes the low expression barrier and more than 100% normal human fVIII levels were achieved after transplant of BDDpfVIII-modified sca-1+ cells under myeloablative and reduced-intensity TBI-based conditioning regimens.4 In the current study using primarily non-TBI-based reduced-intensity conditioning regimens, we again show that the BDDpfVIII construct overcomes the low expression barrier and demonstrate sustained curative fVIII levels in recipient animals. Further studies are required to determine if similar expression levels can be achieved from human hematopoietic cells and, therefore, testing human CD34+ cells both in vitro and in nonobese diabetic/severe combined immunodeficient (NOD/SCID) mouse models is warranted.
In the majority of mice, we observed maximum fVIII activity within the first month after transplant (Figures 4B,F and 6D,F). During this period, fVIII expression is likely derived from integrated and nonintegrated transgene-containing cells and from genetically modified lineage-committed progenitor cells, short-term repopulating cells, and HSCs. It would be predicted that this would lead to a burst of fVIII activity following transplantation, followed by fluctuations during the initial weeks after transplantation as different cell populations expand and then become extinct, thereby altering the plasma fVIII levels, which was observed. In the absence of immunosuppression, as observed in the BU or BU + CY groups, the exposure to BDDpfVIII likely led to immune activation, inhibitory antibody formation, and rejection of genetically modified cells. However, we showed that with the incorporation of additional immunosuppression into the conditioning protocol (ie, costimulation blockade or ATS) the recipient animals became nonresponsive to the BDDpfVIII transgene product and were able to sustain high levels of fVIII activity without anti-BDDpfVIII immune activation. Additionally, these mice were nonresponsive to hfVIII when challenged 5 months after transplantation. The observation that T cells are not depleted from lymph nodes and are only mildly decreased in spleen is consistent with human studies but still perplexing as to the mechanism of tolerance induction in the current study. Our ongoing studies designed to determine the mechanisms of tolerance induction in this model should provide further insight into the mechanism of fVIII immunogenicity. Because no anti-fVIII immune response was observed, it is possible that the mechanism of tolerance in our model is distinct from the mechanism involved in fVIII immune tolerance induction therapy in which large doses of hfVIII are administered in an effort to exhaust the patient's fVIII-specific immune response.32 In addition, because costimulation blockade has little effect on T-cell numbers, it is possible that tolerance through the use of ATS and costimulation blockade are through different mechanisms. These data suggest that multiple routes of immune tolerance induction may be possible in the clinical use of gene-modified HSCT.
Patients with inhibitory antibodies against fVIII are extremely difficult and costly to manage, and represent the patient population that could most benefit from a novel therapeutic alternative. Our previous studies in preimmunized mice10 indicated that with sufficient myeloablation, engraftment of BDDpfVIII-transduced HSCs could be achieved, and no further inhibitor antibody development was observed. These results were somewhat expected based on previous studies on the effects of TBI on the B-cell compartment.33 In the current study, we show that reduced-intensity conditioning with BU + ATS is sometimes successful when applied to mice with pre-existing immunity to hfVIII and that these mice were nonresponsive to both BDDpfVIII and hfVIII. We, therefore, conclude that the goal of nonmyeloablative conditioning and HSCT gene therapy is feasible in this patient population, but more aggressive preconditioning may be required for engraftment of genetically modified cells and sustained expression of fVIII in the presensitized setting.
The goal of the current study was to determine if HSCT incorporating BDDpfVIII gene-modified cells could be used effectively to treat hemophilia A under conditions of reduced-intensity transplantation. Mice conditioned with 35 mg/kg BU or 3 Gy TBI combined with transient immunosuppression at the time of transplantation, either with administration of costimulation blockade or ATS, exhibited sustained high-level expression of BDDpfVIII without inhibitory antibody formation. These results demonstrate that reduced-intensity conditioning and transplantation of BDDpfVIII-modified hematopoietic stem cells is a compelling treatment for hemophilia A.
Supplementary Material
Acknowledgments
This work was supported by a fellowship to L.M.I. by the PhRMA foundation, the National Heart, Lung, and Blood Institute of the National Institutes of Health to H.T.S. (HL081165) and C.B.D. (HL083531), and by a grant from Children's Healthcare of Atlanta to the Aflac Cancer Center and Blood Disorders Service's Gene Therapy Program.
We thank Pete Lollar for his suggestions and helpful discussions. We also thank the members of his laboratory, John Healey, Ernie Parker, and Rachel Barrow for their technical help.
Footnotes
The online version of this manuscript contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: L.I., K.-Y.C., C.D., and H.T.S. designed the research. L.I. and B.G. performed the research. L.I., C.D., and H.T.S. analyzed data. L.I. and H.T.S. wrote the paper, and all authors checked the final version.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: H. Trent Spencer, 2015 Uppergate Dr, Emory Children's Center Room 420, Emory University, Atlanta, GA 30322; e-mail: hspence@emory.edu.
References
- 1.Lollar P. Pathogenic antibodies to coagulation factors. Part one: factor VIII and factor IX. J Thromb Haemost. 2004;2:1082–1095. doi: 10.1111/j.1538-7836.2004.00802.x. [DOI] [PubMed] [Google Scholar]
- 2.High K. Gene transfer for hemophilia: can therapeutic efficacy in large animals be safely translated to patients? J Thromb Haemost. 2005;3:1682–1691. doi: 10.1111/j.1538-7836.2005.01460.x. [DOI] [PubMed] [Google Scholar]
- 3.Doering CB, Healey JF, Parker ET, Barrow RT, Lollar P. Identification of porcine coagulation factor VIII domains responsible for high level expression via enhanced secretion. J Biol Chem. 2004;279:6546–6552. doi: 10.1074/jbc.M312451200. [DOI] [PubMed] [Google Scholar]
- 4.Gangadharan B, Parker ET, Ide LM, Spencer HT, Doering CB. High-level expression of porcine factor VIII from genetically modified bone marrow-derived stem cells. Blood. 2006;107:3859–3864. doi: 10.1182/blood-2005-12-4961. [DOI] [PubMed] [Google Scholar]
- 5.Wognum AW, Eaves AC, Thomas TE. Identification and isolation of hematopoietic stem cells. Arch Med Res. 2003;34:461–475. doi: 10.1016/j.arcmed.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 6.Moayeri M, Hawley TS, Hawley RG. Correction of murine hemophilia A by hematopoietic stem cell gene therapy. Mol Ther. 2005;12:1034–1042. doi: 10.1016/j.ymthe.2005.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Manno CS, Pierce GF, Arruda VR, et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med. 2006;12:342–347. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- 8.Bi L, Lawler AM, Antonarakis SE, High KA, Gearhart JD, Kazazian HH., Jr Targeted disruption of the mouse factor VIII gene produces a model of haemophilia A. Nat Genet. 1995;10:119–121. doi: 10.1038/ng0595-119. [DOI] [PubMed] [Google Scholar]
- 9.Nakanishi T, Kuroiwa A, Yamada S, et al. FISH analysis of 142 EGFP transgene integration sites into the mouse genome. Genomics. 2002;80:564–574. doi: 10.1006/geno.2002.7008. [DOI] [PubMed] [Google Scholar]
- 10.Doering CB, Gangadharan B, Dukart HZ, Spencer HT. Hematopoietic stem cells encoding porcine factor VIII induce pro-coagulant activity in hemophilia A mice with pre-existing factor VIII immunity. Mol Ther. 2007;15:1093–1099. doi: 10.1038/sj.mt.6300146. [DOI] [PubMed] [Google Scholar]
- 11.Doering CB, Parker ET, Healey JF, Craddock HN, Barrow RT, Lollar P. Expression and characterization of recombinant murine factor VIII. Thromb Haemost. 2002;88:450–458. [PubMed] [Google Scholar]
- 12.Doering CB, Parker ET, Nichols CE, Lollar P. Decreased factor VIII levels during acetaminophen-induced murine fulminant hepatic failure. Blood. 2003;102:1743–1744. doi: 10.1182/blood-2003-03-0826. [DOI] [PubMed] [Google Scholar]
- 13.Kasper CK, Aledort L, Aronson D, et al. Proceedings: a more uniform measurement of factor VIII inhibitors. Thromb Diath Haemorrh. 1975;34:612. [PubMed] [Google Scholar]
- 14.Parker ET, Healey JF, Barrow RT, Craddock HN, Lollar P. Reduction of the inhibitory antibody response to human factor VIII in hemophilia A mice by mutagenesis of the A2 domain B-cell epitope. Blood. 2004;104:704–710. doi: 10.1182/blood-2003-11-3891. [DOI] [PubMed] [Google Scholar]
- 15.Kang EM, Hsieh MM, Metzger M, et al. Busulfan pharmacokinetics, toxicity, and low-dose conditioning for autologous transplantation of genetically modified hematopoietic stem cells in the rhesus macaque model. Exp Hematol. 2006;34:132–139. doi: 10.1016/j.exphem.2005.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Andersson G, Illigens BM, Johnson KW, et al. Nonmyeloablative conditioning is sufficient to allow engraftment of EGFP-expressing bone marrow and subsequent acceptance of EGFP-transgenic skin grafts in mice. Blood. 2003;101:4305–4312. doi: 10.1182/blood-2002-06-1649. [DOI] [PubMed] [Google Scholar]
- 17.Miao CH, Ye P, Thompson AR, Rawlings DJ, Ochs HD. Immunomodulation of transgene responses following naked DNA transfer of human factor VIII into hemophilia A mice. Blood. 2006;108:19–27. doi: 10.1182/blood-2005-11-4532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adams AB, Durham MM, Kean L, et al. Costimulation blockade, busulfan, and bone marrow promote titratable macrochimerism, induce transplantation tolerance, and correct genetic hemoglobinopathies with minimal myelosuppression. J Immunol. 2001;167:1103–1111. doi: 10.4049/jimmunol.167.2.1103. [DOI] [PubMed] [Google Scholar]
- 19.Russell JA, Turner AR, Larratt L, et al. Adult recipients of matched related donor blood cell transplants given myeloablative regimens including pretransplant antithymocyte globulin have lower mortality related to graft-versus-host disease: a matched pair analysis. Biol Blood Marrow Transplant. 2007;13:299–306. doi: 10.1016/j.bbmt.2006.10.017. [DOI] [PubMed] [Google Scholar]
- 20.Mandel MA, Asofsky R. The effects of heterologous anti-thymocyte sera in mice. 3: high susceptibility of germfree mice to the suppressive effects of IgG from rabbit anti-mouse thymocyte serum. J Exp Med. 1969;129:1203–1216. doi: 10.1084/jem.129.6.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Preville X, Flacher M, LeMauff B, et al. Mechanisms involved in antithymocyte globulin immunosuppressive activity in a nonhuman primate model. Transplantation. 2001;71:460–468. doi: 10.1097/00007890-200102150-00021. [DOI] [PubMed] [Google Scholar]
- 22.Hoots WK, Nugent DJ. Evidence for the benefits of prophylaxis in the management of hemophilia A. Thromb Haemost. 2006;96:433–440. doi: 10.1160/th06-02-0125. [DOI] [PubMed] [Google Scholar]
- 23.Lenz G, Dreyling M, Schiegnitz E, et al. Moderate increase of secondary hematologic malignancies after myeloablative radiochemotherapy and autologous stem-cell transplantation in patients with indolent lymphoma: results of a prospective randomized trial of the German Low Grade Lymphoma Study Group. J Clin Oncol. 2004;22:4926–4933. doi: 10.1200/JCO.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 24.Baum C, Kustikova O, Modlich U, Li Z, Fehse B. Mutagenesis and oncogenesis by chromosomal insertion of gene transfer vectors. Hum Gene Ther. 2006;17:253–263. doi: 10.1089/hum.2006.17.253. [DOI] [PubMed] [Google Scholar]
- 25.Laufs S, Gentner B, Nagy KZ, et al. Retroviral vector integration occurs in preferred genomic targets of human bone marrow-repopulating cells. Blood. 2003;101:2191–2198. doi: 10.1182/blood-2002-02-0627. [DOI] [PubMed] [Google Scholar]
- 26.Becker PS. The current status of gene therapy in autologous transplantation. Acta Haematol. 2005;114:188–197. doi: 10.1159/000088409. [DOI] [PubMed] [Google Scholar]
- 27.Shi Q, Wilcox DA, Fahs SA, et al. Lentivirus-mediated platelet-derived factor VIII gene therapy in murine haemophilia A. J Thromb Haemost. 2007;5:352–361. doi: 10.1111/j.1538-7836.2007.02346.x. [DOI] [PubMed] [Google Scholar]
- 28.Ohmori T, Mimuro J, Takano K, et al. Efficient expression of a transgene in platelets using simian immunodeficiency virus-based vector harboring glycoprotein Ibalpha promoter: in vivo model for platelet-targeting gene therapy. FASEB J. 2006;20:1522–1524. doi: 10.1096/fj.05-5161fje. [DOI] [PubMed] [Google Scholar]
- 29.Tiede A, Eder M, von Depka M, et al. Recombinant factor VIII expression in hematopoietic cells following lentiviral transduction. Gene Ther. 2003;10:1917–1925. doi: 10.1038/sj.gt.3302093. [DOI] [PubMed] [Google Scholar]
- 30.Klug CA, Cheshier S, Weissman IL. Inactivation of a GFP retrovirus occurs at multiple levels in long-term repopulating stem cells and their differentiated progeny. Blood. 2000;96:894–901. [PubMed] [Google Scholar]
- 31.Moayeri M, Ramezani A, Morgan RA, Hawley TS, Hawley RG. Sustained phenotypic correction of hemophilia a mice following oncoretroviral-mediated expression of a bioengineered human factor VIII gene in long-term hematopoietic repopulating cells. Mol Ther. 2004;10:892–902. doi: 10.1016/j.ymthe.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 32.Astermark J, Morado M, Rocino A, et al. Current European practice in immune tolerance induction therapy in patients with haemophilia and inhibitors. Haemophilia. 2006;12:363–371. doi: 10.1111/j.1365-2516.2006.01296.x. [DOI] [PubMed] [Google Scholar]
- 33.Slifka MK, Antia R, Whitmire JK, Ahmed R. Humoral immunity due to long-lived plasma cells. Immunity. 1998;8:363–372. doi: 10.1016/s1074-7613(00)80541-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}