

Abstract
Bipolar disorder is a serious psychiatric condition that has been treated for over fifty years with lithium. Lithium is a well established glycogen synthase kinase-3 (GSK-3) inhibitor, suggesting that manipulating GSK-3 may have therapeutic value in treating bipolar disorder. GSK-3 is regulated by a wide variety of mechanisms including phosphorylation, binding with protein complexes, phosphorylation state of its substrates, cellular localization and autoregulation, thus providing a wide number of potential therapeutic mechanisms. Mounting evidence suggests that GSK-3 regulation can be used to manage bipolar disorder symptoms. Although GSK-3 mutations have not been detected amongst the general bipolar population, they have been correlated with females with bipolar II and most of the drugs used for successful bipolar disorder treatment regulate GSK-3. These drugs produce a weak antidepressant-like and a strong antimania-like effect in a wide range of animal models tested, mirroring their utility in treating bipolar disorder symptoms. Taken together the evidence suggests that targeting GSK-3 may be a means to control the symptoms of bipolar disorder.
Keywords: Animal Models, Anti-depressant, Anti-Mania, Bipolar Disorder, GSK-3, Lithium, Valproate
1. Introduction
Bipolar mood disorder is a serious chronic illness characterized by persistent mood changes involving both elevated and depressed mood states. The two classes of bipolar disorder, bipolar I and bipolar II, are differentiated by the length and degree of mood elevation and the presence of major depression. Bipolar I is characterized by mania (one week of abnormally elevated or irritable mood) with or without major depression. Bipolar II is defined by hypomania (at least 4 days of elevation different from a normal mood state) with major depression (depressed mood, loss of interest/pleasure, weight change, insomnia, psychomotor retardation, fatigue, feelings of worthlessness, suicidal ideology, diminished thought or concentration capacity) (American Psychiatric Association, 1994).
The toll that bipolar mood disorder takes on its sufferers is high, having a lifetime prevalence rate of 3.9% in the United States alone. Thus, better ways to treat this disorder are constantly being researched (Kessler et al., 2005). The devastating effect of bipolar mood disorder is combated with a variety of drugs generally divided into three categories: antipsychotics (chlorpromazine, haloperidol), atypical antipsychotics (risperidone, olanzapine, clozapine), and mood stabilizers (lithium carbonate, valproic acid, lamotrigine, carbamazepine) (Sachs et al., 2000). Although these drugs have a reasonably high success rate in treating bipolar disorder, particularly the mania and hypomania symptoms, the biological underpinnings of bipolar disorder and the biochemical mechanisms of how these drugs exert their effects are not yet fully understood. One potential target that has gained interest in recent years is glycogen synthase kinase-3 (GSK-3) (Gould et al., 2006; Manji et al., 1999). Although GSK-3 is a fascinating molecule with diverse functions in and of itself, this review is specifically interested in the following questions: 1) Is bipolar disorder a genetic disease, and, if it is, is GSK-3 mutation causally related to bipolar disorder, and 2) Can GSK-3 modulation be used to treat bipolar disorder symptoms?
2. Functional Roles of GSK-3
GSK-3 regulates over 40 proteins which can be generally divided into metabolic and signaling proteins, structural proteins, and transcription factors (Grimes and Jope, 2001; Jope and Johnson, 2004). GSK-3 exerts many effects on pathways and functions such as neuroplasticity, neurotransmission, metabolic, and growth/neuronal polarity. Clinically, the inhibition of GSK-3 is being investigated for treating multiple disease states. For example, lithium through GSK-3 inhibition has been shown to inhibit tau hyperphosphorylation, Aβ generation, Aβ-induced cytotoxicity and to suppress Aβ-elicited memory deficits in preclinical models, suggesting potential utility of GSK-3 inhibition in treating Alzheimer’s disease (Rockenstein et al., 2007; reviewed in Chuang & Priller, 2006).Other clinical areas where GSK-3 is being investigated for treatment potential include type 2 diabetes (Henriksen and Dokken, 2006), shock and inflammation (Dugo et al., 2007), schizophrenia (Jope and Roh, 2006), and cancer (Polakis, 2007). Discussing all regulatory functions and clinical applications of GSK-3 is beyond the scope of this paper. Therefore, in this review we focus on neuroprotection since it has relevance to bipolar disorder.
The idea that bipolar disorder may be linked to cellular loss was first put forth following findings of a reduced volume of brain grey matter in bipolar afflicted patients. Initial postmortem and structural PET studies found that grey matter volume was reduced by about 40% in the region of the subgenual prefrontal cortex in these patients (Drevets et al., 1997), and this reduction was subsequently linked to a loss in glial cell number and, to a lesser extent, neuronal number or size (Ongur et al., 1998, Rajkowska et al., 1999). Follow-up studies extended these findings to other areas such as the amygdala (Bowley et al., 2002). Apoptosis has also been linked to bipolar disorder due to findings of increased expression of apoptotic genes (Benes et al., 2006) and mitochondrial malfunction, suggesting cellular loss as an underlying cause (Kato and Kato, 2000).
It is well documented that GSK-3 is generally pro-apoptotic and inhibition of GSK-3 is anti-apoptotic under a wide variety of conditions (Beurel and Jope, 2006). In almost all cases, increasing GSK-3 activity induces apoptosis (Pap and Cooper, 1998) and/or sensitizes cells towards other apoptotic stimuli (Kaytor and Orr, 2002). It has also been linked to apoptotic death in numerous phenomena ranging from hypoxia (Roh et al., 2005) to Huntington’s disease (Carmichael et al., 2002). Conversely, inhibiting GSK-3 activity reverses or inhibits apoptosis under a wide variety of conditions (Beurel and Jope, 2006; Liang and Chuang, 2007).
The variety of conditions under which GSK-3 promotes apoptosis has led to the suggestion that GSK-3 may be a fundamental player in apoptotic signaling. This hypothesis is further supported by the number of downstream targets of GSK-3 involved in cell fate. For example, the Wnt pathway is thought to be important for both the maintenance of cellular integrity and prevention of premature phagocytic disposal of cells (Chong and Maiese, 2004). Among the transcription factors phosphorylated and regulated by GSK-3, many are involved in cell survival, including both pro-apoptotic factors such as p53, a regulator of Bcl-2 family members (Watcharasit et al., 2002), and anti-apoptotic factors such as CREB, HSF-1, and AP-1. GSK-3 inhibits CREB-mediated transcription by inhibiting its binding (Bullock and Habener, 1998), and negatively regulates HSF-1 by inhibiting HSF-1 DNA binding and HSF-1-dependent transcription (Bijur and Jope 2000; Xavier et al., 2000). Finally, GSK-3 negatively modulates AP-1 binding activity by phosphorylating c-Jun (Boyle et al., 1991).
3. GSK-3 Regulation
GSK-3 was initially identified as a kinase involved in glycogen metabolism, where it phosphorylates glycogen synthase, the rate limiting enzyme, to inhibit its activity (Embi et al., 1980). It was also identified as a key component of Wnt signaling (Patel et al., 2004). Since then, the list of proteins regulated by GSK-3 has grown almost exponentially and now includes over 40 proteins involved in metabolism and signaling, structure and transcription, as mentioned in the preceding section.
GSK is a serine/theonine kinase and consists of two isoforms, GSK-3α and GSK-3β, with GSK-3β having a splice variant (Mukai et al., 2002). These two isoforms are structurally similar except for an additional glycine-rich N-terminal domain in the α isoform, and have 98% sequence identity except for the end of the C-terminus (Doble and Woodgett, 2003). Less is known about functional differences between the two isoforms, although increasing evidence suggests that their functions are not always identical (Liang and Chuang, 2006). Since GSK-3β is the more abundant and better understood of the two, it will be the focus of this review.
GSK-3 regulation has a number of unusual characteristics. First, GSK-3 is constitutively active and hence is primarily regulated via inactivation through serine phosphorylation rather than the standard activation. GSK-3’s activity is inhibited by N-terminal phosphorylation at Ser-9 in GSK-3β and Ser-21 in GSK-3α (Cohen, 1999; Frame and Cohen, 2001). N-terminal phosphorylation decreases GSK-3 activity via a conformational change whereby the N-terminus folds back upon itself and blocks active sites necessary for substrate binding. In essence, the N-terminus is acting as a competitive pseudo-substrate with GSK-3’s active sites (Dajani et al., 2001). Tyrosine phosphorylation, Tyr216 in GSK3β and Tyr279 in GSK3α, acts in opposition to this by significantly increasing GSK-3 activity, although the manner by which it does so is unclear (Hughes et al., 1993). Proteins capable of phosphorylating GSK-3β are widespread and include, for the Ser-9 site, p70s6k, p90rsk, protein kinase A (PKA), protein kinase B (Akt), protein kinase C (PKC), and integrin-linked kinase (ILK) (Doble and Woodgett, 2003). Known agents capable of phosphorylating Tyr216 include Src-like Fyn kinase, Ca2+-sensitive proline-rich tyrosine kinase 2 (PYK2), zaphod kinase 1 (ZAK1), and mitogen-activated protein kinase kinase 1 (MEK1) (Meijer et al., 2004).
A second unusual aspect of GSK-3 mediated regulation is its preference for a primed substrate. GSK-3 contains a consensus sequence of Ser/Thr-X-X-X-Ser-P/Thr-P. The first Ser/Thr is the target residue, and the last is a priming site (Doble and Woodgett, 2003), meaning that GSK-3-mediated phosphorylation has a preference for targets which have already been phosphorylated or “primed” such that it is 100-1000 fold more efficient on primed substrates (Thomas et al., 1999). For example, phosphorylation of glycogen synthase by GSK-3 is dependent upon pre-priming via casein kinase 2 (CK2) (Fiol et al., 1988); thus factors regulating CK2 will in effect also be regulating GSK-3.
In addition to phosphorylation, a number of protein complexes can either stimulate or inhibit GSK-3 activity. Scaffolding proteins, such as axin, bring GSK-3 together with its substrates, hence increasing its activity, while GSK-3 binding proteins (GBP) inhibit GSK-3 when they form protein complexes with it (Thomas et al., 1999). Protein complex regulation may also allow localized regulation and differentially inhibit GSK-3’s ability to phosphorylate specific substrates (Jope and Johnson, 2004).
GSK-3 is primarily located in the cytosol; however, it is also located in organelles such as the mitochondria and nucleus and can show alterations in activity levels. Although little is known about the role of GSK-3 outside the cytosol, both the levels and activity of GSK-3 in these organelles change in accordance to cellular cues, suggesting that these compartments are actively involved in the kinase regulation (Jope and Johnson, 2004). Recent research indicates that the protein Frequently Rearranged in Advanced T-cell lymphomas 1,2 (FRAT 1,2) (Franca-Koh et al., 2002) results in the removal of GSK-3 from the nucleus, while the protein Latent Nuclear Antigen (LANA) sequesters it there (Fujimuro and Hayward 2003), providing at least one mechanism whereby GSK-3 may be regulated by subcellular localization.
In addition to outside influences, GSK-3 autoregulates it’s phosphorylation status via the Protein Phosphatase 1/Inhibitor-2 (PP-1/I-2) complex (Szatmari et al., 2005; Zhang et al., 2003). GSK-3 activates PP-1 by phosphorylating I-2, suppressing its inhibitory activity. Activated PP-1 in turn stimulates GSK-3 by dephosphorylating Ser-9. Functionally, this magnifies GSK-3 inhibition, as GSK-3 inhibition leads to less GSK-3 stimulation through the PP-1/I-2 complex (Marin et al., 1994). Table 1 summarizes the multiple mechanisms whereby GSK-3 activity can be regulated.
Table 1. Regulation of GSK-3 Activity by Multiple Mechanisms.
GSK-3 is regulated by a wide variety of mechanisms. Inhibitory mechanisms include N-terminal phosphorylation and sequestration of GSK-3 by binding proteins. Stimulatory mechanisms include tyrosine phosphorylation and increased substrate access through scaffolding proteins. GSK-3 shows a marked preference for phosphorylated substrates and several molecules can change GSK-3 intracellular location, although the functional consequences of this are still under investigation. Finally, GSK-3 can autoregulate through the PP-1/I-2 complex I where stimulus-induced changes in GSK-3 activity can be magnified.
| Regulation Method | Location of Regulation | Activity Change | Detailed Notes |
|---|---|---|---|
| Protein phosphorylation | N-terminal phosphorylation | Reduced activity | Ser9 in GSK-3β and Ser21 in GSK-3α |
| Protein phosphorylation | Tyrosine phosphorylation | Increased activity | Tyr216 in GSK3β and Tyr279 in GSK3α |
| Protein complex | GSK-3 binding proteins | Reduced activity | Sequester GSK-3 |
| Protein complex | Scaffolding proteins | Increased activity | Catalyze GSK-3 and substrate binding |
| Substrate preference | Phosphorylated substrates | Increased activity | 100-1000 times more efficient on primed substrates |
| Cellular localization | Nucleus | Unknown | Latent nuclear antigen |
| Cellular localization | Cytosol | Unknown | FRAT 1,2 |
| Autoregulation | PP-1 to Ser9 | Amplified activity | PP-1/I-2 complex I |
Given the number of different mechanisms of regulating GSK-3, it is not surprising that a large number of proteins and pathways are involved. As well documented and established pathways, Wnt and phosphatidylinositol 3-kinase (PI3K)/Akt will be discussed in some detail. These two pathways certainly do not represent a complete list of regulatory pathways or even molecules as shown by the previous discussion.
The Wnt-1 pathway, as shown in Figure 1, is an important target of GSK-3. GSK-3 targets this pathway through β-catenin, a proto-oncogene (Ryves and Harwood, 2003). Wnt proteins bind to frizzleds, a family of extracellular receptors (Liu et al., 1999, Malbon and Wang, 2006). GSK-3 is then inhibited when frizzleds activate the intracellular protein Dishevelled 1 (Boutros and Mlodzik, 1999). GSK-3β forms part of a β-catenin destruction complex that phosphorylates and thus facilitates the degradation of β-catenin. As a result, GSK-3β inhibition produces an increase in the levels of β-catenin that in turn interacts with the transcription factor Tcf/Lef (T-cell factor/lymphoid enhancer factor) (Barker et al., 2000). The Tcf/Lef-β-catenin complex is translocated to the nucleus and activates the transcription of diverse genes that include several involved in the inhibition of apoptosis (Barker et al., 2000).
Figure 1. Wnt-1 Pathway.
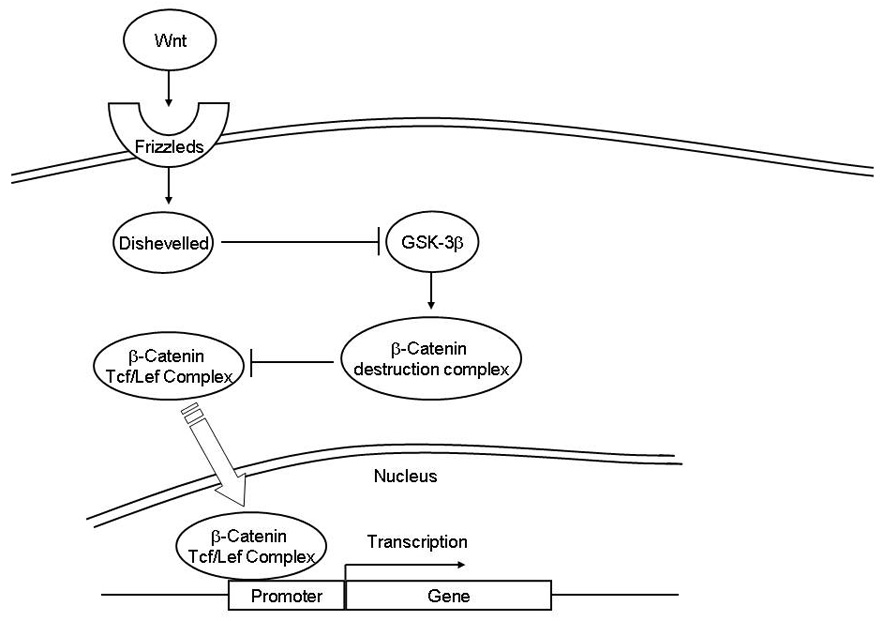
Wnt proteins bind to their receptors Frizzleds. Frizzleds in turn activate disheveled 1 which inhibits GSK-3β. GSK-3β binds to other proteins to form a β-catenin destruction complex which signals the degradation of β-catenin through phosphorylation. When not degraded, β-catenin activates transcription factors Tcf/Lef by binding to them. This complex is then translocated to the nucleus where it is involved in the gene expression of proteins including several anti-apoptotic proteins.
A second well known pathway that tightly regulates by GSK-3 is the PI3K/Akt pathway, as depicted in Figure 2. The PI3K/Akt pathway is an important regulator of cell survival (Hanada et al., 2004). This pathway begins with phosphorylation of tyrosine receptor kinase (Trk). For GSK-3, this has been most closely examined with brain derived neurotrophic factor (BDNF). BDNF binds to the TrkB receptor and leads to phosphorylation of an adaptor protein Shc, which then recruits the Grb2—SOS complex. At this point the pathway splits, although both pathways result in PI3K activation. In one path, the Grb2—SOS complex activates Ras, which in turn stimulates PI3K; in the other, Grb2 forms a complex with Gab1, which then recruits and activates PI3K. Once activated, PI3K produces 3-phosphoinositides, which recruit phospholipid-dependent kinases (PDKs). PDKs activate Akt-1 by phosphorylation of Ser-473 and/or Thr-308. Finally, Akt-1 acts on several targets including GSK-3β. In addition to BDNF other pathways use PI3K/Akt-mediated signaling to regulate GSK-3. The most widely known is the insulin signaling pathway, which regulates GSK-3 through Trk/PI3K/Akt via insulin peripherally and insulin-like growth factor in the brain (Bondy and Cheng, 2004).
Figure 2. PI3K-Akt-GSK-3 Signaling Pathways.

BDNF binds to the TrkB receptor. This leads to the phosphorylation of Shc. Phosphorylated Shc recruits the Grb2-SOS complex. This in turn activates separately Ras and Gab1 followed by Gab2. Both paths lead to activation of PI3K. Activated PI3K produces 3-phosphoinositides that recruit PDK which in turn activates Akt-1 through phosphorylation. Akt-1 then inhibits GSK-3β by phosphorylation.
4. Is GSK-3 a Therapeutic Target in Bipolar Disorder
A) GSK-3 polymorphisms and bipolar disorder
It is widely accepted that bipolar disorder has a strong genetic component with heritability estimates ranging as high as 80% (Kieseppa et al., 2004). Efforts to identify bipolar disorder’s genetic component have produced a plethora of candidates. From 2004 to 2006, over 35 candidate genes have been positively identified as being linked to bipolar disorder (Kato, 2007). Few of these reports are consistent and at present there is no widely accepted gene which either causes or predisposes one to bipolar disorder (Kato, 2007). A few genome scans found candidate loci in the general region of 3q13.3 GSK-3β (Badenhop et al., 2002; Bailer et al., 2002) the location of GSK-3β (Shaw et al., 1998). However, the lack of consistency in the field of bipolar disorder genome scans forces reliance on DNA polymorphism studies, which have specifically targeted GSK-3 for conclusions about its genetic influence on the presence of bipolar disorder.
Several studies have examined common GSK-3 single nucleotide polymorphisms (SNPs) for their relationship to bipolar disorder. In a Korean population, the −1727 A/T and −50 C/T SNPs of GSK-3 showed no difference between bipolar patients and controls (Lee et al., 2006) with similar results found in Caucasian populations (Nishiguchi et al., 2006). In an Italian population, the −50 C/T SNP also showed no difference between bipolar and controls, but did influence the age of onset of bipolar disorder (Benedetti et al., 2004). A fourth research study has found a general trend towards an association between the C allele, T/C polymorphism and bipolar disorder. A closer examination of the data found a strong link between the T-50C polymorphism and females with bipolar II (Szczepankiewicz et al., 2006b). These four studies indicate that GSK-3 mutations are not linked to the presence of bipolar disorder with the possible exception of females with bipolar II.
Even if GSK-3 mutations do not predict the occurrence of bipolar disorder, they may still predict the treatment efficiency, which would argue that GSK-3 manipulation is involved in the treatments. The first study that examined this issue found that the −50 C/T SNP mutation improved the recurrence index, (frequency of episodes pre- and post-lithium treatment) following lithium administration (Benedetti et al., 2005). However subsequent studies found that genotype and allele frequencies did not predict a lithium response in bipolar I patients (Michelon et al., 2006) nor was the T-50C polymorphism related to lithium prophylaxis (Szczepankiewicz et al., 2006a). It should be noted that both the negative studies divided the subject’s response to lithium into generalized categories of responders and hence may have missed the subtler differences found by Benedetti and co-workers. Taken together these studies suggest that GSK-3 mutations may influence the efficacy of lithium treatment in regards to the recurrence index in bipolar patients, and hence, GSK-3 should be considered as a potential target for treatment, but more detailed work is needed before strong conclusions can be drawn.
A second insight into the importance of GSK-3 in bipolar disorder may be found from reviewing the effects of other successful bipolar treatments on GSK-3. Do bipolar disorder treatments directly or indirectly regulate GSK-3 phosphorylation, protein levels, or downstream targets? If successful treatments of bipolar mood disorder do regulate GSK-3, then this suggests that GSK-3 can be a target for treating the symptoms, even though they are not the direct underlying biological cause of the disorder. Successful treatments for bipolar disorder in which their effects on GSK-3 activity have been investigated include lithium, valproate, lamotrigine, chlorpromazine, clozapine and carbamazepine.
B) Effects of lithium treatment on GSK-3
Lithium has a long history in the management of mood disorders and in particular bipolar disorder (Bech 2006). Even today it is the most common and successful treatment for bipolar disorder. This is critical to the theory that GSK-3 can have therapeutic relevance in bipolar disorder since the link between lithium and GSK-3 is well established. Not only does lithium inhibit GSK-3, but it does so both directly and indirectly. Although lithium inhibits both GSK-3α and GSK-3β (Klein and Melton, 1996; Stambolic et al., 1996), only GSK-3β has been rigorously examined, and hence only GSK-3β will be discussed. Lithium directly inhibits GSK-3β activity by competitively inhibiting Mg2+ binding to the active site of the enzyme (Klein and Melton, 1996, Ryves and Harwood, 2001). However, two discrepancies exist between lithium’s ability to directly inhibit GSK-3 and the known results of lithium administration. These two discrepancies imply that direct inhibition of GSK-3 alone cannot fully explain lithium’s neuroprotective and behavioral effects. The first discrepancy concerns treatment time. Direct inhibition of GSK-3 is rapid, while many of lithium’s effects require chronic treatment (Klein and Melton, 1996). The second discrepancy concerns dosage. The IC50 for direct in vitro inhibition, 1.5–2 mM, is greater than the therapeutic doses (e.g. 0.5–1 mM) typically used in clinics and experiments. This indicates that neuroprotective effects are seen when lithium has directly produced less than 50% GSK-3β inhibition.
As previously mentioned, GSK-3 is regulated by serine phosphorylation. Lithium increases phosphorylation levels of GSK-3β Ser-9 and GSK-3α Ser-21, subsequently inhibiting GSK-3 activity (Chalecka-Franaszek and Chuang, 1999; De Sarno et al., 2002; Zhang et al., 2003). The means by which this phosphorylation occurs is not yet fully delineated, but it is believed that multiple pathways play a role. One prominent pathway is the PI3K/Akt pathway. Lithium induces BDNF (Fukumoto et al.; 2001; Hashimoto et al., 2002), which, via the Trk B receptor, then stimulates the PI3K/Akt and MEK/ERK pathways (Chalecka-Franaszek and Chuang, 1999; Einat et al., 2003; Kopnisky et al., 2003). Lithium’s regulation of GSK-3 may also be partially through PKC, as PKC inhibitors reduce lithium-induced GSK-3 Ser-9 phosphorylation and lithium induces PKC activity in vivo (Kirshenboim et al., 2004). A third mechanism, autoregulation, perhaps should not be considered as a separate mechanism, but rather as a property of GSK-3 regulation integrated with other regulatory mechanisms. As previously indicated, GSK-3 inhibition is enhanced via the PP-1/I-2 complex. This system provides a theoretical explanation to counter the criticisms offered against the clinical significance of lithium’s direct inhibition of GSK-3β. Notably, the neuroprotective effects of lithium against excitotoxicity are mimicked by treatments with other GSK-3 inhibitors or transfection with GSK-3 isoform-specific siRNA or dominant-negative mutants (Liang and Chuang, 2007), suggesting that at least this aspect of lithium’s action is mediated through GSK-3 inhibition.
C) Effects of valproate and anti-psychotic drugs
Valproate is a short chain fatty acid with anti-convulsant properties used in the treatment of epilepsy and bipolar disorder, and has recently been shown to be neuroprotective (Leng and Chuang, 2006; Li et al., 2002; Ren et al., 2004) and have neurotrophic properties (Chen et al., 2006). Valproate has been approved by the United States Food and Drug Administration for treatment of seizures and acute mania and is considered an option for maintenance treatment of bipolar disorder. Clinical examination of valproate has found it to be highly efficient in treating the mania symptoms of bipolar disorder, but showed only modest effects alleviating the depressive aspects (Nasrallah et al., 2006).
The effects of valproate on GSK-3 have been extensively studied, but the results remain inconclusive. In support of valproate modulation of GSK-3, research has found that this drug inhibits GSK-3 through many different mechanisms, including elevation in GSK-3 serine phosphorylation levels (Chen et al., 1999; Kim et al., 2005; Kozlovsky et al., 2006) and inhibits GSK-3 activity in SH-SY5Y cells overexpressing GSK-3β as measured by its ability to phosphorylate tau (Grimes and Jope, 2001). Additionally, valproate has metabolites that may act as potent GSK-3 inhibitors in vivo (Werstuck et al., 2004). However, others have been unable to replicate these findings (Eickholt et al., 2005; Hall et al., 2002, Jin et al., 2005; Phiel et al., 2001). This discrepancy is likely to be an issue with the different experimental conditions and techniques, but until resolved, it is premature to conclude the effect that valproate has on GSK-3 in the clinical population. Even if valproate does not directly alter GSK-3 activity, it may interact with GSK-3-mediated pathways and produce many of the same results as GSK-3 inhibition. For example, valproate administration and GSK-3 inhibition both activate the Wnt pathway and increase β-catenin mRNA and protein levels (Chen et al., 1999; Phiel et al., 2001).
Far less work has been done on other mood stabilizers or anti-psychotic drugs used to treat bipolar disorder and schizophrenia, with only a few studies examining their effects on GSK-3. The results have been promising with many drugs tested showing a reduction in GSK-3 enzymatic activity, protein levels, or regulatory targets. Lamotrigine inhibits both heat-shock and staurosporine-induced caspase-3 activation in neuroblastoma SH-SY5Y cells overexpressing GSK-3β (Li et al., 2002). In a single study, chlorpromazine and clozapine were found to increase levels of phospho-Akt and decrease the levels of active, non-phosphorylated GSK-3β protein in Neuro-2A cells (Basta-Kaim et al., 2006). Independently, the levels of phospho-Ser-9-GSK-3β were found to be elevated by treatment with clozapine, but reduced with haloperidol in the frontal cortex of rats (Kozlovsky et al., 2006). Olanzapine was reported to decrease Akt and GSK-3 phosphorylation in L6 myotubes, while leaving their protein levels unchanged (Engl et al., 2005). A recent study shows that acute treatment of mice with olazepine, risperidone, clozapine, quetipine and ziprasidone, but not hapoperidol, markedly increases levels of phosphor-Ser-9-GSK-3β in the cortex, hippocampus, striatum and cerebellum (Li et al., 2007). Interestingly, combined treatment with risperidone and an anti-depressant, imipramine or flouxetine, produces a larger increase in brain levels of phospho-Ser-9-GSK-3β than either anti-depressant alone. A preliminary report shows that chronic treatment of rats with clazepine or haloperidol enhances the serine phosphorylation states of GSK-3β and Akt in the cortex and of GSK-3β in the hippocampus (Koros et al., 2006). Treatment with carbamazepine fails to alter GSK-3 activity as measured directly in neocortical cells (Ryves et al., 2005), or indirectly through caspase-3 activity in GSK-3β overexpressing cells (Li et al., 2002), although this drug enhances levels of ERK 1/2 phosphorylation in SH-SY5Y cells (Mai et al., 2002). A summary of the effects of the aforementioned mood stabilizers (except lithium) and anti-psychotic drugs on GSK-3 and related signaling are shown in Table 2.
Table 2. Effects of Mood Stabilizers and Anti-psychotic Drugs on GSK-3 and Related Signalings.
A wide variety of drugs or manipulations are used to treat bipolar disorder. While interactions between lithium and GSK-3 have been well studied, this is not the case for other bipolar disorder treatments. The table summarizes the reported effects of mood stabilizers and anti-psychotic drugs used to treat bipolar disorder on GSK-3 activity and GSK-3-related signalings. It is well established that lithium inhibits GSK-3 and hence lithium has not been included in the table.
| Treatment | Results of Drug Administration | References |
|---|---|---|
| | ||
| ECS | Biphasic Ser-9 alteration | Roh et al., 2003 |
| | ||
| Carbamazepine | Increased ERK ½ phosphorylation | Li et al., 2002 |
| | ||
| Increased phospho - Akt | Basta-Kaim et al., 2006 | |
| Chlorpromazine | ||
| Decreased total GSK-3β | Basta-Kaim et al., 2006 | |
| | ||
| Increased phospho - Akt | Basta-Kaim et al., 2006 | |
| Decreased total GSK-3β | Basta-Kaim et al., 2006 | |
| Clozapine | Increased GSK-3β Ser-9 phosphorylation | Korus et al., 2006 |
| Decreased GSK-3β protein levels | Kozlovsky et al., 2006 | |
| Increased GSK-3β Ser-9 phosphorylation | Li et al., 2007 | |
| | ||
| Increased GSK-3β Ser-9 phosphorylation | Korus et al., 2006 | |
| Haloperidol | Decreased GSK-3β protein levels | Kozlovsky et al., 2006 |
| Decreased GSK-3β Ser-9 phosphorylation | Li et al.; 2007 | |
| | ||
| Lamotrigine | Inhibited induced capsase-3 activation | Mai et al., 2002 |
| | ||
| Decreased Akt phosphorylation | Engl et al., 2005 | |
| Olanzapine | Decreased GSK-3 Ser-9 phosphorylation | Engl et al., 2005 |
| Increased GSK-3β Ser-9 phosphorylation | Li et al., 2007 | |
| | ||
| Risperidone | Increased GSK-3β Ser-9 phosphorylation | Li et al.; 2007 |
| | ||
| Increased β-catenin levels | Chen et al., 1999 | |
| Inhibited GSK-3 activity | Chen et al., 1999 | |
| Inhibited over expressed GSK-3β activity | Grimes and Jope, 2001 | |
| Inhibited GSK-3 activity | Kim et al., 2005 | |
| Decreased GSK-3α and β activity | Werstuck et al., 2004 | |
| Valproate | Increased GSK-3 protein levels | Kozlovsky et al., 2006 |
| No change in activity | Eickholt et al., 2005 | |
| No change in activity | Hall et al., 2002 | |
| Failed to inhibit GSK-3 activation | Jin et al., 2005 | |
| Failed to inhibit tau phosphorylation | Phiel et al., 2001 | |
| Activates Wnt pathway | Phiel et al., 2001 | |
| | ||
| Ziprasidone | Increased GSK-3β Ser-9 phosphorylation | Li et al., 2007 |
Although only a limited number of studies with each drug have been conducted, the fact that so many structurally different drugs all successfully treat bipolar disorder and all alter GSK-3 suggests that GSK-3 must be considered a target of high priority for further research and drug development. To date, research indicates that bipolar disorder is only associated with GSK-3 mutations amongst female bipolar II patients and fails to consistently find alterations in GSK-3 activity. This failure is offset by findings that GSK-3 mutations are linked to the onset of bipolar disorder and that the majority of bipolar treatments can alter GSK-3 pathways and produce similar functional outcomes. This suggests that while GSK-3 abnormalities may not be causing bipolar disorder, but treatments targeting GSK-3 pathways may be of therapeutic relevance.
5. GSK-3 in Animal Models of Depression and Mania
A) Depression Models
In the clinical population, bipolar disorder consists of bouts of mania and depression. In vivo models of bipolar disorder have been unable to replicate the cyclic nature of these mood changes, but rather, model depression and mania separately. In depression, GSK-3 appears to function normally, having normal levels of expression, phosphorylation, and mRNA (Kozlovsky et al., 2001, 2006). Lithium monotherapy is not generally used for the treatment of depression, although it was once the first choice for augmentation therapy and has a large body of work supporting its effectiveness in this role (Bauer et al., 2003; Bschor and Bauer, 2006). Unfortunately, the methodology of this work has been questioned and most of it is prior to the advent of modern serotonin-selective reuptake inhibitors (SSRIs), making it of limited use in evaluating lithium’s effectiveness in recent depression treatment paradigms (Nierenberg et al., 2003). The loss of interest in lithium as an augmenter stems primarily from practical concerns (e.g. therapeutic index, side effects, etc), rather than lack of effectiveness, and despite the criticisms, lithium augmentation is accepted by clinical guidelines. Therefore, while an in depth examination of combination therapies with lithium and modern SSRIs is needed, present evidence indicates that lithium is a highly successful augmenter.
From a treatment standpoint, the case for GSK-3’s involvement in depression is strong. Depression is regularly treated with drugs that modify the serotonin system, and in the most resistant cases, electroconvulsive therapy (ECT). The serotonin system also regulates GSK-3 in vivo (Li et al., 2002), and suicide victims have decreased Akt activity and increased GSK-3β activity (Karege et al., 2006).
Two types of animal models have been used to examine the relevance of GSK-3 to depressive symptoms. The first is the Porsolt, or forced swim test, the most widely used measure of a drug’s antidepressant-like properties. Unfortunately, the results of the forced swim test vary greatly with minor changes in parameters, and hence the literature is mired with seemingly contradictory results. In mice evaluated with the forced swim test, treatment with specific GSK-3 inhibitors, such as GSK-3 peptide inhibitor (Kaidanovich-Beilin et al., 2004) or chronic lithium (O’Brien et al., 2004) produced antidepressant-like effects.
Studies of GSK-3 mutant mice support these findings. GSK-3β has been disrupted via targeted deletion to produce a heterozygous, GSK-3β+/−, mouse. This mouse showed behavioral effects similar to lithium administration, including antidepressant-like effects in the forced swim test (O’Brien et al., 2004.) In rats, lithium seemed to produce either no effect (Hata et al., 1995; Kitamura et al., 2002; Wegener et al., 2003) or increased immobility, a depressant-like effect (Tomasiewicz et al., 2006). A novel GSK-3 inhibitor, acting as an ATP competitor, produced antidepressant-like effects (Gould et al., 2004). Finally, acute lithium administration was effective in mice, if injected 30 minutes prior to testing (Redrobe and Bourin, 1999).
Other drugs that are used to treat bipolar mood disorder have shown little effect in the forced swim test. Short-term valproate treatment produced no effect from doses of 30 mg/kg to 300 mg/kg in rats (Tomasiewicz et al., 2006) or doses of 1–32 mg/kg in mice (Redrobe and Bourin, 1999). Chronic dietary valproate treatment produced an antidepressant-like effect in CD-1 mice (Rowe et al., 2006). This effect was very sensitive to experimental parameters and was not seen following acute valproate treatment in different mouse strains such as C57, or following the mild stress of daily injections. Transgenic mouse models of GSK-3 overexpression have been examined in the forced swim test where the animals showed a generalized increase in activity level. Hence, GSK-3 overexpression is best viewed as not producing a resistance to depression-like behaviors, but rather as inducing behavioral changes suggestive of mania-like effects (Prickaerts et al., 2006).
Another animal model of depression treatment is electroconvulsive seizures (ECS), a manipulation used to model ECT in animals. ECS has been found to directly alter GSK-3 activity in rats (Roh et al., 2003). Following ECS, GSK-3β phosphorylation at Ser-9 was altered. Of particular interest, this change was biphasic. ECS resulted in an immediate dephosphorylation, which was followed by a reversal, such that hyperphosphorylation was present within a three-hour time period after ECS (Roh et al., 2003). This data is due to different responses in GSK-3 regulatory mechanisms following multiple ECS treatments. The initial response to ECS involved a reduction in kinase-based phosphorylation signals including Akt, GSK-3, and CREB and an increase in PP2A activity. This was followed by an increase in kinase-based activity that was overridden, if additional ECS was administered to reactivate PP2A (Kang et al., 2005). This biphasic response may explain the biphasic phosphorylation patterns preserved following ECS treatment. The level of β-catenin is altered by ECS in the expected manner, further reinforcing the idea that ECS regulates GSK-3 (Madsen et al., 2003).
Taken together, the evidence for GSK-3 producing an antidepressant-like effect in animal models is weak. GSK-3 inhibitors produce unreliable results in the forced swim test and seemingly minor variations in experimental procedure drastically alter results. While GSK-3 is altered in ECS, the results need further confirmation and the importance of this result to the antidepressant-like effects of ECS is still unknown. While this may seem an unsatisfying assessment of the potential of GSK-3 as a candidate for bipolar disorder treatment, it must be stressed that the forced swim test is a measure for antidepressant-like properties. The standard drugs discussed above alleviate the mania phases of bipolar disorder much better than the depressive phases, and in fact antidepressant-like drugs are frequently prescribed, in addition to the targeted bipolar mood disorder stabilizers. These results follow the clinical data well. Thus, a reasonable conclusion is that GSK-3 inhibitors have mild antidepressant-like effects.
B) Mania Models
To correlate with the clinical data GSK-3 inhibitors must show both strong antimania-like, as well as weak antidepressant-like properties. Unfortunately, the antimania-like properties have not been as closely investigated as the antidepressant-like effects. That said, existing evidence is positive, suggests that this is an area that needs further investigation. A drug’s antimania-like properties are most commonly shown via the amphetamine-induced hyperactivity test where drugs are suggested to have antimania-like properties if they reduce the hyperactivity. It has been found that reducing GSK-3 activity through both genetic deletion and pharmacological inhibition reduces dopamine-induced locomotor activity (Beaulieu et al., 2004). This line of investigation found that dopamine, through the D2 receptor, inactivates Akt via phosphorylation of Thr-308, which in turn activates GSK-3 signaling, leading to hyperactivity. Interestingly, GSK-3β did not seem to be involved in the animal’s response to amphetamine in the first 30 minutes, but small changes in GSK-3 activity had a large effect on behavior when examined over the entire time-course. A confirmation of GSK-3’s ability to regulate mania was seen when a novel selective ATP-competitive GSK-3 inhibitor successfully reduced amphetamine-induced hyperactivity (Gould et al., 2004). Transgenic mice overexpressing GSK-3 were found to have increased activity and decreased habituation resembling the hyperactivity found in mania (Prickaerts et al., 2006). The authors went as far as to suggest that these animals could be used as models for the mania aspect of bipolar mood disorder. Finally, hyperactivity of rats resulting from treatment with a MEK inhibitor, SL327, was prevented by chronic lithium pretreatment (Einat et al., 2003). As the MAP kinase pathway, through p90-RSK, and lithium both regulate GSK-3, lithium’s ability to inhibit hyperactivity is likely GSK-3 related (Rowe and Chuang, 2004).
6. Conclusion
The preponderance of data suggests that selective targeting of GSK-3 may be used successfully to treat bipolar disorder symptoms, particularly the manic symptoms, even if it does not cure the underlying cause of the disorder. This conclusion is drawn from three findings. 1) The majority of drug treatments classified as mood stabilizers and to a lesser extent atypical antipsychotics which are used to treat bipolar disorder manipulate GSK-3. Not only has lithium long been known to regulate GSK-3 through a variety of mechanisms, but the majority of atypical drugs have been shown to alter GSK-3 protein levels or activity, and valproate, in at least a few studies, has also been reported to influence GSK-3. While these drugs’ behavioral properties have not been casually linked to GSK-3 manipulation, the sheer coincidence of so many different drugs all producing the same behavioral effect and regulating the same molecule is highly suggestive. 2) Genetic linkage studies suggest that GSK-3 mutation is not linked to bipolar disorder, outside of female bipolar II patients. If GSK-3 is not the biological cause of bipolar disorder, but its manipulation treats this disorder, GSK-3 may be the target for treating symptoms, but not curing the biological cause of the disorder. This is further supported by the studies showing that GSK-3 mutations can predict aspects of bipolar disorder such as the recurrence index. 3) Finally in both animal models and the clinical population, GSK-3 manipulation appears to have weak antidepressant-like, but strong antimania-like effects. Therefore when looking to find novel treatments of the mania symptoms of bipolar disorder, GSK-3 is an excellent choice.
Acknowledgments
We would like to thank the Intramural Research Program of NIMH, NIH for support and the NIH Editorial Board, Dr. Min-Huei Liang and Peter Leeds of the Molecular Neurobiology section of NIMH for their editorial assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. fourth ed. Washington D.C.: American Psychiatric Association; 1994. [Google Scholar]
- Badenhop RF, Moses MJ, Scimone A, Mitchell PB, Ewen-White KR, Rosso A, Donald JA, Adams LJ, Schofield PR. A genome screen of 13 bipolar affective disorder pedigrees provides evidence for susceptibility loci on chromosome 3 as well as chromosomes 9, 13 and 19. Mol Psychiatry. 2002;7:851–859. doi: 10.1038/sj.mp.4001114. [DOI] [PubMed] [Google Scholar]
- Bailer U, Leisch F, Meszaros K, Lenzinger E, Willinger U, Strobl R, Heiden A, Gebhardt C, Doge E, Fuchs K, Sieghart W, Kasper S, Hornik K, Aschauer HN. Genome scan for susceptibility loci for schizophrenia and bipolar disorder. Biol Psychiatry. 2002;52:40–52. doi: 10.1016/s0006-3223(02)01320-3. [DOI] [PubMed] [Google Scholar]
- Barker N, Morin PJ, Clevers H. The Yin-Yang of TCF/beta-catenin signaling. Adv Cancer Res. 2000;77:1–24. doi: 10.1016/s0065-230x(08)60783-6. [DOI] [PubMed] [Google Scholar]
- Basta-Kaim A, Budziszewska B, Jaworska-Feil L, Tetich M, Kubera M, Leskiewicz M, Otczyk M, Lason W. Antipsychotic drugs inhibit the human corticotropin-releasing-hormone gene promoter activity in neuro-2A cells-an involvement of protein kinases. Neuropsychopharmacology. 2006;31:853–865. doi: 10.1038/sj.npp.1300911. [DOI] [PubMed] [Google Scholar]
- Bauer M, Adli M, Baethge C, Berghofer A, Sasse J, Heinz A, Bschor T. Lithium augmentation therapy in refractory depression: clinical evidence and neurobiological mechanisms. Can J Psychiatry. 2003;48:440–448. doi: 10.1177/070674370304800703. [DOI] [PubMed] [Google Scholar]
- Beaulieu JM, Sotnikova TD, Yao WD, Kockeritz L, Woodgett JR, Gainetdinov RR, Caron MG. Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc Natl Acad Sci U S A. 2004;101:5099–5104. doi: 10.1073/pnas.0307921101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bech P. The full story of lithium. A tribute to Mogens Schou (1918–2005) Psychother Psychosom. 2006;75:265–269. doi: 10.1159/000093947. [DOI] [PubMed] [Google Scholar]
- Benedetti F, Bernasconi A, Lorenzi C, Pontiggia A, Serretti A, Colombo C, Smeraldi E. A single nucleotide polymorphism in glycogen synthase kinase 3-beta promoter gene influences onset of illness in patients affected by bipolar disorder. Neurosci Lett. 2004;355:37–40. doi: 10.1016/j.neulet.2003.10.021. [DOI] [PubMed] [Google Scholar]
- Benedetti F, Serretti A, Pontiggia A, Bernasconi A, Lorenzi C, Colombo C, Smeraldi E. Long-term response to lithium salts in bipolar illness is influenced by the glycogen synthase kinase 3-beta -50 T/C SNP. Neurosci Lett. 2005;376:51–55. doi: 10.1016/j.neulet.2004.11.022. [DOI] [PubMed] [Google Scholar]
- Benes FM, Matzilevich D, Burke RE, Walsh J. The expression of proapoptosis genes is increased in bipolar disorder, but not in schizophrenia. Mol Psychiatry. 2006;11:241–251. doi: 10.1038/sj.mp.4001758. [DOI] [PubMed] [Google Scholar]
- Beurel E, Jope RS. The paradoxical pro- and anti-apoptotic actions of GSK3 in the intrinsic and extrinsic apoptosis signaling pathways. Prog Neurobiol. 2006;79:173–189. doi: 10.1016/j.pneurobio.2006.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bijur GN, Jope RS. Opposing actions of phosphatidylinositol 3-kinase and glycogen synthase kinase-3beta in the regulation of HSF-1 activity. J Neurochem. 2000;75:2401–2408. doi: 10.1046/j.1471-4159.2000.0752401.x. [DOI] [PubMed] [Google Scholar]
- Bondy CA, Cheng CM. Signaling by insulin-like growth factor 1 in brain. Eur J Pharmacol. 2004;490:25–31. doi: 10.1016/j.ejphar.2004.02.042. [DOI] [PubMed] [Google Scholar]
- Boutros M, Mlodzik M. Dishevelled: at the crossroads of divergent intracellular signaling pathways. Mech Dev. 1999;83:27–37. doi: 10.1016/s0925-4773(99)00046-5. [DOI] [PubMed] [Google Scholar]
- Bowley MP, Drevets WC, Ongur D, Price JL. Low glial numbers in the amygdala in major depressive disorder. Biol Psychiatry. 2002;52:404–412. doi: 10.1016/s0006-3223(02)01404-x. [DOI] [PubMed] [Google Scholar]
- Boyle WJ, Smeal T, Defize LH, Angel P, Woodgett JR, Karin M, Hunter T. Activation of protein kinase C decreases phosphorylation of c-Jun at sites that negatively regulate its DNA-binding activity. Cell. 1991;64:573–584. doi: 10.1016/0092-8674(91)90241-p. [DOI] [PubMed] [Google Scholar]
- Bschor T, Bauer M. Efficacy and mechanisms of action of lithium augmentation in refractory major depression. Curr Pharm Des. 2006;12:2985–2992. doi: 10.2174/138161206777947650. [DOI] [PubMed] [Google Scholar]
- Bullock BP, Habener JF. Phosphorylation of the cAMP response element binding protein CREB by cAMP-dependent protein kinase A and glycogen synthase kinase-3 alters DNA-binding affinity, conformation, and increases net charge. Biochemistry. 1998;37:3795–3809. doi: 10.1021/bi970982t. [DOI] [PubMed] [Google Scholar]
- Carmichael J, Sugars KL, Bao YP, Rubinsztein DC. Glycogen synthase kinase-3beta inhibitors prevent cellular polyglutamine toxicity caused by the Huntington’s disease mutation. J Biol Chem. 2002;277:33791–33798. doi: 10.1074/jbc.M204861200. [DOI] [PubMed] [Google Scholar]
- Chalecka-Franaszek E, Chuang DM. Lithium activates the serine/threonine kinase Akt-1 and suppresses glutamate-induced inhibition of Akt-1 activity in neurons. Proc Natl Acad Sci U S A. 1999;96:8745–8750. doi: 10.1073/pnas.96.15.8745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Huang LD, Jiang YM, Manji HK. The mood-stabilizing agent valproate inhibits the activity of glycogen synthase kinase-3. J Neurochem. 1999;72:1327–1330. doi: 10.1046/j.1471-4159.2000.0721327.x. [DOI] [PubMed] [Google Scholar]
- Chen PS, Peng GS, Li G, Yang S, Wu X, Wang CC, Wilson B, Lu RB, Gean PW, Chuang D-M, Hong JS. Valproate protects dopaminergic neurons in midbrain neuron/glia cultures by stimulating the release of neurotrophic factors from astrocytes. Mol Psychiatry. 2006;11:1116–1125. doi: 10.1038/sj.mp.4001893. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Maiese K. Targeting WNT, protein kinase B, and mitochondrial membrane integrity to foster cellular survival in the nervous system. Histol Histopathol. 2004;19:495–504. doi: 10.14670/hh-19.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang DM, Priller J. Potential use of lithium in neurodegenerative disorders. In: Bauer M, Grof P, Muller-Oerlingausen B, editors. Lithium in Neuropsychiatry:, The comprehensive Guide. London, UK: Taylor & Francis Books Ltd.; 2006. pp. 381–397. [Google Scholar]
- Cohen P. The Croonian Lecture 1998. Identification of a protein kinase cascade of major importance in insulin signal transduction. Philos Trans R Soc Lond B Biol Sci. 1999;354:485–495. doi: 10.1098/rstb.1999.0399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dajani R, Fraser E, Roe SM, Young N, Good V, Dale TC, Pearl LH. Crystal structure of glycogen synthase kinase 3 beta: structural basis for phosphate-primed substrate specificity and autoinhibition. Cell. 2001;105:721–732. doi: 10.1016/s0092-8674(01)00374-9. [DOI] [PubMed] [Google Scholar]
- De Sarno P, Li X, Jope RS. Regulation of Akt and glycogen synthase kinase-3 beta phosphorylation by sodium valproate and lithium. Neuropharmacology. 2002;43:1158–1164. doi: 10.1016/s0028-3908(02)00215-0. [DOI] [PubMed] [Google Scholar]
- Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116:1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drevets WC, Price JL, Simpson JR, Jr, Todd RD, Reich T, Vannier M, Raichle ME. Subgenual prefrontal cortex abnormalities in mood disorders. Nature. 1997;386:824–827. doi: 10.1038/386824a0. [DOI] [PubMed] [Google Scholar]
- Dugo L, Collin M, Thiemermann C. Glycogen synthase kinase 3beta as a target for the therapy of shock and inflammation. Shock. 2007;27:113–123. doi: 10.1097/01.shk.0000238059.23837.68. [DOI] [PubMed] [Google Scholar]
- Eickholt BJ, Towers GJ, Ryves WJ, Eikel D, Adley K, Ylinen LM, Chadborn NH, Harwood AJ, Nau H, Williams RS. Effects of valproic acid derivatives on inositol trisphosphate depletion, teratogenicity, glycogen synthase kinase-3β inhibition, and viral replication: a screening approach for new bipolar disorder drugs derived from the valproic acid core structure. Mol Pharmacol. 2005;67:1426–1433. doi: 10.1124/mol.104.009308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einat H, Yuan P, Gould TD, Li J, Du J, Zhang L, Manji HK, Chen G. The role of the extracellular signal-regulated kinase signaling pathway in mood modulation. J Neurosci. 2003;23:7311–7316. doi: 10.1523/JNEUROSCI.23-19-07311.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Embi N, Rylatt DB, Cohen P. Glycogen synthase kinase-3 from rabbit skeletal muscle. Separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur J Biochem. 1980;107:519–527. [PubMed] [Google Scholar]
- Engl J, Laimer M, Niederwanger A, Kranebitter M, Starzinger M, Pedrini MT, Fleischhacker WW, Patsch JR, Ebenbichler CF. Olanzapine impairs glycogen synthesis and insulin signaling in L6 skeletal muscle cells. Mol Psychiatry. 2005;10:1089–1096. doi: 10.1038/sj.mp.4001729. [DOI] [PubMed] [Google Scholar]
- Fiol CJ, Haseman JH, Wang YH, Roach PJ, Roeske RW, Kowalczuk M, DePaoli-Roach AA. Phosphoserine as a recognition determinant for glycogen synthase kinase-3: phosphorylation of a synthetic peptide based on the G-component of protein phosphatase-1. Arch Biochem Biophys. 1988;267:797–802. doi: 10.1016/0003-9861(88)90089-6. [DOI] [PubMed] [Google Scholar]
- Frame S, Cohen P. GSK3 takes centre stage more than 20 years after its discovery. Biochem J. 2001;359:1–16. doi: 10.1042/0264-6021:3590001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franca-Koh J, Yeo M, Fraser E, Young N, Dale TC. The regulation of glycogen synthase kinase-3 nuclear export by Frat/GBP. J Biol Chem. 2002;277:43844–43848. doi: 10.1074/jbc.M207265200. [DOI] [PubMed] [Google Scholar]
- Fujimuro M, Hayward SD. The latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus manipulates the activity of glycogen synthase kinase-3β. J Virol. 2003;7:8019–8030. doi: 10.1128/JVI.77.14.8019-8030.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumoto T, Morinobu S, Okamoto Y, Kagaya A, Yamawaki S. Chronic lithium treatment increases the expression of brain-derived neurotrophic factor in the rat brain. Psychopharmacology (Berl) 2001;158:100–106. doi: 10.1007/s002130100871. [DOI] [PubMed] [Google Scholar]
- Gould TD, Einat H, Bhat R, Manji HK. AR-A014418, a selective GSK-3 inhibitor, produces antidepressant-like effects in the forced swim test. Int J Neuropsychopharmacol. 2004;7:387–390. doi: 10.1017/S1461145704004535. [DOI] [PubMed] [Google Scholar]
- Gould TD, Picchini AM, Einat H, Manji HK. Targeting glycogen synthase kinase-3 in the CNS: implications for the development of new treatments for mood disorders. Curr Drug Targets. 2006;7:1399–1409. doi: 10.2174/1389450110607011399. [DOI] [PubMed] [Google Scholar]
- Grimes CA, Jope RS. CREB DNA binding activity is inhibited by glycogen synthase kinase-3 beta and facilitated by lithium. J Neurochem. 2001;78:1219–1232. doi: 10.1046/j.1471-4159.2001.00495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall AC, Brennan A, Goold RG, Cleverley K, Lucas FR, Gordon-Weeks PR, Salinas PC. Valproate regulates GSK-3-mediated axonal remodeling and synapsin I clustering in developing neurons. Mol Cell Neurosci. 2002;20:257–270. doi: 10.1006/mcne.2002.1117. [DOI] [PubMed] [Google Scholar]
- Hanada M, Feng J, Hemmings BA. Structure, regulation and function of PKB/AKT--a major therapeutic target. Biochim Biophys Acta. 2004;1697:3–16. doi: 10.1016/j.bbapap.2003.11.009. [DOI] [PubMed] [Google Scholar]
- Hashimoto R, Takei N, Shimazu K, Christ L, Lu B, Chuang DM. Lithium induces brain-derived neurotrophic factor and activates TrkB in rodent cortical neurons: an essential step for neuroprotection against glutamate excitotoxicity. Neuropharmacology. 2002;43:1173–1179. doi: 10.1016/s0028-3908(02)00217-4. [DOI] [PubMed] [Google Scholar]
- Hata T, Itoh E, Nishikawa H. Behavioral characteristics of SART-stressed mice in the forced swim test and drug action. Pharmacol Biochem Behav. 1995;51:849–853. doi: 10.1016/0091-3057(95)00057-4. [DOI] [PubMed] [Google Scholar]
- Henriksen EJ, Dokken BB. Role of glycogen synthase kinase-3 in insulin resistance and type 2 diabetes. Curr Drug Targets. 2006;7:1435–1441. doi: 10.2174/1389450110607011435. [DOI] [PubMed] [Google Scholar]
- Hughes K, Nikolakaki E, Plyte SE, Totty NF, Woodgett JR. Modulation of the glycogen synthase kinase-3 family by tyrosine phosphorylation. Embo J. 1993;12:803–808. doi: 10.1002/j.1460-2075.1993.tb05715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin N, Kovacs AD, Sui Z, Dewhurst S, Maggirwar SB. Opposite effects of lithium and valproic acid on trophic factor deprivation-induced glycogen synthase kinase-3 activation, c-Jun expression and neuronal cell death. Neuropharmacology. 2005;48:576–583. doi: 10.1016/j.neuropharm.2004.11.010. [DOI] [PubMed] [Google Scholar]
- Jope RS, Johnson GV. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci. 2004;29:95–102. doi: 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Jope RS, Roh MS. Glycogen synthase kinase-3 (GSK3) in psychiatric diseases and therapeutic interventions. Curr Drug Targets. 2006;7:1421–1434. doi: 10.2174/1389450110607011421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaidanovich-Beilin O, Milman A, Weizman A, Pick CG, Eldar-Finkelman H. Rapid antidepressive-like activity of specific glycogen synthase kinase-3 inhibitor and its effect on beta-catenin in mouse hippocampus. Biol Psychiatry. 2004;55:781–784. doi: 10.1016/j.biopsych.2004.01.008. [DOI] [PubMed] [Google Scholar]
- Kang UG, Jeon WJ, Kim Y, Chung CK, Park JB, Juhnn YS, Kim YS. Transient activation of protein phosphatase 2A induced by electroconvulsive shock in the rat frontal cortex. Neurosci Lett. 2005;390:171–175. doi: 10.1016/j.neulet.2005.08.020. [DOI] [PubMed] [Google Scholar]
- Karege F, Perroud N, Burkhardt S, Schwald M, Ballmann E, La Harpe R, Malafosse A. Alteration in Kinase Activity But Not in Protein Levels of Protein Kinase B and Glycogen Synthase Kinase-3beta in Ventral Prefrontal Cortex of Depressed Suicide Victims. Biol Psychiatry. 2006 doi: 10.1016/j.biopsych.2006.04.036. (in press) [DOI] [PubMed] [Google Scholar]
- Kato T, Kato N. Mitochondrial dysfunction in bipolar disorder. Bipolar Disord. 2000;2:180–190. doi: 10.1034/j.1399-5618.2000.020305.x. [DOI] [PubMed] [Google Scholar]
- Kato T. Molecular genetics of bipolar disorder and depression. Psychiatry Clin Neurosci. 2007;61:3–19. doi: 10.1111/j.1440-1819.2007.01604.x. [DOI] [PubMed] [Google Scholar]
- Kaytor MD, Orr HT. The GSK3 beta signaling cascade and neurodegenerative disease. Curr Opin Neurobiol. 2002;12:275–278. doi: 10.1016/s0959-4388(02)00320-3. [DOI] [PubMed] [Google Scholar]
- Kessler RC, Berglund P, Demler O, Jin R, Merikangas KR, Walters EE. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62:593–602. doi: 10.1001/archpsyc.62.6.593. [DOI] [PubMed] [Google Scholar]
- Kieseppa T, Partonen T, Haukka J, Kaprio J, Lonnqvist J. High concordance of bipolar I disorder in a nationwide sample of twins. Am J Psychiatry. 2004;161:1814–1821. doi: 10.1176/ajp.161.10.1814. [DOI] [PubMed] [Google Scholar]
- Kim AJ, Shi Y, Austin RC, Werstuck GH. Valproate protects cells from ER stress-induced lipid accumulation and apoptosis by inhibiting glycogen synthase kinase-3. J Cell Sci. 2005;118:89–99. doi: 10.1242/jcs.01562. [DOI] [PubMed] [Google Scholar]
- Kirshenboim N, Plotkin B, Shlomo SB, Kaidanovich-Beilin O, Eldar-Finkelman H. Lithium-mediated phosphorylation of glycogen synthase kinase-3beta involves PI3 kinase-dependent activation of protein kinase C-alpha. J Mol Neurosci. 2004;24:237–245. doi: 10.1385/JMN:24:2:237. [DOI] [PubMed] [Google Scholar]
- Kitamura Y, Araki H, Gomita Y. Influence of ACTH on the effects of imipramine, desipramine and lithium on duration of immobility of rats in the forced swim test. Pharmacol Biochem Behav. 2002;71:63–69. doi: 10.1016/s0091-3057(01)00625-6. [DOI] [PubMed] [Google Scholar]
- Klein PS, Melton DA. A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci U S A. 1996;93:8455–8359. doi: 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopnisky KL, Chalecka-Franaszek E, Gonzalez-Zulueta M, Chuang DM. Chronic lithium treatment antagonizes glutamate-induced decrease of phosphorylated CREB in neurons via reducing protein phosphatase 1 and increasing MEK activities. Neuroscience. 2003;116:425–435. doi: 10.1016/s0306-4522(02)00573-0. [DOI] [PubMed] [Google Scholar]
- Koros E, Dorner-Ciossek C, Mueller E, Sams-Dodd F. Glycogen synthase kinase (GSK-3B) signaling in schizophrenia; Abstracts of the Society for Neuroscience Annual Meeting; 2006. 589.5. [Google Scholar]
- Kozlovsky N, Belmaker RH, Agam G. Low GSK-3 activity in frontal cortex of schizophrenic patients. Schizophr Res. 2001;52:101–105. doi: 10.1016/s0920-9964(00)00174-2. [DOI] [PubMed] [Google Scholar]
- Kozlovsky N, Amar S, Belmaker RH, Agam G. Psychotropic drugs affect Ser9-phosphorylated GSK-3 beta protein levels in rodent frontal cortex. Int J Neuropsychopharmacol. 2006;9:337–342. doi: 10.1017/S1461145705006097. [DOI] [PubMed] [Google Scholar]
- Lee KY, Ahn YM, Joo EJ, Jeong SH, Chang JS, Kim SC, Kim YS. No association of two common SNPs at position −1727 A/T, −50 C/T of GSK-3 beta polymorphisms with schizophrenia and bipolar disorder of Korean population. Neurosci Lett. 2006;395:175–178. doi: 10.1016/j.neulet.2005.10.059. [DOI] [PubMed] [Google Scholar]
- Leng Y, Chuang DM. Endogenous alpha-synuclein is induced by valproic acid through histone deacetylase inhibition and participates in neuroprotection against glutamate-induced excitotoxicity. J Neurosci. 2006;26:7502–7512. doi: 10.1523/JNEUROSCI.0096-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Bijur GN, Jope RS. Glycogen synthase kinase-3beta, mood stabilizers, and neuroprotection. Bipolar Disord. 2002;4:137–144. doi: 10.1034/j.1399-5618.2002.40201.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Rosborough KM, Friedman AB, Zhu W, Rota KA. Regulation of mouse brain glycogen synthase kinase-3 by atypical antipsychotics. Internat J. Neuropsychopharmacol. 2007;10:7–19. doi: 10.1017/S1461145706006547. [DOI] [PubMed] [Google Scholar]
- Liang MH, Chuang DM. Differential roles of glycogen synthase kinase-3 isoforms in the regulation of transcriptional activation. J Biol Chem. 2006;281:30479–30484. doi: 10.1074/jbc.M607468200. [DOI] [PubMed] [Google Scholar]
- Liang MH, Chuang DM. Regulation and function of glycogen synthase kinase-3 isoforms in neuronal survival. J Biol Chem. 2007;282:3904–3917. doi: 10.1074/jbc.M605178200. [DOI] [PubMed] [Google Scholar]
- Liu X, Liu T, Slusarski DC, Yang-Snyder J, Malbon CC, Moon RT, Wang H. Activation of a frizzled-2/beta-adrenergic receptor chimera promotes Wnt signaling and differentiation of mouse F9 teratocarcinoma cells via Galphao and Galphat. Proc Natl Acad Sci U S A. 1999;96:14383–14388. doi: 10.1073/pnas.96.25.14383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen TM, Newton SS, Eaton ME, Russell DS, Duman RS. Chronic electroconvulsive seizure up-regulates beta-catenin expression in rat hippocampus: role in adult neurogenesis. Biol Psychiatry. 2003;54:1006–10014. doi: 10.1016/s0006-3223(03)00700-5. [DOI] [PubMed] [Google Scholar]
- Mai L, Jope RS, Li X. BDNF-mediated signal transduction is modulated by GSK3β and mood stabilizing agents. J Neurochem. 2002;82:75–83. doi: 10.1046/j.1471-4159.2002.00939.x. [DOI] [PubMed] [Google Scholar]
- Malbon CC, Wang HY. Dishevelled: a mobile scaffold catalyzing development. Curr Top Dev Biol. 2006;72:153–166. doi: 10.1016/S0070-2153(05)72002-0. [DOI] [PubMed] [Google Scholar]
- Manji HK, McNamara R, Chen G, Lenox RH. Signalling pathways in the brain: cellular transduction of mood stabilisation in the treatment of manic-depressive illness. Aust N Z J Psychiatry. 1999;33:S65–S83. doi: 10.1111/j.1440-1614.1999.00670.x. [DOI] [PubMed] [Google Scholar]
- Marin O, Meggio F, Sarno S, Andretta M, Pinna LA. Phosphorylation of synthetic fragments of inhibitor-2 of protein phosphatase-1 by casein kinase-1 and -2. Evidence that phosphorylated residues are not strictly required for efficient targeting by casein kinase-1. Eur J Biochem. 1994;223:647–653. doi: 10.1111/j.1432-1033.1994.tb19037.x. [DOI] [PubMed] [Google Scholar]
- Meijer L, Flajolet M, Greengard P. Pharmacological inhibitors of glycogen synthase kinase 3. Trends Pharmacol Sci. 2004;25:471–480. doi: 10.1016/j.tips.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Michelon L, Meira-Lima I, Cordeiro Q, Miguita K, Breen G, Collier D, Vallada H. Association study of the INPP1, 5HTT, BDNF, AP-2beta and GSK-3beta GENE variants and restrospectively scored response to lithium prophylaxis in bipolar disorder. Neurosci Lett. 2006;403:288–293. doi: 10.1016/j.neulet.2006.05.001. [DOI] [PubMed] [Google Scholar]
- Mukai F, Ishiguro K, Sano Y, Fujita SC. Alternative splicing isoform of tau protein kinase I/glycogen synthase kinase 3beta. J Neurochem. 2002;81:1073–1083. doi: 10.1046/j.1471-4159.2002.00918.x. [DOI] [PubMed] [Google Scholar]
- Nasrallah HA, Ketter TA, Kalali AH. Carbamazepine and valproate for the treatment of bipolar disorder: a review of the literature. J Affect Disord. 2006;95:69–78. doi: 10.1016/j.jad.2006.04.030. [DOI] [PubMed] [Google Scholar]
- Nierenberg AA, Papakostas GI, Petersen T, Kelly KE, Iacoviello BM, Worthington JJ, Tedlow J, Alpert JE, Fava M. Nortriptyline for treatment-resistant depression. J Clin Psychiatry. 2003;64:35–39. doi: 10.4088/jcp.v64n0108. [DOI] [PubMed] [Google Scholar]
- Nishiguchi N, Breen G, Russ C, St Clair D, Collier D. Association analysis of the glycogen synthase kinase-3beta gene in bipolar disorder. Neurosci Lett. 2006;394:243–245. doi: 10.1016/j.neulet.2005.10.042. [DOI] [PubMed] [Google Scholar]
- O’Brien WT, Harper AD, Jove F, Woodgett JR, Maretto S, Piccolo S, Klein PS. Glycogen synthase kinase-3beta haploinsufficiency mimics the behavioral and molecular effects of lithium. J Neurosci. 2004;24:6791–6798. doi: 10.1523/JNEUROSCI.4753-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ongur D, Drevets WC, Price JL. Glial reduction in the subgenual prefrontal cortex in mood disorders. Proc Natl Acad Sci U S A. 1998;95:13290–13295. doi: 10.1073/pnas.95.22.13290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pap M, Cooper GM. Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-Kinase/Akt cell survival pathway. J Biol Chem. 1998;273:19929–19932. doi: 10.1074/jbc.273.32.19929. [DOI] [PubMed] [Google Scholar]
- Patel S, Doble B, Woodgett JR. Glycogen synthase kinase-3 in insulin and Wnt signalling: a double-edged sword? Biochem Soc Trans. 2004;32:803–808. doi: 10.1042/BST0320803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem. 2001;276:36734–36741. doi: 10.1074/jbc.M101287200. [DOI] [PubMed] [Google Scholar]
- Polakis P. The many ways of Wnt in cancer. Curr Opin Genet Dev. 2007;17:45–51. doi: 10.1016/j.gde.2006.12.007. [DOI] [PubMed] [Google Scholar]
- Prickaerts J, Moechars D, Cryns K, Lenaerts I, van Craenendonck H, Goris I, Daneels G, Bouwknecht JA, Steckler T. Transgenic mice overexpressing glycogen synthase kinase 3beta: a putative model of hyperactivity and mania. J Neurosci. 2006;26:9022–9029. doi: 10.1523/JNEUROSCI.5216-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajkowska G, Miguel-Hidalgo JJ, Wei J, Dilley G, Pittman SD, Meltzer HY, Overholser JC, Roth BL, Stockmeier CA. Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression. Biol Psychiatry. 1999;45:1085–1098. doi: 10.1016/s0006-3223(99)00041-4. [DOI] [PubMed] [Google Scholar]
- Redrobe JP, Bourin M. Evidence of the activity of lithium on 5-HT1B receptors in the mouse forced swimming test: comparison with carbamazepine and sodium valproate. Psychopharmacology (Berl) 1999;141:370–377. doi: 10.1007/s002130050846. [DOI] [PubMed] [Google Scholar]
- Ren M, Leng Y, Jeong M, Leeds PR, Chuang DM. Valproic acid reduces brain damage induced by transient focal cerebral ischemia in rats: potential roles of histone deacetylase inhibition and heat shock protein induction. J Neurochem. 2004;89:1358–1367. doi: 10.1111/j.1471-4159.2004.02406.x. [DOI] [PubMed] [Google Scholar]
- Rockenstein E, Torrance M, Adame A, Mante M, Bar-on P, Rose JB, Crews L, Masliah E. Neuroprotective effects of regulators of the glycogen synthase kinase-3beta signaling pathway in a transgenic model of Alzheimer's disease are associated with reduced amyloid precursor protein phosphorylation. J Neurosci. 2007;27:1981–1991. doi: 10.1523/JNEUROSCI.4321-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh MS, Kang UG, Shin SY, Lee YH, Jung HY, Juhnn YS, Kim YS. Biphasic changes in the Ser-9 phosphorylation of glycogen synthase kinase-3beta after electroconvulsive shock in the rat brain. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:1–5. doi: 10.1016/s0278-5846(02)00307-x. [DOI] [PubMed] [Google Scholar]
- Roh MS, Eom TY, Zmijewska AA, De Sarno P, Roth KA, Jope RS. Hypoxia activates glycogen synthase kinase-3 in mouse brain in vivo: protection by mood stabilizers and imipramine. Biol Psychiatry. 2005;57:278–286. doi: 10.1016/j.biopsych.2004.10.039. [DOI] [PubMed] [Google Scholar]
- Rowe MR, Chuang DM. Lithium neuroprotection: molecular mechanisms and clinical implications. Expert Rev Mol Med. 2004;6:1–18. doi: 10.1017/S1462399404008385. [DOI] [PubMed] [Google Scholar]
- Rowe M, Wiest C, Chuang DM. Histone deacetylase inhibitors valproate and pivanex in the forced swim test: strain dependent outcomes; Abstract of the Society for Neuroscience Annual Meeting; 2006. Abstract number 290.20. [Google Scholar]
- Ryves WJ, Harwood AJ. Lithium inhibits glycogen synthase kinase-3 by competition for magnesium. Biochem Biophys Res Commun. 2001;280:720–725. doi: 10.1006/bbrc.2000.4169. [DOI] [PubMed] [Google Scholar]
- Ryves WJ, Harwood AJ. The interaction of glycogen synthase kinase-3 (GSK-3) with the cell cycle. Prog Cell Cycle Res. 2003;5:489–495. [PubMed] [Google Scholar]
- Ryves JW, Dalton EC, Harwood AJ, Williams RS. GSK-3 activity in neocortical cells is inhibited by lithium but not carbamazepine or valproic acid. Bipolar Disord. 2005;7:260–265. doi: 10.1111/j.1399-5618.2005.00194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs GS, Koslow CL, Ghaemi SN. The treatment of bipolar depression. Bipolar Disord. 2000;2:256–260. doi: 10.1034/j.1399-5618.2000.20306.x. [DOI] [PubMed] [Google Scholar]
- Shaw PC, Davies AF, Lau KF, Garcia-Barcelo M, Waye MM, Lovestone S, Miller CC, Anderton BH. Isolation and chromosomal mapping of human glycogen synthase kinase-3 alpha and -3 beta encoding genes. Genome. 1998;41:720–727. [PubMed] [Google Scholar]
- Stambolic V, Ruel L, Woodgett JR. Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr Biol. 1996;6:1664–1668. doi: 10.1016/s0960-9822(02)70790-2. [DOI] [PubMed] [Google Scholar]
- Szatmari E, Habas A, Yang P, Zheng JJ, Hagg T, Hetman M. A positive feedback loop between glycogen synthase kinase 3beta and protein phosphatase 1 after stimulation of NR2B NMDA receptors in forebrain neurons. J Biol Chem. 2005;280:37526–37535. doi: 10.1074/jbc.M502699200. [DOI] [PubMed] [Google Scholar]
- Szczepankiewicz A, Rybakowski JK, Suwalska A, Skibinska M, Leszczynska-Rodziewicz A, Dmitrzak-Weglarz M, Czerski PM, Hauser J. Association study of the glycogen synthase kinase-3β gene polymorphism with prophylactic lithium response in bipolar patients. World J Biol Psychiatry. 2006a;7:158–161. doi: 10.1080/15622970600554711. [DOI] [PubMed] [Google Scholar]
- Szczepankiewicz A, Skibinska M, Hauser J, Slopien A, Leszczynska-Rodziewicz A, Kapelski P, Dmitrzak-Weglarz M, Czerski PM, Rybakowski JK. Association analysis of the GSK-3β T-50C gene polymorphism with schizophrenia and bipolar disorder. Neuropsychobiology. 2006b;53:51–56. doi: 10.1159/000090704. [DOI] [PubMed] [Google Scholar]
- Thomas GM, Frame S, Goedert M, Nathke I, Polakis P, Cohen P. A GSK3-binding peptide from FRAT1 selectively inhibits the GSK3-catalysed phosphorylation of axin and beta-catenin. FEBS Lett. 1999;458:247–251. doi: 10.1016/s0014-5793(99)01161-8. [DOI] [PubMed] [Google Scholar]
- Tomasiewicz HC, Mague SD, Cohen BM, Carlezon WA., Jr Behavioral effects of short-term administration of lithium and valproic acid in rats. Brain Res. 2006;1093:83–94. doi: 10.1016/j.brainres.2006.03.102. [DOI] [PubMed] [Google Scholar]
- Watcharasit P, Bijur GN, Zmijewski JW, Song L, Zmijewska A, Chen X, Johnson GV, Jope RS. Direct, activating interaction between glycogen synthase kinase-3beta and p53 after DNA damage. Proc Natl Acad Sci U S A. 2002;99:7951–7955. doi: 10.1073/pnas.122062299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegener G, Bandpey Z, Heiberg IL, Mork A, Rosenberg R. Increased extracellular serotonin level in rat hippocampus induced by chronic citalopram is augmented by subchronic lithium: neurochemical and behavioural studies in the rat. Psychopharmacology (Berl) 2003;166:188–194. doi: 10.1007/s00213-002-1341-6. [DOI] [PubMed] [Google Scholar]
- Werstuck GH, Kim AJ, Brenstrum T, Ohnmacht SA, Panna E, Capretta A. Examining the correlations between GSK-3 inhibitory properties and anticonvulsant efficacy of valproate and valproate-related compounds. Bioorg Med Chem Lett. 2004;14:5465–5467. doi: 10.1016/j.bmcl.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Xavier IJ, Mercier PA, McLoughlin CM, Ali A, Woodgett JR, Ovsenek N. Glycogen synthase kinase 3beta negatively regulates both DNA-binding and transcriptional activities of heat shock factor 1. J Biol Chem. 2000;275:29147–29152. doi: 10.1074/jbc.M002169200. [DOI] [PubMed] [Google Scholar]
- Zhang F, Phiel CJ, Spece L, Gurvich N, Klein PS. Inhibitory phosphorylation of glycogen synthase kinase-3 (GSK-3) in response to lithium. Evidence for autoregulation of GSK-3. J Biol Chem. 2003;278:33067–33077. doi: 10.1074/jbc.M212635200. [DOI] [PubMed] [Google Scholar]