

Abstract
Paramutations represent heritable epigenetic alterations that cause departures from Mendelian inheritance. While the mechanism responsible is largely unknown, recent results in both mouse and maize suggest paramutations are correlated with RNA molecules capable of affecting changes in gene expression patterns. In maize, multiple required to maintain repression (rmr) loci stabilize these paramutant states. Here we show rmr1 encodes a novel Snf2 protein that affects both small RNA accumulation and cytosine methylation of a proximal transposon fragment at the Pl1-Rhoades allele. However, these cytosine methylation differences do not define the various epigenetic states associated with paramutations. Pedigree analyses also show RMR1 does not mediate the allelic interactions that typically establish paramutations. Strikingly, our mutant analyses show that Pl1-Rhoades RNA transcript levels are altered independently of transcription rates, implicating a post-transcriptional level of RMR1 action. These results suggest the RNA component of maize paramutation maintains small heterochromatic-like domains that can affect, via the activity of a Snf2 protein, the stability of nascent transcripts from adjacent genes by way of a cotranscriptional repression process. These findings highlight a mechanism by which alleles of endogenous loci can acquire novel expression patterns that are meiotically transmissible.
Author Summary
Genetics is founded on the principle that heritable changes in genes are caused by mutations and that the regulatory state of gene pairs (alleles) is passed on to progeny unchanged. An exception to this rule, paramutations—which reflect the outcome of interactions between alleles—produce changes in gene control that are stably inherited without altering the DNA sequence. It is currently thought that these allelic interactions cause structural alterations to the chromatin surrounding the gene. Recent work in both maize and mice suggests that RNA molecules may be responsible for paramutations. Several genes are required to maintain the repressed paramutant state of a maize purple plant1 (pl1) allele, and here we report that one of these genes encodes a protein (RMR1) with similarity to a protein previously implicated in facilitating genomic DNA modifications via small RNA molecules. Genetic and molecular experiments support a similar role for RMR1 acting at a repeated sequence found adjacent to this pl1 gene. Although loss of these DNA modifications leads to heritable changes in gene regulation, the data indicate these changes do not represent the heritable feature responsible for paramutation. These findings highlight an unusual but dynamic role for repeated genomic features and small RNA molecules in affecting heritable genetic changes independent of the DNA template.
Paramutations are heritable epigenetic changes, which, in maize, are stabilized by multiple genes, one of which expresses a novel Snf2 protein. This protein affects the stability of transcripts from adjacent genes by a cotranscriptional repression process.
Introduction
The term “paramutation” describes a genetic behavior in which the regulatory state of specific alleles is heritably altered through interactions with their homologous partners in trans [1,2]. This behavior presents an exception to the Mendelian principle that alleles segregate from a heterozygous state unchanged [3]. Paramutations have been best characterized at loci encoding transcriptional regulators of pigment biosynthesis in maize, but similar behaviors have been described in other plant and animal systems, most recently in mice [4,5]. While the broader roles of paramutation in genome-wide regulation and evolution remain to be seen, the Pl1-Rhoades allele of the maize purple plant1 (pl1) locus presents a tractable system to study the paramutation process.
The pl1 locus encodes a Myb-like protein that acts as a transcriptional activator of genes required for anthocyanin pigment production [6]. Inheritance patterns illustrate that the Pl1-Rhoades allele can exist in quantitatively distinct regulatory states, reflected by differences in plant color. When individuals with a highly expressed reference state of Pl1-Rhoades, termed Pl-Rh, are crossed with plants having a repressed state, referred to as Pl′, only progeny with weak pigmentation are produced [7,8]. Pl-Rh states invariably change to Pl′ in Pl-Rh/Pl′ heterozygotes [7]; this is a typical hallmark of paramutation. Relative to Pl-Rh, the Pl′ state displays reductions in both Pl1-Rhoades RNA levels (∼10-fold) and transcription rate (∼3-fold) that are associated with a reduction in plant pigment [8]. This repressed Pl′ state is meiotically stable when maintained in a Pl1-Rhoades homozygote, with no reversion to Pl-Rh seen to date. Pl′ can, however, revert to Pl-Rh when heterozygous with some pl1 alleles other than Pl1-Rhoades, when maintained in a hemizygous condition, or in the presence of specific recessive mutations [9–12].
Genetic screens for ethyl methanesulfonate (EMS)–induced recessive mutations identify at least ten loci, including required to maintain repression1 (rmr1), rmr2, rmr6, and mediator of paramutation1 (mop1), whose normal functions maintain the repressed Pl′ state ([10,11,13]; J. B. H., unpublished data). These rmr mutations specifically affect the expression of Pl1-Rhoades and not other pl1 alleles [10,11], indicating that the Pl1-Rhoades allele is a direct and specific target of paramutation-based epigenetic changes. mop1 was recently identified [14,15] as encoding the putative ortholog of the Arabidopsis protein RDR2, a presumed RNA-dependent RNA polymerase involved in siRNA-based maintenance of de novo cytosine methylation [16]. Recessive mutations defining rmr1, rmr2, and rmr6 destabilize the repressed Pl′ state, resulting in darkly pigmented plant tissues, an increase in pl1 RNA levels, and meiotic transmission of Pl-Rh revertant states [10,11]. To date, the molecular identity of these rmr factors remains unknown.
In this report we identify rmr1 as encoding a novel Snf2 protein that represents a founding member of a subgroup of factors similar to proteins involved in plant small RNA metabolism. Our analyses show that RMR1 affects both pl1 RNA transcript stability as well as small interfering RNA (siRNA) accumulation and DNA methylation patterns at Pl1-Rhoades. These results support a model in which maintenance of paramutant states is dependent on a repression mechanism similar to the recently proposed cotranscriptional gene silencing mechanism in fission yeast [17,18]. To our knowledge, RMR1 is the first protein identified that maintains trans-generationally repressed states established by paramutation.
Results
rmr1 Defects Affect pl1 RNA Stability
The rmr1 locus is defined by four recessive mutations (Protocol S1) characterized by a darkly pigmented plant phenotype that results from loss of Pl′ repression. Previous RNase protection experiments showed a 26-fold increase in pl1 RNA in floret tissue between rmr1–1 mutant plants and heterozygous siblings [10]. However, these experiments did not address if changes in pl1 transcript abundance correlated with changes in actual transcription at the pl1 locus.
In vitro transcription assays using nuclei isolated from husk leaf tissue revealed there was no statistically significant change in relative transcription rates of the Pl1-Rhoades allele between rmr1–1 mutants and heterozygous siblings (Figure S1). However, transcription rates of anthocyaninless1 (a1), a direct target of the PL1 transcriptional activator [7,19], were ∼4-fold greater in rmr1–1 mutants (Figure S1), reflecting significantly increased PL1 activity. Transcription rates from colored plant1 (b1)—a locus encoding a basic helix-loop-helix factor genetically required for a1 transcription— remained unchanged. These results were recapitulated in comparisons between nuclei isolated from rmr1–3 mutants and heterozygous siblings in which in vitro transcription assays revealed no significant change in transcription rate of Pl1-Rhoades (Figures 1A and S1; n = 4, two-tailed two-sample t-test, t = 0.8, p = 0.5) while RNase protection experiments showed a 5.7-fold increase in pl1 RNA for rmr1–3 mutants (Figure 1B and 1C; n = 2, two-tailed two-sample t-test, t = 10.8, p < 0.01) using RNA isolated from the same tissues of the same individuals. Similar comparisons from identical tissues but in a different genetic background again showed that transcription rates at pl1 remained unchanged while pl1 RNA levels increased 7.52-fold in rmr1–3 mutants compared to heterozygous siblings (n = 1; see Protocol S1).
Figure 1. Comparison of pl1 Expression between rmr1 Mutants and Heterozygous Siblings.
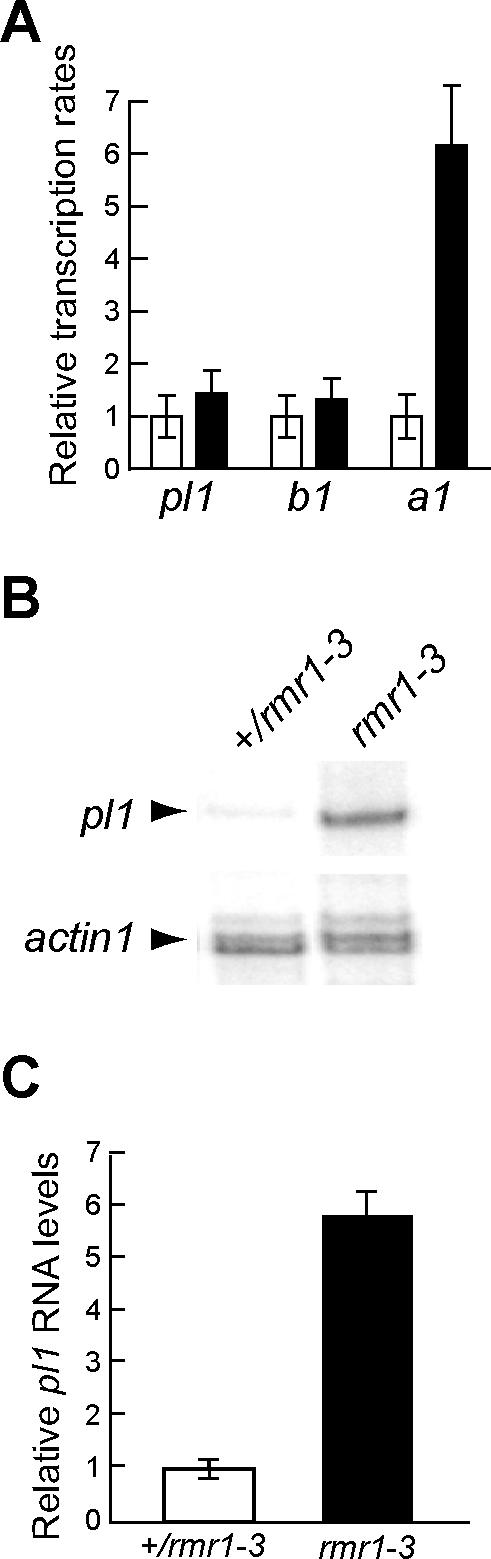
pl1 RNA levels increase significantly in rmr1 mutants, while transcription rates of paramutant alleles are unaffected.
(A) The relative mean transcription rates from four independent sets of +/rmr1–3 (open) and rmr1–3/rmr1–3 (filled) siblings (± standard error of the mean) at the indicated loci.
(B) RNase protection analysis comparing pl1 and actin1 RNA levels in the same individual plants and tissues used for in vitro transcription analysis.
(C) Quantification of relative pl1 RNA levels from analyses as represented in (B).
These RNA expression results sharply contrast those of previous reports using identical in vitro transcription assays that detected significant differences in Pl1-Rhoades transcription rates between Pl′ and Pl-Rh states and between rmr6 mutants and non-mutants [8,11]. This indicates our in vitro results represent an accurate assessment of transcription rates and not a limitation of the assay to detect rate differences at the pl1 locus. Combined, these results imply an increase of pl1 RNA abundance disproportionate to insignificant changes in transcription rate in rmr1 mutants, the most direct interpretation being that RMR1 functions at a post-transcriptional level to stabilize Pl1-Rhoades RNA.
rmr1 Encodes a Novel Protein with a Snf2 Domain
To better understand Rmr1 function and the paramutation mechanism, we used a map-based approach to identify the rmr1 gene. Using a polymorphic F2 population we looked for genetic linkage between the mutant phenotype and previously mapped chromosome markers [20]. The dark-color phenotype of rmr1–1 homozygotes showed invariant cosegregation with the mutant parent polymorphism of SSLP markers bnlg1174a (680 chromosomes tested; <0.15 cM) and npi252 (60 chromosomes tested; <1.7 cM), indicating rmr1 was tightly linked to those markers in bin 6.05 on Chromosome 6. We used the high degree of synteny between this region and rice Chromosome 5 to identify candidate rmr1 orthologs (Figure 2A and 2B).
Figure 2. Map-Based Cloning of rmr1 .

(A and B) Rice Chromosome 5 (http://rice.tigr.org/) (A) and maize Chromosome 6 (B) (2005 FPC map, contig 285; http://www.genome.arizona.edu/fpc/maize/) with synteny of annotated rice loci and orthologous maize markers (gray boxes) highlighted. Black boxes indicate the rice rmr1 ortholog, Os05g32610, and the SSLP marker npi252. Neither rmr1 nor SSLP marker bnlg1174a are represented on the FPC map, though both can be amplified from a BAC (c0007N19), identified by GenBank accession AY109873, which maps to the region identified by the black line.
(C) Gene structure of rmr1 with exons in black; EMS-derived mutations are noted.
(D) Gray boxes highlight conserved Pfam SNF2_N (E-value = 1.3 × 10−8; amino acids 851 to 1214) and Helicase_C (E-value = 1.1 × 10−11; amino acids 1255 to 1334) profiles in the RMR1 protein. Predicted translational consequences of each rmr1 mutation are indicated.
Within the syntenic rice region we identified a gene model, Os05g32610 (http://rice.tigr.org/), predicted to encode a Snf2 protein. The Snf2 protein family is composed of members similar to Saccharomyces cerevisiae Snf2p with a bipartite helicase domain containing Pfam SNF2_N and Helicase_C profiles, and includes many proteins involved in ATP-dependent chromatin remodeling [21,22]. While there was no public maize expressed sequence tag for this candidate, we used BLAST searches to identify genomic survey sequence similar to Os05g32610. Oligonucleotide primers were designed from these sequences and used to generate PCR amplicons spanning the maize Os05g32610 ortholog, which were sequenced from individuals homozygous for Rmr1 progenitor alleles and mutant derivatives (see Materials and Methods and Dataset S1). The maize sequence generated from each of the homozygous mutants revealed single unique transition-type base pair changes consistent with EMS mutagenesis relative to the progenitor (Figure 2C). The amino acid change associated with the rmr1–1 allele is predicted to prevent proper folding of the helicase domain [23], while the non-conservative amino acid substitutions associated with the rmr1–2 and rmr1–4 alleles occur at highly conserved residues in the SNF2_N profile (Figure 2D). The rmr1–3 allele is associated with a nonsense mutation predicted to truncate the peptide before the conserved helicase domain. CAPS markers were designed to the potential rmr1–1 and rmr1–3 lesions and used to show that the base pair polymorphisms at each of the probable lesions invariably cosegregate with the mutant phenotype (see Materials and Methods). These results support these polymorphisms as bona fide molecular lesions in the rmr1 gene. Based upon molecular genetic mapping data, DNA sequencing results, and the relevance of the fact that Snf2 proteins affect chromatin environments, we conclude the rmr1 locus encodes a protein containing a Snf2 helicase domain.
Os05g32610 gene models and our cDNA sequencing analysis (see Materials and Methods) indicate rmr1 encodes a 1,435-amino-acid protein. In addition to having the conserved Snf2 helicase domain, the protein has a large N-terminal region with no significant identity to any known or predicted proteins. Phylogenetic comparison with other known Snf2 proteins in maize, rice, Arabidopsis, and budding yeast shows RMR1 is a member of a Rad54-like subfamily defined by DRD1 (Figure 3). Arabidopsis DRD1 is a putative chromatin remodeling factor affecting RNA-directed DNA methylation (RdDM) patterns [24–26]. In the emerging RdDM pathway model, DNA sequences are targeted for de novo cytosine methylation by complementary siRNA molecules generated from “aberrant” RNA transcripts. The putative MOP1 ortholog in Arabidopsis, RDR2, is required in this pathway to presumably generate double-stranded RNA from these transcripts and provide a substrate for siRNA biogenesis through activity of a Dicer-like enzyme [27]. DRD1 is thought to be a downstream effector protein that facilitates de novo methylation of targeted DNA sequences, possibly by modulating chromatin architecture to provide access to de novo methyltransferases [24–26,28]. The DRD1 subfamily also includes the recently identified CLSY1 protein implicated in the systemic spreading of siRNA-mediated silencing in Arabidopsis [29].
Figure 3. RMR1 Defines a Monophyletic Clade Distinct from DRD1.

Distance tree with bootstrap values produced from alignment (Figure S2) of the predicted Snf2 domain with other Snf2 proteins: the tree shows that RMR1, CLSY1, and DRD1 (highlighted in gray) are members of a Rad54-like subfamily of Snf2 proteins. Three distinct monophyletic groups compose this subfamily, numbered 1 to 3. Prefixes: At, Arabidopsis; Os, rice; Sc, S. cerevisiae.
Multiple sequence alignments (Figure S2) indicate RMR1 is not the structural ortholog of either DRD1 or CLSY1. The DRD1 subfamily can be divided into three distinct monophyletic groups, with RMR1, DRD1, and CLSY1 defining different groups (Figure 3). The presumed maize ortholog of DRD1 is likely one of two proteins in the DRD1 subgroup, Chromatin remodeling complex subunit R 127 (CHR127) (http://chromdb.org/), a partial protein predicted from maize expressed sequence tag sequences, or CHR156, a full-length protein predicted from maize genomic sequence (see Materials and Methods). RMR1 is more similar to Arabidopsis proteins predicted from At1g05490 and At3g24340. RNA interference knockdowns of these putative Arabidopsis orthologs are known to have little to no effect in response to DNA damage [30].
Taking into account the phylogenetic analysis of the predicted coding sequence, it is possible RMR1 function may be similar to, but distinct from, that of DRD1 and CLSY1. The three proteins may fulfill a similar role in RdDM, but perhaps function under different conditions or in distinct genomic contexts. Alternatively, they could perform different roles within an RdDM pathway, or function in separate epigenetic mechanisms altogether. Given the results of our pl1 RNA expression analyses, it is possible that RMR1 represents a Snf2 protein that links chromatin organization to RNA transcript stability.
RMR1 Maintains Cytosine Methylation and Small RNA Accumulation at Pl1-Rhoades
In the described Arabidopsis RdDM pathway, DRD1 maintains cytosine methylation at nonsymmetrical CNN sequences represented by siRNAs [24–26]. Many endogenous genomic targets of DRD1 appear to be repetitive elements [31]. At Pl1-Rhoades there is a 402-bp terminal fragment of a CACTA-like type II DNA transposon, similar to doppia, 129 bp upstream of the translational start site [8,32,33]. Assuming analogous functional roles of RMR1 and DRD1 we compared DNA methylation patterns at this upstream repetitive element in rmr1 mutants and non-mutant siblings.
Previous restriction-enzyme-based comparisons of DNA methylation status between Pl-Rh and Pl′ states found no differences, although few 5′ proximal sites were evaluated [8]. Using Southern blot hybridization analysis following digestion of genomic DNA with methylation-sensitive restriction enzymes, we found that the doppia fragment is hypomethylated at specific sites in plants homozygous for the rmr1–1 mutation compared to heterozygous wild-type siblings (Figures 4A, 4B, and S3). Consistent with findings in Arabidopsis RdDM mutants [16,34–36], the sites hypomethylated in rmr1 mutants were of the CNN context. A relative hypomethylation pattern in 5′ sequences is also present in plants homozygous for mutations at either rmr6 or mop1 (Figures S4 and S5). In rmr6 mutants the extent of hypomethylation was greater than that of either rmr1 or mop1 mutants and encompassed CG methylation sites as well as non-CG targets, suggesting Rmr6 has a broader effect in cytosine methylation maintenance. The presence of these methylation differences in multiple mutant backgrounds indicates that this hypomethylation pattern reflects the chromatin status at doppia in plants where maintenance of repressed paramutant states is compromised.
Figure 4. Cytosine Methylation Patterns and Small RNA Accumulation Are Altered at Pl1-Rhoades in rmr1 Mutants.

(A) Schematic of Pl1-Rhoades locus with exons highlighted in black and the upstream doppia element represented by the gray arrow. The methylation context of sites cut by methylation-sensitive enzymes are shown in parentheses. Open circles denote sites hypomethylated in rmr1–1 mutants while filled circles are sites methylated in both wild-type and rmr1 mutants. BsrI restriction sites and the regions used to generate probes for blot hybridization analysis, denoted A and B, are also shown.
(B and C) Representative Southern blots hybridized with probe A showing methylation status at a StuI site in rmr1 mutants and heterozygous siblings (B), as well as Pl′ and Pl-Rh plants (C) with a larger 2.9-kb band (upper arrow) representative of a fully methylated BsrI fragment, and a 2.1-kb band (lower arrow) indicative of a hypomethylated StuI site. Additional primary blots shown in Figures S3 and S8.
(D) Small RNA northern blot probed with doppia sequence from probe B showing changes in amount of small RNAs between rmr1–1 plants and wild-type (WT) siblings.
(E) Southern blot of genomic DNA digested with BstNI (“B” lanes) and methylation-sensitive PspGI (“P” lanes) hybridized with probe B, showing no bulk changes in doppia methylation genome-wide.
Consistent with the Arabidopsis RdDM model, small RNAs (∼26 nt) with sequence similarity to the doppia element are detected in wild-type Pl′ plants in both sense and antisense orientations (Figures 4D and S6). These small RNAs are undetectable in rmr1 mutants, unlike in wild-type siblings. This result contrasts those in Arabidopsis showing that DRD1 deficiencies do not affect the abundance of endogenous siRNAs representing repetitive elements [31]. However, it has been reported that the abundance of endogenous siRNA and trans-acting siRNA populations are highly reduced in CLSY1 mutants [29].
To test if the doppia fragment hypomethylation was indicative of genome-wide changes we assayed the cytosine methylation status at centromeres and 45S repeat sequences. Cytosine methylation patterns were unaffected in either of these regions in rmr1 mutants as compared to non-mutant siblings (Figure S7). Additionally, we examined the methylation status of doppia-like loci genome-wide (Figure 4E) and found no obvious differences between rmr1 mutants and non-mutant siblings. These results indicate that while RMR1 acts on the doppia sequence upstream of Pl1-Rhoades, doppia elements appear unaffected throughout the genome. This specificity of RMR1 function may be due to its intimate and exclusive involvement with alleles that undergo paramutation, or may be indicative of differential regulation of repetitive elements depending on their genomic and epigenetic context.
If RMR1 is involved in maintaining cytosine methylation patterns characteristic of repressed paramutant states then a prediction would be that the methylation differences seen between mutants and non-mutants would reflect the Pl′ and Pl-Rh regulatory states. Surprisingly, there are no methylation differences at the doppia fragment between Pl-Rh and Pl′ states (Figures 4C and S8). These results suggest that while the upstream doppia element of Pl1-Rhoades is a target of multiple factors involved in maintaining the epigenetic repression associated with paramutation, the actual process of paramutation does not result in similar changes of DNA methylation at this element.
RMR1 Is Not Required for Establishment of Paramutant States
Based on a reverse transcriptase PCR (RT-PCR) expression profile (Figure S9) rmr1 appears to be expressed in all rapidly dividing somatic tissues, consistent with a role in maintaining paramutant states throughout development. However, since the methylation patterns maintained by RMR1 appear unrelated to the paramutant state of Pl1-Rhoades, we questioned whether RMR1 is directly required for paramutation to occur. This process results in the invariable establishment of the Pl′ state in Pl′/Pl-Rh plants, as evidenced by the observation that only Pl′/Pl′ progeny are found when Pl′/Pl-Rh plants are crossed to Pl-Rh/Pl-Rh testers [7,8]. If RMR1 were directly involved in this process we would expect that an rmr1 deficiency might interfere with the Pl′ establishment event. To test this, we tracked the behavior of individual Pl1-Rhoades alleles in test crosses to assess the ability of the Pl′ state to facilitate paramutations in Pl′/Pl-Rh; rmr1–1/rmr1–2 plants. The Pl1-Rhoades allele in a Pl-Rh state was genetically linked (∼1.5 cM) to a T6–9 translocation breakpoint (T6–9). The T6–9 interchange can act as a dominant semi-sterility marker, allowing us to trace specific Pl1-Rhoades alleles through genetic crosses [11]. rmr1 mutants heterozygous for the T6–9 interchange (T6–9 Pl-Rh/Pl′) were crossed to a Pl-Rh/Pl-Rh tester (Figure 5; Table S1). If establishment of the Pl′ state was prevented in rmr1 mutants, we would expect all progeny receiving the interchange to display a Pl-Rh/Pl-Rh phenotype (dark anther pigmentation). We observed that over half the progeny inheriting the interchange displayed a Pl′/Pl′-like phenotype (light anther pigmentation), indicating that paramutation was established in the rmr1 mutant parent. It should also be noted that Pl-Rh/Pl-Rh plants, and those of an intermediate phenotype of partial pigmentation [7], were present in both progeny inheriting the interchange and those inheriting a normal chromosome. These results are consistent with previous work showing Pl′ can revert to a Pl-Rh state in rmr1 mutants [10].
Figure 5. Pl′ Establishment in an rmr1 Mutant Background.

Plants with a T6–9 translocation chromosome carrying Pl1-Rhoades in the Pl-Rh state (dark anther pigmentation) and heterozygous for the rmr1–2 allele were crossed to Pl′ plants (light anther pigmentation) heterozygous for the rmr1–1 allele. Of the resultant progeny with semi-sterile pollen (heterozygous for the T6–9 interchange pair), plants homozygous for a mutation at rmr1 were chosen based on the dark anther phenotype. These plants were then crossed to a Pl-Rh tester with the expectation that in progeny inheriting the interchange, the expression status of the Pl1-Rhoades allele on the T6–9 translocation chromosome (T6–9 Pl(?)) would indicate if establishment of the Pl′ state was affected in the F1. The numbers represent the number of plants displaying a given anther phenotype, indicating that the Pl′ state was established on the interchange chromosome in the rmr1 mutants.
Corresponding analysis of the establishment of paramutant states at the b1 locus generated similar results (Table S2). The repressed B′ state of the B1-Intense allele [37] was established in B′/B-I rmr1 mutants greater than 95% of the time. While it is possible that rmr1 defects affect establishment efficiency, it will be difficult to differentiate any such effects from its clear role in maintenance [11]. These results point to an interesting duality in RMR1 function in which the wild-type protein is necessary for meiotic heritability of repressed epigenetic states, but is not required to establish these states. This duality is markedly different from results generated in the analysis of DRD1, which was shown to be necessary for the maintenance, establishment, and removal of repressive epigenetic marks [24,25].
Discussion
RMR1 is the first protein identified whose function acts to maintain trans-generationally repressed states associated with paramutation, a genetic behavior that affects meiotically heritable epigenetic variation through allelic interactions at endogenous loci. The identification of RMR1 as a Snf2 protein highlights an emerging role of these proteins in establishing and maintaining epigenetic marks. In Arabidopsis the Snf2 proteins DRD1 and DDM1 [38,39] are known to maintain cytosine methylation patterns. Lsh1, the mammalian protein most closely related to DDM1, is also required for normal DNA methylation patterns [40–42]. There are some 42 Snf2 proteins in Arabidopsis and at least as many in maize (http://chromdb.org/). This diversity likely represents great functional specialization amongst these proteins. We have placed RMR1 in an RdDM pathway based on its helicase domain similarity to DRD1 and the recent identification of MOP1 as an RDR2 ortholog [14,15]. Consistent with this proposed pathway, the rmr1 mRNA expression profile (Figure S9) closely matches that of mop1 [15]. Additionally, both RMR1 and MOP1 are necessary to maintain cytosine methylation patterns at silenced transgenes [43], the Pl1-Rhoades doppia sequences, and certain Mutator transposable elements ([15,44]; J. B. H. and D. Lisch, unpublished data). DRD1 is also known to target repetitive elements found in euchromatic contexts through an RdDM pathway [31]. However, the role RMR1 plays to maintain the repressed paramutant states at Pl1-Rhoades appears different than the function of DRD1 in the Arabidopsis RdDM pathway, as RMR1 has, in addition to its requirement for CNN methylation at doppia, a role in the normal accumulation of small RNAs with similarity to that element.
It is unclear how RMR1 mediates the post-transcriptional regulation of pl1 transcripts as suggested by the in vitro transcription and RNase protection assays reported here. It is possible that pl1 transcripts resulting from Pl1-Rhoades in the Pl′ state are less stable than those produced from the Pl-Rh state because of differences in the chromatin environment of Pl1-Rhoades. However, there do not appear to be any Pl′-specific small RNAs produced from the pl1 coding region [12]. In S. pombe it has been shown that the chromatin environment of a locus can affect RNA transcript levels without altering RNA polymerase II occupancy of that locus, leading to the proposal of a cotranscriptional gene silencing mechanism whereby nascent transcripts initiating in a heterochromatic environment are degraded by complexes targeted via heterochromatic small RNAs [17,18]. Chromatin differences in the upstream region of Pl1-Rhoades may favor recruitment of alternative RNA-processing factors or RNA polymerases, which in turn influence the stability of pl1 transcripts. In plants, localization of the large subunit 1a of RNA polymerase IV to loci targeted for RdDM appears necessary for the biogenesis of siRNAs from these loci [28]. When Pl′ repression is disrupted in rmr1 mutants, this alternate genesis or processing of the pl1 transcript may also be lost. Alternatively, our results may highlight a novel role for RMR1-like Snf2 proteins in directly interacting with nascent RNA transcripts via a helicase domain, or in recruiting factors that directly destabilize these transcripts.
Importantly, our analysis of rmr1 mutants calls into question the relationship between RMR1 function and the mechanism of paramutation at Pl1-Rhoades. The mutational screens identifying rmr1, rmr6, and mop1 were designed to discover genetic components necessary to maintain the repressed state of Pl′, not necessarily factors needed to establish this repressed state [10,13]. Therefore, it is possible that loci thus far identified may be indirectly related to the paramutation mechanism. Our results are consistent with a model wherein RMR1 functions in an RdDM pathway, along with an RDR2-like enzyme, MOP1, to maintain a persistent heterochromatic-like chromatin structure at the repetitive element found directly upstream of the pl1 coding region. While it is not clear where RMR1 acts in this pathway it presumably acts coordinately with the maize orthologs of known RdDM components identified in Arabidopsis, namely DCL3 [16,45], the DRM methyltransferases [36], AGO4 [46,47], the RNA polymerase IV subunits, and the maize DRD1 ortholog (Figure 6A). In this model, doppia transcripts, perhaps because of the repetitive nature of the doppia genomic elements and/or the numerous internal subterminal repeats that are present in these elements [32,48], are the source of aberrant RNA that is processed via MOP1 and a DCL3 enzyme into siRNAs. This small RNA production is carried out in a manner that is dependent on RMR1 activity, possibly via direct interaction with a small RNA processing complex or by making the DNA accessible to factors necessary for siRNA precursor generation such as polymerase IVa. These siRNAs, through the activity of AGO4, DRM enzymes, and polymerase IVb, then establish a heterochromatic state at the Pl1-Rhoades doppia-like element that is present in both Pl-Rh and Pl′ states. The methylation effects seen in rmr1 mutants might indicate that this heterochromatization machinery depends on the activity of RMR1 to feed back on the doppia element, or loss of RMR1 may short circuit this pathway and thus affect methylation activity indirectly. An RMR1 defect then affects stability of paramutant states at pl1 because of the chromatin context of the Pl1-Rhoades allele, and not through direct disruption of components required for paramutations to occur. This is in line with a report that MOP1-dependent small RNAs produced at the b1 locus are insufficient to mediate paramutation [49].
Figure 6. Two General Models for RMR1 Action at the Pl1-Rhoades Allele.

RMR1 maintains nonsymmetrical methylation of the doppia element (light gray arrow) upstream of the pl1 coding region (exons in black) via an RdDM pathway. Small RNAs are produced in a RMR1-dependent fashion with homology to the doppia element, and maize orthologs of characterized RdDM proteins, as well as RMR1, then act as effectors of these siRNAs, facilitating cytosine methylation at complementary sequences of the DNA template. In the model shown in (A), the heterochromatic region of doppia is maintained and established independently of the Pl1-Rhoades chromatin state, but derepression of the upstream repetitive element in an rmr1 mutant causes changes in the nearby genic region through processivity of RNA polymerase II or other general transcription factors that bind the upstream elements. In the model shown in (B), the doppia element is repressed by the same RdDM pathway shown in (A), but the Pl′ state represents a spread of the heterochromatic domain beyond the region targeted by the siRNAs for cytosine methylation. This spread might be mediated by RMR1 activity, or by another chromatin modifier. In (B), loss of RMR1 would lead to a loss of the repressive chromatin state at doppia and the ability for it to spread.
The relationship between RMR1 action, the chromatin organization of Pl1-Rhoades, and the repressed Pl′ state is not clearly understood at this time. It is possible that derepression of the upstream repetitive element makes the region more accessible to general transcription factors whose actions could destabilize repressive Pl′ chromatin states that are independent of those maintained at doppia (Figure 6A). Indeed, RNA polymerase processivity can lead to changes in the chromatin environment through histone modifications or histone replacement [50,51]. Alternatively, Pl′ chromatin states may represent a spreading of the heterochromatic domain at doppia into a euchromatic region defined by the Pl1-Rhoades gene space (Figure 6B). In fission yeast, heterochromatic domains nucleated by small RNAs have the ability to spread in cis through successive H3 K9 methylation [52]. In this situation, loss of RMR1 function would alleviate Pl′ repression by disrupting maintenance of this expanded heterochromatic domain. In either of these situations RMR1 affects Pl1-Rhoades paramutations by virtue of its role in maintaining heterochromatic states at a proximal repetitive element.
McClintock was the first to describe derivative alleles in which transposons acted to control the expression patterns of attendant genes [53]. It is now clear that epigenetic modulations of the transposons themselves—what McClintock referred to as “changes in state”—can alter the regulatory properties of individual genes both somatically [54] and trans-generationally [55,56]. Our results indicate that even transient changes in state of the Pl1-Rhoades doppia fragment can have trans-generational effects on pl1 gene expression patterns. These experimental examples, in the context of McClintock's thesis [53], point to a dynamic source of regulatory, and potentially adaptive, variation adjunct to the DNA itself. Precisely how this epi-variation relates to existing genome structure and function, as well as its evolutionary potential, remains a largely unexplored area of investigation.
Currently, well-characterized examples of paramutation are limited to loci where expression states have a clear phenotypic read-out, such as pigment synthesis. cis-Elements required to facilitate paramutation have been functionally identified at specific alleles of b1 and colored1 (r1) [57–59]. To date, there is no evidence that the chromatin status of these cis-elements is affected by mutations at trans-acting loci required for maintenance of repressed paramutant states. It appears that paramutations represent a type of emergent system wherein genomic context and maintenance of chromatin states interact to facilitate meiotically heritable epigenetic variation. In this view, it is possible that cis- and trans-elements necessary for maintenance of such variation might not interact in a direct and predictable manner. What remains to be seen is the extent to which this type of system acts throughout the genome. Genome-wide screens for paramutation-like behavior, in which expression states are affected by allele history, remain technologically and conceptually challenging. Recent work by Kasschau et al. [60] suggests that in Arabidopsis, few endogenous genes are regulated by proximal presumed RdDM targets. However, it is tempting to speculate that examples of paramutation represent an exception to this trend, representing a mechanism by which populations can quickly, and heritably, change their transcriptome profile and regulation.
Materials and Methods
Scoring of the Pl1-Rhoades allele expression state and rmr mutants.
Plants were scored as carrying Pl-Rh or Pl′ states through visual inspection of anther pigmentation and assignment of an anther color score as previously described [7]. Pl′/Pl′ (anther color score 1 to 4) anthers show little to no pigmentation while Pl-Rh/Pl-Rh (anther color score 7) anthers are dark red to purple. Mutants were scored in the same way, with rmr and mop mutants showing a Pl-Rh/Pl-Rh-like phenotype, except in the case of the F2 rmr1 mapping populations, in which mutants were chosen on the basis of a dark seedling leaf phenotype [10].
Genetic stocks.
Elite inbred lines (B73, A619, and A632) were provided by the North Central Regional Plant Introduction Station (http://www.ars.usda.gov/main/site_main.htm?modecode=36-25-12–00). Color-converted versions of A619 and A632 inbred lines were created by introgressing the Pl1-Rhoades allele into each [11]. The rmr1–1, rmr1–2, mop1–1, and rmr6–1 alleles have been previously described [8,10,13]. The rmr1–3 allele was derived from identical materials used to isolate rmr1–1 and rmr1–2; rmr1–4 was derived from EMS-treated pollen from an A619 color-converted line applied to a color-converted A632 line [11] (see Protocol S1 and Table S3 for complementation tests). The T6–9 translocation line carrying the Pl1-Rhoades allele used in Pl′ establishment tests has been described previously [11].
pl1 expression analyses.
In vitro transcription assays (rmr1–1 and rmr1–3; Figures 1 and S1) and RNase protection assays (rmr1–3 only; Figure 1) were carried out as described [8] with husk nuclei and RNA isolated from single ears of the same genetic stocks used to measure pl1 RNA differences in rmr1–1 anthers [10]. The b1 and pl1 genotypes of these plants are as follows: B1-Intense (B-I)/B-I; Pl1-Rhoades (Pl′) Rmr1/Pl′ rmr1–1 and B-I/B-I; Pl′ rmr1–1/Pl′ rmr1–1, or B-I/B-I; Pl′ Rmr1/Pl′ rmr1–3 and B-I/B-I; Pl′ rmr1–3/Pl′ rmr1–3. Identical procedures were applied to single ears from plants homozygous for Pl′ and either homozygous or heterozygous for rmr1–3 following a single backcross into the KYS inbred line [12]. Additional details regarding stock syntheses are available upon request.
Genetic mapping of rmr1.
A F2 mapping population was created from inbred (S9) rmr1–1/rmr1–1, Pl′/Pl′, and color-converted A632 inbred (Pl′/Pl′, >93% A632) parents. DNA was isolated using the DNeasy 96 plant kit (Qiagen, http://www1.qiagen.com/) from F2 mutant seedlings, mapping parents, and F1 hybrid leaf tissue. These DNA samples were screened with SSLP markers developed from the Maize Mapping Project (http://www.maizemap.org/; US National Science Foundation award number 9872655; primer sequences and protocol available at http://maizegdb.org/). Initial marker choice was restricted to Chromosomes 6 and 9 because of linkage of rmr1 to a T6–9 breakpoint. In addition to the rmr1–1 mapping population, a second F2 mapping population created with inbred (S7) rmr1–3/rmr1–3, Pl′/Pl′, and color-converted A632 parents showed similar cosegregation with marker bnlg1174a (178 chromosomes tested; <0.56 cM). CAPS [61] markers were designed to test cosegregation of the rmr1–1- and rmr1–3-associated lesions with the rmr1 mutant phenotype (see Protocol S1 for details). No recombinant chromosomes (876 chromosomes tested for rmr1–1, 268 chromosomes tested for rmr1–3) were found using either marker.
Candidate gene selection and sequencing.
A BLAST search using the rice Os05g32610 ORF as a query identified maize GSS and sorghum expressed sequence tag sequences that were used to generate a contig representing the putative maize gene (see Protocol S1 for sequence identifiers). Oligonucleotide primers (Sigma-Genosys, http://www.sigmaaldrich.com/Brands/Sigma_Genosys.html) were designed from these sequences and used in PCR amplification of genomic DNA from three separate individuals homozygous for each rmr1 mutant allele as well as functional reference alleles Rmr1-B73, Rmr1-A632, and Rmr1-A619. PCR amplicons were purified using QIAquick gel extraction kit (Qiagen) and dideoxy sequenced (UC Berkeley DNA Sequencing Facility, http://mcb.berkeley.edu/barker/dnaseq/). To verify the intron/exon structure of rmr1, cDNA was generated from rmr1–1 mutants as well as non-mutant B73 plants as described [15], and rmr1 was amplified via RT-PCR. The resulting products, which were the predicted size for spliced rmr1 transcript, were sequenced to validate the intron/exon structure shown in Figure 2C. See Protocol S1 and Table S4 for all oligonucleotide primer sequences used.
Phylogenetic analysis.
Sequencing reads from genomic and cDNA were aligned and edited with Sequencher (Gene Codes, http://www.genecodes.com/) to create a contig representing rmr1. The N-terminal prediction is based on alignment of RMR1 with the protein model for Os05g32610. A search of the Pfam database (http://www.sanger.ac.uk/Software/Pfam/) with the predicted RMR1 protein sequence was used to identify the conserved SNF2_N and Helicase_C protein profiles of the Snf2 helicase domain. MUSCLE [62] was used to generate an alignment between RMR1 and proteins from Arabidopsis, rice, maize (CHR127 and CHR156), and budding yeast over the helicase domain (Figure S2). Sequences for CHR127 and CHR156 were retrieved from ChromDB (http://www.chromdb.org/). Additional sequence information for CHR156 was identified from BAC CH201-3L17 (GenBank accession AC194602), and gene model prediction was performed using FGENESH+ (Softberry, http://www.softberry.com/) with RMR1 as similar protein support. A distance tree was created and bootstrap values were calculated using PAUP* 4.0 from the above alignment (Sinauer Associates, http://www.sinauer.com/).
Southern blot analysis.
Genomic DNA was isolated as described [13] from the terminal flag leaves of adult plants segregating for rmr1, rmr6, and mop1 mutants and heterozygous siblings as well as Pl′ and Pl-Rh plants as assayed by anther pigmentation [7,8,10,13]. Restriction digest and subsequent Southern blots were carried out as previously described [13], using the restriction enzymes listed in Figure 4 (New England Biolabs, http://www.neb.com/). The probes specific to pl1 are shown in Figure 4; the 45S and centromere probes are as described [13].
Small RNA northern blots.
Small RNAs were prepared from 10-mm immature ear tissue and used to generate small RNA northern blots as previously described [63]. In Figure 4D the small RNAs were run with a 27-bp DNA oligonucleotide containing doppia sequence that hybridized with the riboprobe used to identify the small RNAs. The riboprobe was synthesized as described [63] from a plasmid containing the region denoted probe B in Figure 4A linearized at an AseI site so as to contain only doppia sequence.
Pl′ establishment tests.
Establishment of the Pl′ state in rmr1 mutants was assayed essentially as described previously [11]. When the T6–9 interchange pair is heterozygous with structurally normal chromosomes, the plants display ∼50% pollen sterility due to meiotic-segregation-induced aneuploidy in the resulting gametes. Pollen sterility was assayed in the field using a pocket microscope. rmr1 mutants were crossed to Pl-Rh/Pl-Rh A619 or A632 inbreds (Table S1), and the resultant progeny were scored with respect to Pl1-Rhoades expression state.
Supporting Information
(42 KB PDF)
In vitro assays with isolated husk nuclei show no differences in pl1 transcription rates between rmr1 mutants and non-mutant heterozygotes.
(A) In vitro radiolabeled RNAs corresponding to specific genes from isolated husk nuclei of sibling plants detected with slotblot hybridizations (pBS, bacterial plasmid DNA; pl1, purple plant1; b1, colored plant1; a1, anthocyaninless1; uq, ubiquitin2).
(B) Quantification of relative mean transcription rates from five independent sets of +/rmr1–1 (open) and rmr1–1/rmr1–1 (closed) siblings (± standard error of the mean) showing no significant difference between pl1 transcription rates.
(C) In vitro radiolabeled RNAs from isolated husk nuclei of rmr1–3 mutants and heterozygous siblings used to generate quantification in Figure 1A.
(708 KB TIF)
Multiple species alignment of RMR1 helicase domain with other known and predicted Snf2 proteins.
(101 KB TIF)
Additional Southern blots comparing the DNA methylation status of the Pl1-Rhoades upstream region in rmr1–1 mutants and heterozygous siblings.
(A) Genomic digests of an rmr1–1 mutant (−) and heterozygous sibling (+) using methylation-sensitive restriction enzymes in concert with BsrI, hybridized with probe A (Figure 4A). These results were used to generate the methylation profile shown in Figure 4A.
(B) Blot hybridized with probe A comparing rmr1–1 mutants to heterozygous siblings with respect to methylation at a PspGI site.
(2.4 MB TIF)
Southern blots comparing the DNA methylation status of the Pl1-Rhoades upstream region in rmr6–1 mutants and heterozygous siblings.
(A) Methylation profile similar to that shown in Figure 4A showing sites at the Pl1-Rhoades locus hypomethylated (open circle) in a rmr6–1 mutant as compared to heterozygous siblings.
(B and C) Blots shown in (B) and (C) were used to generate this profile and are analogous to the blots shown for rmr1 mutants in Figure S3A and S3B, respectively.
(2.2 MB TIF)
Southern blots comparing the DNA methylation status of the Pl1-Rhoades upstream region in mop1–1 mutants and heterozygous siblings.
(A) Methylation profile similar to that shown in Figure 4A showing sites at the Pl1-Rhoades locus hypomethylated (open circle) in a mop1–1 mutant as compared to heterozygous siblings.
(B and C) Blots shown in (B) and (C) were used to generate this profile and are analogous to the blots shown for rmr1 mutants in Figure S3A and S3B, respectively.
(2.2 MB TIF)
Additional small RNA northern blots showing that doppia small RNAs of both sense and antisense orientations are absent in rmr1 mutants. Small RNA northern blots were probed with probe B (Figure 4A) in both the sense (A) and antisense (B) orientations, showing small RNAs (∼26 nt) with doppia sequence similarity are present in sense and antisense orientation in rmr1–1 heterozygotes (+) and are lost in rmr1–1 mutants. DNA oligonucleotides (22 and 21 nt) used as sizing standards are also shown.
(1.3 MB TIF)
Southern blots show that rmr1 mutations do not affect methylation levels at centromeric sequences and 45S ribosomal DNA repeats. Genomic DNA from four rmr1–3 mutants and non-mutant siblings digested with BstNI (“B” lanes) and a CNG methylation-sensitive enzyme, PspGI (“P” lanes), which has the same recognition site, probed with (A) radiolabeled centromere sequence and (B) 45S repeat sequence. The comparison between the PspGI digests in mutant and non-mutant individuals reveals no gross methylation differences.
(6.2 MB TIF)
Additional Southern blots show no changes in Pl1-Rhoades methylation status between the Pl-Rh and Pl′ states.
(A) The methylation status of upstream PspGI sites is compared for Pl′/Pl′ and Pl-Rh/Pl-Rh plants with the Pl1-Rhoades allele introgressed (>98%) into distinct A619 and A632 backgrounds via hybridization with probe A. The blot reveals no methylation differences at this site between the two Pl1-Rhoades regulatory states. The “C” lanes indicate control lanes where the digest was carried out with BstNI, a methylation-insensitive restriction enzyme.
(B) Analogous to blot shown in Figure 4C, though the plants are from a different background (A619 introgression) than the plants used in Figure 4C (A632 introgression), showing that there are no methylation differences at the StuI site in either background.
(1.9 MB TIF)
RT-PCR expression profile shows rmr1 is expressed primarily in tissues with high mitotic index. Tissue samples represented in the analysis include seedling leaf, adult leaf, shoot apical meristem, immature tassel, and immature ear. RT-PCR was carried out using primers that span the first and second introns of rmr1.
(358 KB TIF)
(47 KB DOC)
Test cross results measuring acquisition of paramutagenicity by Pl-Rh in T Pl-Rh rmr1–2/+ Pl′ rmr1–1 plants. Genetic assay used for this experiment is detailed in Results and Figure 5. Table details individual anther phenotypes using a 1–7 graded anther color score for specific test cross progeny. Progeny structural genotypes refer to the presence or absence of the reference T6–9 interchange chromosome.
(127 KB DOC)
Table details individual progeny plant phenotypes from crosses of rmr1/rmr1; B-I/B′ plants to Rmr1 b1 testers. Two different mutant rmr1 alleles are assayed. The B-I and B′ plant phenotypes represent darkly pigmented and light or variegated pigment, respectively. With four exceptions, 206 test cross progeny had a B′ phenotype.
(38 KB DOC)
Table details individual progeny anther phenotypes graded using a 1–7 anther color score from crosses designed to test genetic complementation of various rmr mutations.
(70 KB DOC)
(45 KB DOC)
Acknowledgments
Thanks to all members of the Hollick lab for help and insightful comments on this manuscript and the corresponding research, with special thanks to Agnes Choi for assistance with the small RNA blots, and Negar Yaghooti for work on the sequencing of rmr1.
Abbreviations
- EMS
ethyl methanesulfonate
- RdDM
RNA-directed DNA methylation
- RT-PCR
reverse transcriptase PCR
- siRNA
small interfering RNA
Footnotes
Author contributions. CJH, JLS, and JBH conceived and designed the experiments. All authors performed the experiments and analyzed the data. CJH, JLS, SMG, and JBH contributed reagents/materials/analysis tools. CJH and JBH wrote the paper.
Funding. This work was supported by the National Research Initiative of the USDA Cooperative State Research, Education and Extension Service (99–35301-7753, 2001-35301-10641, 2005-35301-15891) and the National Science Foundation (MCB-0419909). The views expressed are solely those of the authors and are not endorsed by the sponsors of this work..
Competing interests. The authors have declared that no competing interests exist.
References
- Brink RA. Paramutation at the R locus in maize. Cold Spring Harb Symp Quant Biol. 1958;23:379–391. doi: 10.1101/sqb.1958.023.01.036. [DOI] [PubMed] [Google Scholar]
- Hollick JB, Dorweiler JE, Chandler VL. Paramutation and related allelic interactions. Trends Genet. 1997;13:302–308. doi: 10.1016/s0168-9525(97)01184-0. [DOI] [PubMed] [Google Scholar]
- Brink RA. Paramutation. Annu Rev Genet. 1973;7:129–152. doi: 10.1146/annurev.ge.07.120173.001021. [DOI] [PubMed] [Google Scholar]
- Chandler VL, Stam M. Chromatin conversations: Mechanisms and implications of paramutation. Nat Rev Genet. 2004;5:532–544. doi: 10.1038/nrg1378. [DOI] [PubMed] [Google Scholar]
- Rassoulzadegan M, Grandjean V, Gounon P, Vincent S, Gillot I, et al. RNA-mediated non-mendelian inheritance of an epigenetic change in the mouse. Nature. 2006;441:469–474. doi: 10.1038/nature04674. [DOI] [PubMed] [Google Scholar]
- Cone KC, Cocciolone SM, Burr FA, Burr B. Maize anthocyanin regulatory gene pl is a duplicate of c1 that functions in the plant. Plant Cell. 1993;5:1795–1805. doi: 10.1105/tpc.5.12.1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollick JB, Patterson GI, Coe EH, Jr, Cone KC, Chandler VL. Allelic interactions heritably alter the activity of a metastable maize pl allele. Genetics. 1995;141:709–719. doi: 10.1093/genetics/141.2.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollick JB, Patterson GI, Asmundsson IM, Chandler VL. Paramutation alters regulatory control of the maize pl locus. Genetics. 2000;154:1827–1838. doi: 10.1093/genetics/154.4.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollick JB, Chandler VL. Epigenetic allelic states of a maize transcriptional regulatory locus exhibit overdominant gene action. Genetics. 1998;150:891–897. doi: 10.1093/genetics/150.2.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollick JB, Chandler VL. Genetic factors required to maintain repression of a paramutagenic maize pl1 allele. Genetics. 2001;157:369–378. doi: 10.1093/genetics/157.1.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollick JB, Kermicle JL, Parkinson SE. Rmr6 maintains meiotic inheritance of paramutant states in Zea mays . Genetics. 2005;171:725–740. doi: 10.1534/genetics.105.045260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross SM, Hollick JB. Multiple trans-sensing interactions affect meiotically heritable epigenetic states at the maize pl1 locus. Genetics. 2007;176:829–839. doi: 10.1534/genetics.107.072496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorweiler JE, Carey CC, Kubo KM, Hollick JB, Kermicle JL, et al. Mediator of paramutation1 is required for establishment and maintenance of paramutation at multiple maize loci. Plant Cell. 2000;12:2101–2118. doi: 10.1105/tpc.12.11.2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alleman M, Sidorenko L, McGinnis K, Seshadri V, Dorweiler JE, et al. An RNA-dependent RNA polymerase is required for paramutation in maize. Nature. 2006;442:295–298. doi: 10.1038/nature04884. [DOI] [PubMed] [Google Scholar]
- Woodhouse MR, Freeling M, Lisch D. Initiation, establishment, and maintenance of heritable MuDR transposon silencing in maize are mediated by distinct factors. PLoS Biol. 2006;4:e339. doi: 10.1371/journal.pbio.0040339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SW, Zilberman D, Xie Z, Johansen LK, Carrington JC, et al. RNA silencing genes control de novo DNA methylation. Science. 2004;303:1336. doi: 10.1126/science.1095989. [DOI] [PubMed] [Google Scholar]
- Buhler M, Verdel A, Moazed D. Tethering RITS to a nascent transcript initiates RNAi- and heterochromatin-dependent gene silencing. Cell. 2006;125:873–886. doi: 10.1016/j.cell.2006.04.025. [DOI] [PubMed] [Google Scholar]
- Buhler M, Haas W, Gygi SP, Moazed D. RNAi-dependent and -independent RNA turnover mechanisms contribute to heterochromatic gene silencing. Cell. 2007;129:707–721. doi: 10.1016/j.cell.2007.03.038. [DOI] [PubMed] [Google Scholar]
- Sainz MB, Grotewold E, Chandler VL. Evidence for direct activation of an anthocyanin promoter by the maize C1 protein and comparison of DNA binding by related Myb domain proteins. Plant Cell. 1997;9:611–625. doi: 10.1105/tpc.9.4.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence CJ, Schaeffer ML, Seigfried TE, Campbell DA, Harper LC. MaizeGDB's new data types, resources and activities. Nucleic Acids Res. 2007;35:D895–D900. doi: 10.1093/nar/gkl1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaus A, Martin DM, Barton GJ, Owen-Hughes T. Identification of multiple distinct Snf2 subfamilies with conserved structural motifs. Nucleic Acids Res. 2006;34:2887–2905. doi: 10.1093/nar/gkl295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durr H, Flaus A, Owen-Hughes T, Hopfner KP. Snf2 family ATPases and DExx box helicases: Differences and unifying concepts from high-resolution crystal structures. Nucleic Acids Res. 2006;34:4160–4167. doi: 10.1093/nar/gkl540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DT. GenTHREADER: An efficient and reliable protein fold recognition method for genomic sequences. J Mol Biol. 1999;287:797–815. doi: 10.1006/jmbi.1999.2583. [DOI] [PubMed] [Google Scholar]
- Kanno T, Mette MF, Kreil DP, Aufsatz W, Matzke M, et al. Involvement of putative SNF2 chromatin remodeling protein DRD1 in RNA-directed DNA methylation. Curr Biol. 2004;14:801–805. doi: 10.1016/j.cub.2004.04.037. [DOI] [PubMed] [Google Scholar]
- Kanno T, Aufsatz W, Jaligot E, Mette MF, Matzke M, et al. A SNF2-like protein facilitates dynamic control of DNA methylation. EMBO Rep. 2005;6:649–655. doi: 10.1038/sj.embor.7400446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SW, Henderson IR, Zhang X, Shah G, Chien JS, et al. RNAi, DRD1, and histone methylation actively target developmentally important non-CG DNA methylation in Arabidopsis . PLoS Genet. 2006;2:e83. doi: 10.1371/journal.pgen.0020083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieu O, Bender J. RNA-directed DNA methylation. J Cell Sci. 2004;117:4881–4888. doi: 10.1242/jcs.01479. [DOI] [PubMed] [Google Scholar]
- Pontes O, Li CF, Nunes PC, Haag J, Ream T, et al. The Arabidopsis chromatin-modifying nuclear siRNA pathway involves a nucleolar RNA processing center. Cell. 2006;126:79–92. doi: 10.1016/j.cell.2006.05.031. [DOI] [PubMed] [Google Scholar]
- Smith LM, Pontes O, Searle I, Yelina N, Yousafzai FK, et al. An SNF2 protein associated with nuclear RNA silencing and the spread of a silencing signal between cells in Arabidopsis . Plant Cell. 2007;19:1507–1521. doi: 10.1105/tpc.107.051540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaked H, Avivi-Ragolsky N, Levy AA. Involvement of the Arabidopsis SWI2/SNF2 chromatin remodeling gene family in DNA damage response and recombination. Genetics. 2006;173:985–994. doi: 10.1534/genetics.105.051664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huettel B, Kanno T, Daxinger L, Aufsatz W, Matzke AJ, et al. Endogenous targets of RNA-directed DNA methylation and Pol IV in Arabidopsis . EMBO J. 2006;25:2828–2836. doi: 10.1038/sj.emboj.7601150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cone KC, Cocciolone SM, Moehlenkamp CA, Weber T, Drummond BJ, et al. Role of the regulatory gene pl in the photocontrol of maize anthocyanin pigmentation. Plant Cell. 1993;5:1807–1816. doi: 10.1105/tpc.5.12.1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker EL, Robbins TP, Bureau TE, Kermicle J, Dellaporta SL. Transposon-mediated chromosomal rearrangements and gene duplications in the formation of the maize R-r complex. EMBO J. 1995;14:2350–2363. doi: 10.1002/j.1460-2075.1995.tb07230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X, Jacobsen SE. Locus-specific control of asymmetric and CpNpG methylation by the DRM and CMT3 methyltransferase genes. Proc Natl Acad Sci U S A. 2002;99:16491–16498. doi: 10.1073/pnas.162371599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X, Jacobsen SE. Role of the Arabidopsis DRM methyltransferases in de novo DNA methylation and gene silencing. Curr Biol. 2002;12:1138–1144. doi: 10.1016/s0960-9822(02)00925-9. [DOI] [PubMed] [Google Scholar]
- Cao X, Aufsatz W, Zilberman D, Mette MF, Huang MS, et al. Role of the DRM and CMT3 methyltransferases in RNA-directed DNA methylation. Curr Biol. 2003;13:2212–2217. doi: 10.1016/j.cub.2003.11.052. [DOI] [PubMed] [Google Scholar]
- Patterson GI, Thorpe CJ, Chandler VL. Paramutation, an allelic interaction, is associated with a stable and heritable reduction of transcription of the maize b regulatory gene. Genetics. 1993;135:881–894. doi: 10.1093/genetics/135.3.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeddeloh JA, Stokes TL, Richards EJ. Maintenance of genomic methylation requires a SWI2/SNF2-like protein. Nat Genet. 1999;22:94–97. doi: 10.1038/8803. [DOI] [PubMed] [Google Scholar]
- Brzeski J, Jerzmanowski A. Deficient in DNA methylation 1 (DDM1) defines a novel family of chromatin-remodeling factors. J Biol Chem. 2003;278:823–828. doi: 10.1074/jbc.M209260200. [DOI] [PubMed] [Google Scholar]
- Raabe EH, Abdurrahman L, Behbehani G, Arceci RJ. An SNF2 factor involved in mammalian development and cellular proliferation. Dev Dyn. 2001;221:92–105. doi: 10.1002/dvdy.1128. [DOI] [PubMed] [Google Scholar]
- Dennis K, Fan T, Geiman T, Yan Q, Muegge K. Lsh, a member of the SNF2 family, is required for genome-wide methylation. Genes Dev. 2001;15:2940–2944. doi: 10.1101/gad.929101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourc'his D, Bestor TH. Helicase homologues maintain cytosine methylation in plants and mammals. Bioessays. 2002;24:297–299. doi: 10.1002/bies.10078. [DOI] [PubMed] [Google Scholar]
- McGinnis KM, Springer C, Lin Y, Carey CC, Chandler V. Transcriptionally silenced transgenes in maize are activated by three mutations defective in paramutation. Genetics. 2006;173:1637–1647. doi: 10.1534/genetics.106.058669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisch D, Carey CC, Dorweiler JE, Chandler VL. A mutation that prevents paramutation in maize also reverses Mutator transposon methylation and silencing. Proc Natl Acad Sci U S A. 2002;99:6130–6135. doi: 10.1073/pnas.052152199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Johansen LK, Gustafson AM, Kasschau KD, Lellis AD, et al. Genetic and functional diversification of small RNA pathways in plants. PLoS Biol. 2004;2:e104. doi: 10.1371/journal.pbio.0020104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilberman D, Cao X, Jacobsen SE. ARGONAUTE4 control of locus-specific siRNA accumulation and DNA and histone methylation. Science. 2003;299:716–719. doi: 10.1126/science.1079695. [DOI] [PubMed] [Google Scholar]
- Zilberman D, Cao X, Johansen LK, Xie Z, Carrington JC, et al. Role of Arabidopsis ARGONAUTE4 in RNA-directed DNA methylation triggered by inverted repeats. Curr Biol. 2004;14:1214–1220. doi: 10.1016/j.cub.2004.06.055. [DOI] [PubMed] [Google Scholar]
- Bercury SD, Panavas T, Irenze K, Walker EL. Molecular analysis of the doppia transposable element of maize. Plant Mol Biol. 2001;47:341–351. doi: 10.1023/a:1011606529513. [DOI] [PubMed] [Google Scholar]
- Chandler VL. Paramutation: From maize to mice. Cell. 2007;128:641–645. doi: 10.1016/j.cell.2007.02.007. [DOI] [PubMed] [Google Scholar]
- Eissenberg JC, Shilatifard A. Leaving a mark: The many footprints of the elongating RNA polymerase II. Curr Opin Genet Dev. 2006;16:184–190. doi: 10.1016/j.gde.2006.02.004. [DOI] [PubMed] [Google Scholar]
- Farris SD, Rubio ED, Moon JJ, Gombert WM, Nelson BH, et al. Transcription-induced chromatin remodeling at the c-myc gene involves the local exchange of histone H2A.Z. J Biol Chem. 2005;280:25298–25303. doi: 10.1074/jbc.M501784200. [DOI] [PubMed] [Google Scholar]
- Irvine DV, Zaratiegui M, Tolia NH, Goto DB, Chitwood DH, et al. Argonaute slicing is required for heterochromatic silencing and spreading. Science. 2006;313:1134–1137. doi: 10.1126/science.1128813. [DOI] [PubMed] [Google Scholar]
- McClintock B. Chromosome organization and genic expression. Cold Spring Harb Symp Quant Biol. 1951;16:13–47. doi: 10.1101/sqb.1951.016.01.004. [DOI] [PubMed] [Google Scholar]
- Martienssen R, Barkan A, Taylor WC, Freeling M. Somatically heritable switches in the DNA modification of mu transposable elements monitored with a suppressible mutant in maize. Genes Dev. 1990;4:331–343. doi: 10.1101/gad.4.3.331. [DOI] [PubMed] [Google Scholar]
- Fedoroff NV. The Suppressor-mutator element and the evolutionary riddle of transposons. Genes Cells. 1999;4:11–19. doi: 10.1046/j.1365-2443.1999.00233.x. [DOI] [PubMed] [Google Scholar]
- Blewitt ME, Vickaryous NK, Paldi A, Koseki H, Whitelaw E. Dynamic reprogramming of DNA methylation at an epigenetically sensitive allele in mice. PLoS Genet. 2006;2:e49. doi: 10.1371/journal.pgen.0020049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kermicle JL, Eggleston WB, Alleman M. Organization of paramutagenicity in R-stippled maize. Genetics. 1995;141:361–372. doi: 10.1093/genetics/141.1.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panavas T, Weir J, Walker EL. The structure and paramutagenicity of the R-marbled haplotype of Zea mays . Genetics. 1999;153:979–991. doi: 10.1093/genetics/153.2.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stam M, Belele C, Ramakrishna W, Dorweiler JE, Bennetzen JL, et al. The regulatory regions required for B′ paramutation and expression are located far upstream of the maize b1 transcribed sequences. Genetics. 2002;162:917–930. doi: 10.1093/genetics/162.2.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasschau KD, Fahlgren N, Chapman EJ, Sullivan CM, Cumbie JS, et al. Genome-wide profiling and analysis of Arabidopsis siRNAs. PLoS Biol. 2007;5:e57. doi: 10.1371/journal.pbio.0050057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konieczny A, Ausubel FM. A procedure for mapping Arabidopsis mutations using co-dominant ecotype-specific PCR-based markers. Plant J. 1993;4:403–410. doi: 10.1046/j.1365-313x.1993.04020403.x. [DOI] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotkin RK, Freeling M, Lisch D. Heritable transposon silencing initiated by a naturally occurring transposon inverted duplication. Nat Genet. 2005;37:641–644. doi: 10.1038/ng1576. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(42 KB PDF)
In vitro assays with isolated husk nuclei show no differences in pl1 transcription rates between rmr1 mutants and non-mutant heterozygotes.
(A) In vitro radiolabeled RNAs corresponding to specific genes from isolated husk nuclei of sibling plants detected with slotblot hybridizations (pBS, bacterial plasmid DNA; pl1, purple plant1; b1, colored plant1; a1, anthocyaninless1; uq, ubiquitin2).
(B) Quantification of relative mean transcription rates from five independent sets of +/rmr1–1 (open) and rmr1–1/rmr1–1 (closed) siblings (± standard error of the mean) showing no significant difference between pl1 transcription rates.
(C) In vitro radiolabeled RNAs from isolated husk nuclei of rmr1–3 mutants and heterozygous siblings used to generate quantification in Figure 1A.
(708 KB TIF)
Multiple species alignment of RMR1 helicase domain with other known and predicted Snf2 proteins.
(101 KB TIF)
Additional Southern blots comparing the DNA methylation status of the Pl1-Rhoades upstream region in rmr1–1 mutants and heterozygous siblings.
(A) Genomic digests of an rmr1–1 mutant (−) and heterozygous sibling (+) using methylation-sensitive restriction enzymes in concert with BsrI, hybridized with probe A (Figure 4A). These results were used to generate the methylation profile shown in Figure 4A.
(B) Blot hybridized with probe A comparing rmr1–1 mutants to heterozygous siblings with respect to methylation at a PspGI site.
(2.4 MB TIF)
Southern blots comparing the DNA methylation status of the Pl1-Rhoades upstream region in rmr6–1 mutants and heterozygous siblings.
(A) Methylation profile similar to that shown in Figure 4A showing sites at the Pl1-Rhoades locus hypomethylated (open circle) in a rmr6–1 mutant as compared to heterozygous siblings.
(B and C) Blots shown in (B) and (C) were used to generate this profile and are analogous to the blots shown for rmr1 mutants in Figure S3A and S3B, respectively.
(2.2 MB TIF)
Southern blots comparing the DNA methylation status of the Pl1-Rhoades upstream region in mop1–1 mutants and heterozygous siblings.
(A) Methylation profile similar to that shown in Figure 4A showing sites at the Pl1-Rhoades locus hypomethylated (open circle) in a mop1–1 mutant as compared to heterozygous siblings.
(B and C) Blots shown in (B) and (C) were used to generate this profile and are analogous to the blots shown for rmr1 mutants in Figure S3A and S3B, respectively.
(2.2 MB TIF)
Additional small RNA northern blots showing that doppia small RNAs of both sense and antisense orientations are absent in rmr1 mutants. Small RNA northern blots were probed with probe B (Figure 4A) in both the sense (A) and antisense (B) orientations, showing small RNAs (∼26 nt) with doppia sequence similarity are present in sense and antisense orientation in rmr1–1 heterozygotes (+) and are lost in rmr1–1 mutants. DNA oligonucleotides (22 and 21 nt) used as sizing standards are also shown.
(1.3 MB TIF)
Southern blots show that rmr1 mutations do not affect methylation levels at centromeric sequences and 45S ribosomal DNA repeats. Genomic DNA from four rmr1–3 mutants and non-mutant siblings digested with BstNI (“B” lanes) and a CNG methylation-sensitive enzyme, PspGI (“P” lanes), which has the same recognition site, probed with (A) radiolabeled centromere sequence and (B) 45S repeat sequence. The comparison between the PspGI digests in mutant and non-mutant individuals reveals no gross methylation differences.
(6.2 MB TIF)
Additional Southern blots show no changes in Pl1-Rhoades methylation status between the Pl-Rh and Pl′ states.
(A) The methylation status of upstream PspGI sites is compared for Pl′/Pl′ and Pl-Rh/Pl-Rh plants with the Pl1-Rhoades allele introgressed (>98%) into distinct A619 and A632 backgrounds via hybridization with probe A. The blot reveals no methylation differences at this site between the two Pl1-Rhoades regulatory states. The “C” lanes indicate control lanes where the digest was carried out with BstNI, a methylation-insensitive restriction enzyme.
(B) Analogous to blot shown in Figure 4C, though the plants are from a different background (A619 introgression) than the plants used in Figure 4C (A632 introgression), showing that there are no methylation differences at the StuI site in either background.
(1.9 MB TIF)
RT-PCR expression profile shows rmr1 is expressed primarily in tissues with high mitotic index. Tissue samples represented in the analysis include seedling leaf, adult leaf, shoot apical meristem, immature tassel, and immature ear. RT-PCR was carried out using primers that span the first and second introns of rmr1.
(358 KB TIF)
(47 KB DOC)
Test cross results measuring acquisition of paramutagenicity by Pl-Rh in T Pl-Rh rmr1–2/+ Pl′ rmr1–1 plants. Genetic assay used for this experiment is detailed in Results and Figure 5. Table details individual anther phenotypes using a 1–7 graded anther color score for specific test cross progeny. Progeny structural genotypes refer to the presence or absence of the reference T6–9 interchange chromosome.
(127 KB DOC)
Table details individual progeny plant phenotypes from crosses of rmr1/rmr1; B-I/B′ plants to Rmr1 b1 testers. Two different mutant rmr1 alleles are assayed. The B-I and B′ plant phenotypes represent darkly pigmented and light or variegated pigment, respectively. With four exceptions, 206 test cross progeny had a B′ phenotype.
(38 KB DOC)
Table details individual progeny anther phenotypes graded using a 1–7 anther color score from crosses designed to test genetic complementation of various rmr mutations.
(70 KB DOC)
(45 KB DOC)