

Abstract
Background
The natural course of Sturge-Weber syndrome (SWS) is poorly understood, although neurological symptoms are often progressive.
Aims
To track longitudinal changes in brain glucose metabolism measured with positron emission tomography (PET) and their relation to clinical changes during the early course of SWS.
Methods
Fourteen children (age 3 months to 3.9 years at enrollment) with SWS and unilateral leptomeningeal angioma underwent two consecutive glucose metabolism PET scans with a mean follow-up time of 1.2 years. Longitudinal changes of the extent of cortical glucose hypometabolism on the angioma side were measured and correlated with age, clinical seizure frequency and hemiparesis.
Result
An increase in the size of the hypometabolic cortex was seen in 6 children, coinciding with an age-related increase in cortical glucose metabolism measured in unaffected contralateral cortex. These 6 patients were younger both at the initial (mean age 0.75 vs. 2.8 years; p<0.001) and the second scan (mean age 1.8 vs. 4.2 years; p=0.001) than those with no change in the extent of hypometabolic cortex (N=6). The area of cortical hypometabolism decreased in the two remaining children, and this was associated with resolution of an initial hemiparesis in one of them. Seizure frequency between the two scans was higher in children who showed progressive enlargement of cortical hypometabolism, as compared to those with no progression (p = 0.008).
Conclusions
In SWS, detrimental metabolic changes occur before 3 years of age coinciding with a sharp increase of developmentally regulated cerebral metabolic demand. Progressive hypometabolism is associated with high seizure frequency in these children. However, metabolic abnormalities may remain limited or even partially recover later in some children with well-controlled seizures. Metabolic recovery accompanied by neurological improvement suggests a window for therapeutic intervention in children with unilateral SWS.
Keywords: Sturge-Weber syndrome, positron emission tomography, brain glucose metabolism, seizures, longitudinal study, brain development
INTRODUCTION
Sturge-Weber syndrome (SWS) is a phakomatosis characterized by a port wine stain (PWS) over the trigeminal area, leptomeningeal angiomatosis and ocular abnormalities. The cerebral vascular malformation consists of abnormally developed venous vessels on the brain surface, resulting in an impairment of cerebral blood flow, hypoxia and chronic ischemia in the underlying brain tissue. Neurological outcome in children with SWS is highly variable, ranging from minimal or no neurological signs to a devastating impairment with uncontrolled seizures, hemiparesis, visual field defect and progressive mental retardation.1
Neuroimaging studies help establish the diagnosis, assess severity and follow the progression of brain involvement in SWS. MRI detects the leptomeningeal angioma and also can determine the extent of structural brain abnormalities,2-5 which are correlated, to a certain extent, with the severity of neurological symptoms.6,7 However, MRI abnormalities are not reliable predictors of neurological deficit in the early stage of the disease. Positron emission tomography (PET) scanning with 2-deoxy-2[18F]fluoro-d-glucose (FDG) often shows hypometabolic cortex extending beyond the apparent structural abnormalities.8,9 Progression of abnormalities in metabolism and perfusion often occur during the course of the disease8,10 and can be associated with neurologic deterioration.11
Although it is generally believed that SWS has an irremissive progression, the natural course of the disease is poorly characterized, largely because of the lack of longitudinal studies that could quantify changes of structural or functional brain abnormalities. Therefore, we have performed a longitudinal neurological and neuroimaging study of children with SWS to test the hypothesis that the majority of progression in the extent of glucose hypometabolism occurs during the critical period when metabolic demand in the brain increases sharply due to normal maturational processes.12 We also assessed whether the observed hypometabolism is irreversible and how metabolic changes are related to seizures and hemiparesis.
METHODS
Subjects
Fourteen children (9 girls, age 3 months to 3.9 years at the time of the first scan; mean age: 1.9 years) with the clinical and radiological diagnosis of Sturge-Weber syndrome with unilateral hemispheric involvement (based on location of leptomeningeal angioma on post-contrast MRI) and a history of seizures were included in the study (Table 1). All patients underwent FDG PET scanning twice; the interval between the two PET scans was within 3 years (range: 0.6 – 2.8 years, mean 1.2 ± 0.7 years). These patients were either prospectively recruited to participate in a longitudinal imaging study with yearly follow-ups or had a previous PET scan as part of a presurgical evaluation but were not operated and were re-scanned later. Frequency of clinical seizures was estimated for the period before the first PET scan (since the onset of the first seizure) as well as between the two PET scans by interviewing the parents and also by reviewing clinical charts. An estimated yearly seizure frequency was then calculated; this included all clinical seizures observed by the parents. None of the patients had status epilepticus. Age at the first clinical seizure (in months) was also recorded. Motor strength was clinically evaluated by one of the co-investigators (H.T.C.). Because some of the patients were enrolled retrospectively, and they had no detailed, standardized motor evaluation available, motor strength was categorized on a robust 3-point scale (0 : normal motor strength; 1: mild/moderate weakness in the arm, hand and/or leg; 2: severe hemiparesis). Based on these categories, longitudinal change of gross motor functions between the two scans was categorized as: (1) no significant change; (2) progression, when the patient developed hemiparesis between the two scans or the degree of paresis worsened, and (3) improvement, when the paresis observed at the first time was not seen at the second evaluation or when the degree of hemiparesis decreased. The study was approved by the Human Investigation Committee at Wayne State University, and written informed consent of the parent or legal guardian was obtained.
Table 1.
Clinical data of the 14 patients. Yearly seizure frequency is shown, calculated for the period between the two PET scans.
| No. | G | Age
Scan 1 (yr) |
Age
Scan 2 (yr) |
Inter-scan
period (yr) |
AED
Scan 1 |
AED
Scan 2 |
Seizure
Frequency |
Age at
seizure onset (mo) |
Paresis
Scan 1 |
Paresis
Scan 2 |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F | 0.2 | 1.7 | 1.5 | none | CBZ | ~1000 | 5 | - | + |
| 2 | F | 0.4 | 1.2 | 0.8 | CBZ | CBZ, PHB | ~1200 | 1 | + | + |
| 3 | F | 0.4 | 1.2 | 0.8 | none | OXC, VPA | 6 | 2 | - | + |
| 4 | F | 0.7 | 1.3 | 0.6 | PHB, LAM | PHB | 3 | 4 | - | + |
| 5 | M | 0.9 | 2.7 | 1.8 | PHB | OXC | 15 | 3 | + | + |
| 6 | F | 1.8 | 2.8 | 1 | OXC | LEV | 1 | 18 | + | - |
| 7 | M | 1.9 | 2.5 | 0.6 | TPX | TPX, PHB | 4 | 6 | + | ++ |
| 8 | M | 1.9 | 3 | 1.1 | CBZ | CBZ | 0 | 6 | + | + |
| 9 | M | 2.3 | 3.3 | 1 | PHB, OXC | PHB, OXC | 3 | 5 | + | + |
| 10 | F | 2.5 | 3.5 | 1 | LAM | LAM | 0 | 23 | - | - |
| 11 | F | 3.1 | 5.5 | 2.4 | PHB | PHB | 0 | 0.5 | + | + |
| 12 | M | 3.5 | 6.3 | 2.8 | CBZ, LAM, CLO | CBZ, LEV | 0 | 9 | - | - |
| 13 | F | 3.6 | 4.7 | 1.1 | OXC, LEV | OXC, LEV | 15 | 10 | - | - |
| 14 | F | 3.9 | 4.9 | 1 | OXC | OXC | 3 | 5 | + | + |
G: Gender; Age scan 1 and 2: age, in years, at the first and second scan; AED scan 1 and 2: antiepileptic drug(s) taken at the time of the first and second scan, respectively; CBZ: carbamazepine, OXC: oxcarbazepine, TPX: topiramate, LEV: levetiracetam, PHB: phenobarbital, LAM: lamotrigine, VPA: valproate, CLO: clobazam, Paresis scan 1 and 2: - indicates normal motor strength, + indicates mild/moderate paresis in the hand, arm or leg, and ++ indicates severe hemiparesis.
PET image acquisition
The PET protocol was performed as previously described.9,13 In brief, all patients underwent their PET scans using the CTI/Siemens EXACT/HR PET scanner. This scanner has a 15 cm field of view and generates 47 image planes with a slice thickness of 3 mm. The reconstructed image in-plane resolution obtained is 5.5 ± 0.35 mm at full-width-at-half-maximum and 6.0 ± 0.49 mm in the axial direction (reconstruction parameters: Shepp-Logan filter with 0.3 cycles/pixel cutoff frequency). FDG was produced using a Siemens RDS-11 cyclotron, applying the synthesis module purchased from Siemens/CTI (Knoxville, TN). Subjects fasted for 4 hours prior to the PET procedure. A venous line was established for injection of FDG (0.143 mCi/kg). The lights were dimmed and interactions were discouraged in order to reflect a resting awake state. All PET scans were done at least 24 hours after the last clinical seizure and the median time between the last seizure and the PET scan was 9 and 7 weeks for the first and second scans, respectively (first scans: 2 weeks – 2 years; second scans: 1 day – 4 years). All scans were acquired in the interictal state as demonstrated by scalp EEG recorded throughout the uptake period. Thus, ictal/post-ictal states were not confounding factors for interpretation of the PET results. After 40 minutes of FDG uptake, patients were positioned in the scanner and a static 20-minute emission scan of brain was performed. Children below 2 years of age were sedated with chloral hydrate (50-100 mg/kg by mouth), and children older than 2 years were sedated with nembutal (3 mg/kg), followed by fentanyl (1 μg/kg).
PET image analysis
Calculated attenuation correction was applied to the brain images using automated threshold fits to the sinogram data. The hemispheric extent of cortical glucose hypometabolism on the side of the angioma was obtained by using a semiautomated software package as described previously.9,14 This software marks the abnormal cortical areas of glucose metabolism based on an asymmetry index (AI) derived from contralateral homotopic areas according to a cutoff threshold selected by the user (AI (%) =(I-C)/[(I+C)/2]×100%, where I represents the radioactivity concentration in the ipsilateral and C represents the concentration in the contralateral homotopic cortical region). A cutoff threshold of AI(%) = 10% was used to mark cortical segments with abnormally decreased glucose metabolism on the side of the angioma.9,15 The marked PET data files were then processed by the 3DTool software package creating a three-dimensional brain surface image to display the marked areas in red. The extent of cortical hypometabolism was measured by delineating and counting the pixel number in the marked hypometabolic area, and dividing this number by the number of all surface pixels representing the ipsilateral hemisphere,13 using software developed in-house. Thus, the size of cortical hypometabolism was described as the percent of total hemispheric surface, and this value was independent of the actual size of the hemisphere. The advantages and limitations of this method have been discussed elsewhere.13 Importantly, since this method does not rely on absolute or normalized metabolic rates, the obtained values are highly independent of global cerebral metabolic changes related to age12 or drug-effects.16 A potential, newly recognized pitfall of this analysis method is that the measured extent of cortical hypometabolism may be overestimated if increased glucose metabolism is present in the contralateral cortex: indeed, this has been observed in the posterior (mostly occipital) cortex of some children with unilateral SWS, presumably reflecting reorganizational changes.17 However, the effect of these increases should be minimal when using the stricter 20% cutoff threshold: >20% asymmetry is consistent with severe hypometabolism and cannot be simply explained by mild or moderate increases of metabolism in the contralateral hemisphere. Therefore, we have reanalyzed the data by using this stricter threshold. Since the results did not change substantially, and interpretation of the findings remained the same, only data with the 10% threshold are reported.
In order to evaluate how small changes in extent of hypometabolism can be detected reliably, we performed test-retest measurements. The measured values were highly reproducible: correlation between corresponding values measured at the first and second measurements was highly significant (r = 0.99, p < 0.001). The mean difference between the test-retest measurements was 1.2 ± 1.3 %, with a maximal individual difference of 4.5% of the hemispheric surface. Therefore, longitudinal changes (increases or decreases) in extent of hypometabolism of ≥5% of the hemispheric surface were considered to represent true progression (increase of extent) or improvement (decrease in extent).
Since follow-up time differed among patients, a monthly rate of metabolic change was calculated by using the following formula: ([PET1 − PET2] / ΔT), where PET1 and PET2 represent the extent of cortical hypometabolism (expressed as the % of total hemispheric surface area) at the first and second scan, respectively, and ΔT represents the time between the two scans (in months).
In order to estimate regional brain glucose metabolism and its relation to the patients’ age (on the first PET scan) in the unaffected hemisphere, standard uptake values (SUVs) were calculated for cortex contralateral to the angioma. The SUV was calculated as the ratio between the average radioactivity concentration obtained from the cortex and the injected FDG dose per weight (mCi/kg). A previous study demonstrated that SUV provides a good estimation of cerebral glucose uptake and allows inter-individual comparisons in young children where arterial input function is not available for quantification of glucose metabolic rates.18 Since the contralateral hemisphere was not directly affected by the pathology, an age-related increase due to brain maturation was expected.12 In order to avoid the confounding effect of increased glucose metabolism observed in the contralateral posterior cortex seen in some children with SWS and severe ipsilateral posterior hypometabolism,17 we have correlated age with SUVs measured in the contralateral frontal cortex.
Statistical analysis
The age of patients with vs. without metabolic progression was compared by an unpaired t-test. The relationship between patient age and monthly rate of metabolic change was assessed using linear and non-linear regression analyses. The most appropriate regression models for the data sets were determined using the curve estimation procedure (SPSS 11.5, Chicago, IL). Age and SUV values measured in the contralateral cortex were correlated by using Pearson’s correlation while SUV values at the time of the first and second scans were compared using a paired t-test. The Mann-Whitney test was used to compare estimated seizure frequency and age at seizure onset between patients with vs. without metabolic progression. Finally, the extent of hypometabolism was compared between patients with vs. without paresis using an unpaired t-test. P<0.05 was considered to be significant.
RESULTS
Hemispheric extent of the hypometabolic area
Three different patterns of metabolic change were observed from the PET data. In 6 children, a robust increase in the extent of the hypometabolic area was found (from an average of 17% [range 0-36%] at the first scan to an average of 58% [range 26-78%] at the second scan), with a progression rate of 4% (1-11%)/month (Figure 1). In 6 other children, the extent of the hypometabolic area remained stable (mean 36% on both the first and the second scan). The remaining 2 patients showed a decrease in the extent of hypometabolism from 38% and 69% to 24 and 60%, respectively, suggesting a diminution (i.e., improvement) in the extent of hypometabolism (Figure 2).
Figure 1.
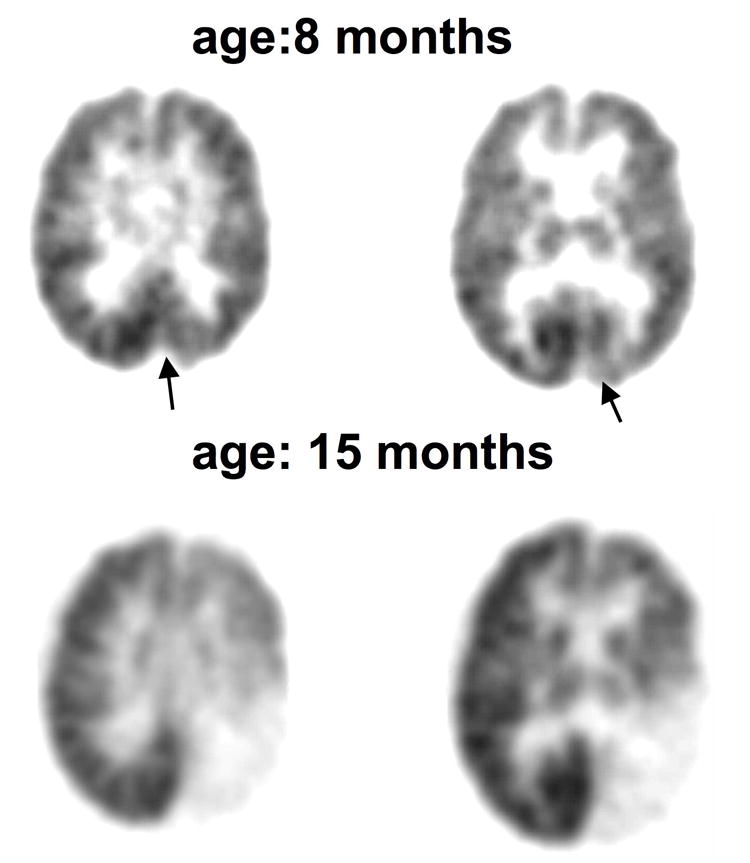
FDG PET scans in a girl (patient #4) with a left posterior angioma showing progression of the metabolic abnormality. A. PET scan at 8 months of age showed a small area of occipital hypometabolism which comprised only 2% of the cortical surface (arrows). B. Scan 2 at15 months of age showed that the hypometabolism became more severe and extended beyond the occipital region occupying almost 80% of the hemispheric surface. During the 7 months between the 2 scans, she developed a right moderate hemiparesis.
Figure 2.
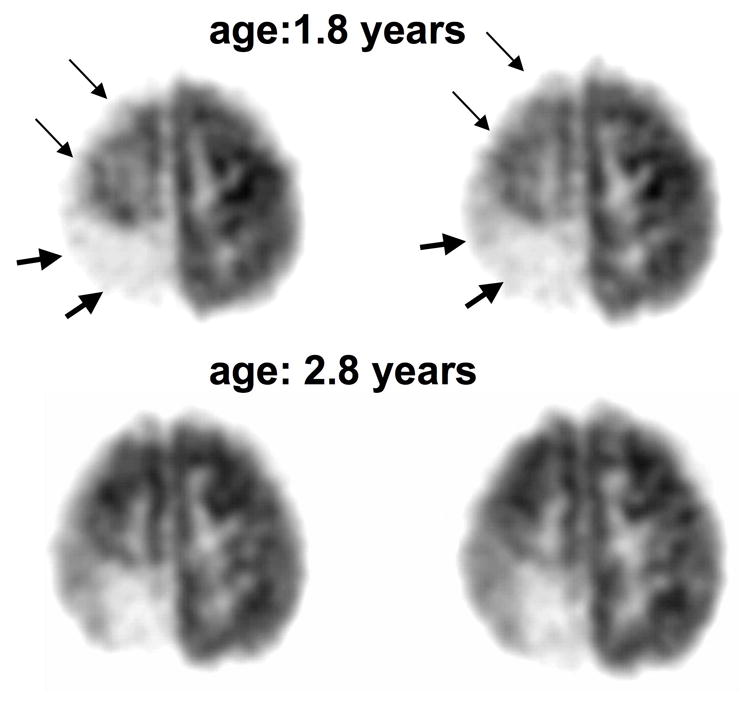
FDG PET scans in girl (patient #6) with a right posterior angioma showing partial recovery after one year. A. The first PET scan at 1.8 years of age showed extensive right hemispheric hypometabolism involving both the right posterior (severe hypometabolism, thick arrows) and superior frontal cortex (mild hypometabolism, thin arrows). B. PET scan one year later showed less extensive hypometabolism with a recovery of frontal cortex glucose metabolism. This improvement in glucose metabolism was accompanied by recovery of hemiparesis in the period between the two scans. Her seizures were relatively well-controlled, with only a single partial seizure during the 1-year period between the two scans.
Age vs. metabolic changes
SUV values for FDG uptake, measured in the contralateral frontal cortex showed a significant positive correlation with age at the time of the first scan (r = 0.70, p = 0.005), consistent with previously reported increases in cortical glucose metabolism with maturation between birth and 3 years of age.12 FDG SUVs at the time of the second scans were higher than at the time of the first scans (p = 0.023), consistent with an age-related increase of cerebral glucose metabolism in the patient group, despite the relatively short period between the two scans.
Patients with increased area of hypometabolic cortex (n=6) were younger both at the initial and the second scan (mean age 0.75 years and 1.8 years, respectively) than those with no change in the extent of hypometabolic cortex (2.8 and 4.2 years) (p<0.001 at the first scan and p=0.001 at the second scan). All patients with progression of cortical metabolic abnormalities were less than 3 years old at the time of both scans. There was a significant non-linear inverse correlation between the age of the patients at the second scan and the rate of change in the extent of hypometabolism (r = 0.69, p = 0.006, logarithmic regression) (Figure 3).
Figure 3.
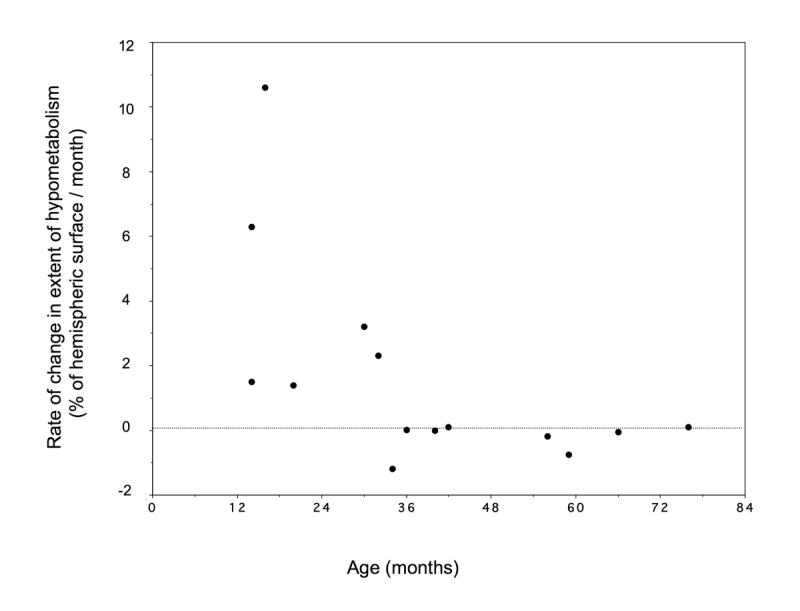
Relationship between age (at the time of the second PET scan) and monthly rate of metabolic change between the two scans. Major change in the extent of hypometabolic cortex occurred in children before 3 years of age. Relatively small metabolic changes were found in older children. The relationship between age and metabolic changes could be described by a logarithmic regression (r=0.69, p = 0.006).
Clinical variables vs. the extent of hypometabolic area
Seizure variables
The extent of cortical hypometabolism (at the first or second scan) did not correlate with seizure frequency (p > 0.7). Seizure frequency in the period between the two scans varied widely (between 0 and 1,200; median: 3/year; Table 1) and was higher in children whose hypometabolic area progressed than in patients with no metabolic progression (p = 0.008). Patients with metabolic progression showed a trend for earlier seizure onset, but this difference did not reach the level of significance (p=0.06).
Hemiparesis
Eight children had hemiparesis at the time of the first scan and new onset paresis or progression of hemiparesis was seen in four children by the time of the second scan (#1, 3, 4, 7). The extent of cortical hypometabolism was larger in children with hemiparesis than those with no motor deficit at the first scan (7% ± 5% vs. 48% ± 34%; p = 0.014), and also at the time of the second scan (13% ± 8% vs. 60% ± 31%; p = 0.01). One child (#6) showed an initial mild hemiparesis, which was no longer apparent at the time of the second scan; this patient showed metabolic improvement (decrease of the extent of cortical hypometabolism) between the two PET scans (see Figure 2). In contrast, all four children who developed or showed progression of hemiparesis between the two scans showed an increase in the area of cortical hypometabolism, which included the motor cortex.
DISCUSSION
Progression of cerebral metabolic and perfusion abnormalities is not unusual and had been described previously in a small group of children with SWS.11 Our present study provides a quantitative assessment of the extent of cortical glucose metabolic changes and demonstrates that the majority of progressive changes occur before 3 years of age. This vulnerable period coincides with a sharp increase of in the metabolic demand of the developing brain. Previous calculations of glucose metabolic rates in cerebral cortex of children showed a peak of 50-65 umol/min/100g around 3-4 years of age, thus exceeding mean adult values by more than two-fold.12,19 Our present data show a similar magnitude of age-related increases in glucose metabolism SUV values in cortex contralateral to the angioma. This rapid increase of metabolic demand coupled with an impairment of cortical blood flow detected as early as 2 months of age in infants with SWS3,20 likely plays an important role in cortical damage and extension of cortical dysfunction indicated by glucose hypometabolism in the affected hemisphere. The time course of the bulk of the damage to cerebral cortex in the first 3 years of life may also account for the observation that most patients with SWS who have epilepsy have their seizure onset within the first 3 years.21 Thus, our data suggest that early presentation of symptoms in SWS may be, at least partly, the result of an age-related increased cerebral metabolic demand in a system of vascular compromise due to the leptomeningeal angioma. Although fine details of motor functions were not available in some patients who were enrolled retrospectively, all patients who developed hemiparesis or showed a considerable progression of degree of hemiparesis during the follow-up showed an unequivocal increase of the extent of cortical hypometabolism, with involvement of their presumed sensorimotor cortex. This suggests that early progression in cortical dysfunction plays a major role in development or progression of neurological deficit, and evaluation of glucose metabolism by PET can help predict motor functions in young children with SWS.
It should be noted that frequent seizures themselves may also contribute to progression of cortical hypometabolism. Our previous studies in non-SWS children with uncontrolled epilepsy have indeed demonstrated a relationship between frequency of seizures and extent of cortical hypometabolism.13,22 Consistent with this notion, other investigators have reported that glucose metabolic abnormalities are relatively rare in both children and adults with new onset epilepsy.23,24 Altogether, these studies strongly suggest that at least some of the cortical hypometabolism seen on PET scans may be the consequence of persistent seizures. The present study provides further support to this notion. Although clinical seizure frequency is an imperfect estimate of seizure burden, the number of seizures observed by the parents and caregivers appears to be a significant factor related to progression of regional metabolic abnormalities in SWS. Since these seizures occur simultaneously with the developmentally regulated increase of cerebral metabolic demand, at this point it would be difficult to differentiate between dysfunction caused by hypoxia due to impaired blood flow vs. seizure-induced abnormalities; both of these mechanisms could play an important role in disease progression in an additive fashion. However, a definitive conclusion would require a much larger patient population with clinical and imaging data with quantitative metabolic measures, allowing multivariate analyses, to sort out the effect of seizures vs. developmental metabolic changes.
A novel and potentially important finding in the present study is that in two children with SWS, an initially large area of hypometabolism had actually decreased on the subsequent scan, thus suggesting a partial functional recovery. Since all of the PET scans were performed at least 24 hours after the last clinical seizures, it is unlikely that the observed extensive hypometabolism on the first scans could have been simply due to a postictal effect. Rather, seizures in both children with improved metabolism became relatively well-controlled during the months before the second scan. Reversibility of cortical hypometabolism has been shown in several previous studies on patients with epilepsy.13,23,25 Adaptive mechanisms, e.g., development of collateral circulation via deep veins, could also play a role in this metabolic improvement and contribute to improvement of hemiparesis. Future studies addressing mechanisms accounting for such recovery of functional impairment may assist in identifying novel therapeutic approaches to facilitate functional recovery.
Despite the inter-individual variations in the timing of metabolic changes, our findings indicate a general course of early evolution of metabolic abnormalities in children with SWS. This suggests that therapeutic attempts aiming at preventing further damage in SWS (e.g., by improving venous circulation, preventing seizures or applying neuroprotection) should be started early, since most of the detrimental metabolic changes occur before 3 years of age. On the other hand, metabolic abnormalities were limited to a small portion of the hemisphere and remained stable in 4 young children, thus not requiring aggressive treatment. It should be noted that extent of cortical glucose hypometabolism is only one (although important) aspect of cerebral pathological changes related to neuro-cognitive functions in SWS. Other changes in brain structure and function, such as damage in the underlying white matter and compensatory changes in remote cortical areas are emerging as potentially important additional predictors of neuro-cognitive outcome.17,26 Therefore, initial extent of cortical involvement is often not a reliable predictor of subsequent neuro-cognitive changes and should be interpreted together with additional variables such as white matter changes and clinical variables. Future studies in larger patient groups undergoing multimodal neuroimaging studies could sort out how these different mechanisms contribute to neuro-cognitive outcome, and could provide guide to clinical management decisions.
Table 2.
Imaging abnormalities of the 14 patients
| No | Angioma
side/location (on MRI) |
Extent
PET 1 |
Extent
PET 2 |
Monthly rate
of change |
|---|---|---|---|---|
| 1 | R PO | 0 | 26% | 1.4% |
| 2 | L PO | 20% | 78% | 6.3% |
| 3 | L PO | 15% | 29% | 1.5% |
| 4 | L PO | 2% | 78% | 10.5% |
| 5 | R POT | 28% | 77% | 2.3% |
| 6 | R POT | 38% | 24% | -1.2% |
| 7 | L PTO F | 36% | 59% | 3.2% |
| 8 | R FP | 3% | 3% | 0% |
| 9 | L POTF | 95% | 95% | 0% |
| 10 | L P | 7% | 8% | 0.1% |
| 11 | R POTF | 94% | 92% | -0.1% |
| 12 | R TO | 11% | 13% | 0.1% |
| 13 | R P | 8% | 6% | -0.2% |
| 14 | L PTO, F | 69% | 60% | -0.8% |
Extent scan 1 and 2: extent of cortical hypometabolism (>10% decrease as compared to the contralateral homotopic area, expressed as the % of hemispheric surface) on the first and second PET scan, respectively, R: right, L: left, T: temporal, F: frontal, O: occipital, P: parietal, s: superior, m: medial; i: inferior, a: anterior, p: posterior.
Acknowledgments
This work was supported by a grant from the National Institutes of Health (NS041922 to CJ). The authors thank Galina Rabkin, CNMT, and Angie Wigeluk, CNMT, for their expert technical assistance in performing the PET studies, and Anna Deboard RN, for performing sedations.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Roach ES, Bodensteiner JB. Neurologic manifestations of Sturge-Weber syndrome. In: Bodensteiner JB, Roach ES, editors. Sturge-Weber syndrome. The Sturge-Weber Foundation: Mt Freedom, NJ; 1999. pp. 27–38. [Google Scholar]
- 2.Benedikt RA, Brown DC, Walker R, Ghaed VN, Mitchell M, Geyer CA. Sturge-Weber syndrome: cranial MR imaging with Gd-DTPA. AJNR Am J Neuroradiol. 1993;14:409–15. [PMC free article] [PubMed] [Google Scholar]
- 3.Griffiths PD, Boodram MB, Blaser S, Armstrong D, Gilday DL, Harwood-Nash D. 99mTechnetium HMPAO imaging in children with the Sturge-Weber syndrome: a study of nine cases with CT and MRI correlation. Neuroradiology. 1997;39:219–24. doi: 10.1007/s002340050398. [DOI] [PubMed] [Google Scholar]
- 4.Cakirer S, Yagmurlu B, Savas MR. Sturge-Weber syndrome: diffusion magnetic resonance imaging and proton magnetic resonance spectroscopy findings. Acta Radiol. 2005;46:407–10. doi: 10.1080/02841850510021274. [DOI] [PubMed] [Google Scholar]
- 5.Mentzel HJ, Dieckmann A, Fitzek C, Brandl U, Reichenbach JR, Kaiser WA. Early diagnosis of cerebral involvement in Sturge-Weber syndrome using high-resolution BOLD MR venography. Pediatr Radiol. 2005;35:85–90. doi: 10.1007/s00247-004-1333-2. [DOI] [PubMed] [Google Scholar]
- 6.Marti-Bonmati L, Menor F, Mulas F. The Sturge-Weber syndrome: correlation between the clinical status and radiological CT and MRI findings. Childs Nerv Syst. 1993;9:107–9. doi: 10.1007/BF00305319. [DOI] [PubMed] [Google Scholar]
- 7.Kelley TM, Hatfield LA, Lin DD, Comi AM. Quantitative analysis of cerebral cortical atrophy and correlation with clinical severity in unilateral Sturge-Weber syndrome. J Child Neurol. 2005;20:867–70. doi: 10.1177/08830738050200110201. [DOI] [PubMed] [Google Scholar]
- 8.Chugani HT, Mazziotta JC, Phelps ME. Sturge-Weber syndrome: a study of cerebral glucose utilization with positron emission tomography. J Pediatr. 1989;114:244–53. doi: 10.1016/s0022-3476(89)80790-5. [DOI] [PubMed] [Google Scholar]
- 9.Lee JS, Asano E, Muzik O, Chugani DC, Juhasz C, Pfund Z, Philip S, Behen M, Chugani HT. Sturge-Weber syndrome: correlation between clinical course and FDG PET findings. Neurology. 2001;57:189–95. doi: 10.1212/wnl.57.2.189. [DOI] [PubMed] [Google Scholar]
- 10.Pinton F, Chiron C, Enjolras O, Motte J, Syrota A, Dulac O. Early single photon emission computed tomography in Sturge-Weber syndrome. J Neurol Neurosurg Psychiatry. 1997;63:616–621. doi: 10.1136/jnnp.63.5.616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maria BL, Neufeld JA, Rosainz LC, et al. Central nervous system structure and function in Sturge-Weber syndrome: evidence of neurologic and radiologic progression. J Child Neurol. 1998;13:606–18. doi: 10.1177/088307389801301204. [DOI] [PubMed] [Google Scholar]
- 12.Chugani HT, Phelps ME, Mazziotta JC. Positron emission tomography study of human brain functional development. Ann Neurol. 1987;22:487–97. doi: 10.1002/ana.410220408. [DOI] [PubMed] [Google Scholar]
- 13.Benedek K, Juhasz C, Chugani DC, Muzik O, Chugani HT. Longitudinal changes in cortical glucose hypometabolism in children with intractable epilepsy. J Child Neurol. 2006;21:26–30. doi: 10.1177/08830738060210011101. [DOI] [PubMed] [Google Scholar]
- 14.Muzik O, Chugani DC, Shen C, et al. Objective method for localization of cortical asymmetries using positron emission tomography to aid surgical resection of epileptic foci. Comput Aided Surg. 1998;3:74–82. doi: 10.1002/(SICI)1097-0150(1998)3:2<74::AID-IGS4>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 15.Muzik O, da Silva EA, Juhasz C, et al. Intracranial EEG versus flumazenil and glucose PET in children with extratemporal lobe epilepsy. Neurology. 2000;54:171–9. doi: 10.1212/wnl.54.1.171. [DOI] [PubMed] [Google Scholar]
- 16.Theodore WH. PET: cerebral blood flow and glucose metabolism - pathophysiology and drug effects. Adv Neurol. 2000;83:121–30. [PubMed] [Google Scholar]
- 17.Batista CEA, Juhász C, Chugani HT. Glucose metabolism in contralateral cortex in children with unilateral Sturge-Weber syndrome. Ann Neurol. 2006;60(suppl 3):S144–5. abstract. [Google Scholar]
- 18.Suhonen-Polvi H, Ruotsalainen U, Kinnala A, Bergman J, Haaparanta M, Teras M, Makela P, Solin O, Wegelius U. FDG-PET in early infancy: simplified quantification methods to measure cerebral glucose utilization. J Nucl Med. 1995;36:1249–54. [PubMed] [Google Scholar]
- 19.Chugani HT. A critical period of brain development: studies of cerebral glucose utilization with PET. Prev Med. 1998;27:184–8. doi: 10.1006/pmed.1998.0274. [DOI] [PubMed] [Google Scholar]
- 20.Evans AL, Widjaja E, Connolly DJ, Griffiths PD. Cerebral perfusion abnormalities in children with Sturge-Weber syndrome shown by dynamic contrast bolus magnetic resonance perfusion imaging. Pediatrics. 2006;117:2119–25. doi: 10.1542/peds.2005-1815. [DOI] [PubMed] [Google Scholar]
- 21.Oakes WJ. The natural history of patients with the Sturge-Weber syndrome. Pediatr Neurosurg. 1992;18:287–90. doi: 10.1159/000120677. [DOI] [PubMed] [Google Scholar]
- 22.Juhász C, Chugani DC, Muzik O, Watson C, Shah J, Shah A, Chugani HT. Electroclinical correlates of flumazenil and fluorodeoxyglucose PET abnormalities in lesional epilepsy. Neurology. 2000;55:825–35. doi: 10.1212/wnl.55.6.825. [DOI] [PubMed] [Google Scholar]
- 23.Matheja P, Kuwert T, Ludemann P, et al. Temporal hypometabolism at the onset of cryptogenic temporal lobe epilepsy. Eur J Nucl Med. 2001;28:625–632. doi: 10.1007/s002590100499. [DOI] [PubMed] [Google Scholar]
- 24.Gaillard WD, Kopylev L, Weinstein S, et al. Low incidence of abnormal (18)FDG-PET in children with new-onset partial epilepsy: a prospective study. Neurology. 2002;58:717–22. doi: 10.1212/wnl.58.5.717. [DOI] [PubMed] [Google Scholar]
- 25.Maeda N, Watanabe K, Negoro T, Aso K, Ohki T, Ito K, Kato T. Evolutional changes of cortical hypometabolism in West’s syndrome. Lancet. 1994;343:1620–3. doi: 10.1016/s0140-6736(94)93065-1. [DOI] [PubMed] [Google Scholar]
- 26.Juhász C, Lai C, Behen ME, Muzik O, Helder EJ, Chugani DC, Chugani HT. White matter volume is a major predictor of cognitive function in Sturge-Weber syndrome. Arch Neurol. doi: 10.1001/archneur.64.8.1169. in press. [DOI] [PubMed] [Google Scholar]