

Abstract
Introduction:
Numerous biofilm models have been described for the study of bacteria associated with the supragingival plaque. However, there are fewer models available for the study of subgingival plaque. The purpose of this study was to develop and validate a model that closely mimicked the composition of the subgingival flora.
Methods:
The model was developed as follows: calcium hydroxyapatite disks were coated overnight with 10% sterile saliva, placed in flat-bottomed tissue culture plates containing trypticase-soy broth, directly inoculated with a small aliquot of dispersed subgingival plaque, incubated anaerobically, and transferred to fresh medium at 48-h intervals until climax (steady-state) biofilms were formed (∼10 days).
Results:
The model, based on samples from eight periodontitis patients and eight healthy subjects, yielded a multi-species, heterogeneous biofilm, consisting of both gram-positive and gram-negative species, and comprising 15−20 cultivable species associated with the subgingival flora. The species present and their proportions were reflective of the initial cultivable subgingival flora. Comparisons of the initial plaque samples from healthy subjects and the mature biofilms showed 81% similarity in species and 70% similarity in the proportions present. Biofilms formed from samples obtained from periodontally diseased subjects were 69% similar in species and 57% similar in the proportions present.
Conclusions:
The biofilm model described here closely reproduces the composition of the cultivable subgingival plaque both in the species present and in their relative proportions. Differences existed between biofilms grown from diseased and non-diseased sites with the former being characterized by the presence of periodontal pathogens at microbially significant levels.
Keywords: bacteria, biofilms, model system, periodontitis, subgingival
Over 100 years ago Robert Koch made one of the most important conceptual breakthroughs in microbiology with the discovery of methods for the production of solid nutrient media and the ability to isolate microorganisms in pure culture. Since this development, the training of microbiologists and the study of microbiology have been based, to a significant degree, on the elucidation of the properties of a microorganism cultivated in a pure culture. Although it has long been acknowledged that pure cultures of bacteria are virtually absent in nature, it has been only in the past few years that the biofilm-mode of growth has been recognized as the default state for most bacteria. It has become accepted that biofilm-grown bacteria express different phenotypes and often exhibit totally different characteristics than do the same bacteria grown planktonically. Bacteria that are sessile (attached to a surface) express different genes and, so, behave differently from free-floating or planktonic bacteria. Notable among these differences is the increased resistance to antimicrobial agents that can be 100- to 1000-fold greater for a species in a mature biofilm relative to that same species grown planktonically. Most common infections of the oral cavity, e.g. caries, gingivitis and periodontitis, are the result of the accumulation of biofilms. However, treatment and control of these diseases has been based on the in vitro study of pure cultures and their response to various antimicrobial agents. Thus, to better control such infections, it is imperative to understand oral biofilm formation and maturation.
Various supragingival plaque biofilm models have been employed for the study of plaque formation, structure and antimicrobial susceptibility. Guggenheim has described a defined multi-species model designed to mimic the composition of the supragingival plaque and has used this model to study structure and antimicrobial susceptibility (9–11). Several investigators have utilized in-mouth splints in healthy subjects in which supragingival plaque formed over time on the splints (2, 45, 46). Wimpenny (44) has described several different laboratory biofilm models that make use of a constant depth film fermenter using a plaque inoculum. The constant depth film fermenter models have been used to study the structure (27, 29, 45) and spatial distribution (2, 15) of viable and non-viable supragingival plaque bacteria.
Attempts to obtain realistic subgingival plaque biofilms have been made by placing various insert materials into the periodontal pockets of periodontitis patients and then analysing the bacterial components that colonized the inserts (37, 42). Most recently, Hope and Wilson have described the development of subgingival plaque on hydroxyapatite disks in a constant depth film fermenter (16). This model used a plaque inoculum and reached a steady state after 4 days. Although this is an excellent model for the study of subgingival plaque structure and viability, the apparatus for maintaining an anaerobic constant depth film fermenter is somewhat complex.
Although all of the above models have greatly increased our understanding of plaque formation and development, none have directly addressed the question of how subgingival plaque matures over time and the sequence of events that leads up to a steady-state or climax biofilm. For our studies, we needed a simple, inexpensive model that was reproducible and that mimicked the in vivo composition of the subgingival plaque. One requirement was that a sufficient period needed to exist before the establishment of a climax biofilm so that the sequel of colonization could be reasonably followed. Ideally this period would be somewhat similar to what occurs in vivo. It was also felt that the model should be applicable to studying the development of subgingival plaque associated with both diseased and non-diseased sites and be able to demonstrate differences in the bacterial composition. We describe an in vitro multi-species biofilm model of subgingival plaque that closely mimics the composition and proportions of the cultivable bacterial taxa recovered from the gingival crevice and/or periodontal pocket and that is relatively representative of the bacteria recovered from both healthy and diseased periodontal sites. In addition, the effect that saliva and subgingival plaque from the same subject (homologous) vs. plaque or saliva from different subjects (heterogeneous) had on biofilm formation was determined.
Materials and methods
Biofilm preparation
Sampling
Following written informed consent, microbial samples of subgingival plaque were collected from eight individuals with no evidence of periodontal disease and from eight individuals with non-aggressive periodontitis. Criteria used for selection of the latter included bleeding on probing, a pocket depth ≥5 mm, and an attachment loss ≥4 mm. Samples were collected by inserting a sterile absorbent paper point (Henry Schein®, Melville, NY) to the depth of the sulcus and moving it laterally along the surface of the tooth and the sulcular epithelial lining. The paper-point sample was immediately placed into a 1-ml aliquot of Amies transport medium (1), supplemented with 0.5% gelatin (Fisher Scientific, Ocala, FL) and 0.1% sodium thioglycollate (Fisher Scientific), and stored overnight at 4°C. Previous studies using this transport medium have verified its ability to maintain the viability and proportions present of relatively sensitive gram-negative anaerobes such as Porphyromonas gingivalis and Prevotella intermedia overnight (6, 12, 13).
Saliva collection and processing
Unstimulated saliva was obtained in 5-ml aliquots from the same subjects who donated subgingival plaque. Each saliva sample was diluted (1 : 10) with sterile Ringer solution, centrifuged for 10 min to remove any particulate matter, and the supernatant was filter sterilized.
Biofilm development
Sterile ceramic calcium hydroxyapatite disks (5-mm diameter by 2-mm thickness; Clarkson Chromatography Products, Williamsport, PA). were coated with 10% sterile saliva overnight at room temperature, placed in the wells of a six- or 12-well tissue culture plate containing either 2 or 4 ml of trypticase-soy broth (BBL®; Becton Dickinson & Co., Sparks, MD) respectively. Each well was inoculated with 50 μl of sonically dispersed subgingival plaque. The disks were incubated in an anaerobic chamber (10% H2, 5% CO2, 85% N2) at 37°C for up to 10 days with change to fresh medium at 48-h intervals. Biofilm-containing disks were removed from the growth media at each of six different time intervals after inoculation: 4 h, 8 h, 24 h, 48 h, 5 days and 10 days. Biofilms from each sample and each time interval were cultivated and processed in triplicate. Representative disks from each time-point were examined by scanning electron microscopy.
Biofilm processing
After incubation, biofilm disks were removed from the growth media and gently rinsed in sterile Ringer solution to remove loosely adherent bacteria. The disks were then transferred to 1 ml pre-reduced, anaerobically-sterilized Ringer solution (14), supplemented with 0.5% Tween-20 (Fisher), and gently sonicated to disrupt the biofilm matrix and disperse the bacterial cells. Sonication of the biofilm from the disk was performed by sonicating for ∼30 s at a low-intensity setting (30% output) using a water-filled cup horn (Model W-370, 375 W; Heat Systems-Ultrasonics, Farmingdale, NY) so that the sample was not exposed to atmospheric air during the sonication process. This sonication procedure has been found adequate to disperse plaque samples without damage to the more sensitive gram-negative anaerobes or to spirochetes (38, 39).
The bacterial dispersions were vortexed, serially diluted 10-fold in Ringer solution, and plated onto trypticase-soy agar supplemented with 5% defibrinated sheep blood, 0.005% hemin and 0.0005% menadione (TSBA-HK). The plates were incubated anaerobically at 37°C for 5−7 days for total viable counts. The biofilms as well as the initial collected plaque samples were characterized by predominant cultivable methodology as described by Moore et al. (23–25). Forty isolates from each were subcultured and identified to genus and species by cellular fatty acid analyses on capillary gas–liquid chromatography (MIDI, Newark, DE) as described by Moore et al. (21). Based on Good's formula of coverage (22), we calculated that 40 isolates yielded between 75 and 90% of the cultivable bacteria present in the sites sampled and between 80 and 90% in the biofilms. DNA–DNA hybridization as described by Socransky (34, 35) was used to verify the initial results.
Scanning electron microscopy
Specimens evaluated under scanning electron microscopy were placed into Trumps fixative (Fisher), for 1 h at room temperature. Each sample was washed in phosphate-buffered saline three times for 10 min, fixed in 1% buffered osmium tetroxide for 1 h under hooded conditions, and immediately buffer washed twice for 10 min each time. Each sample was dehydrated in a graded ethanol series: 25%, 50%, 75%, 95% and 100% for 10 min each, bathed twice in hexamethyldisilazane for 5 min each, and air-dried overnight under a hood. Each was then mounted, sputter-coated with gold/palladium, and viewed with a Hitachi S-4000 field emission scanning electron microscopy at the ICBR Electron Microscopy Core Laboratory of the University of Florida.
Effect of Tween-20 on colony-forming units (CFUs) obtained
In the early stages of developing the model, unrealistically high CFUs were obtained from 10-day-old biofilms. A final concentration of 0.5% Tween-20 was found necessary to prevent the re-coaggregation of the bacteria when removed from the disk. This concentration was determined by testing final concentrations of 0, 0.1, 0.25, 0.5, 1.0 and 2.0% Tween-20 in Ringer solution against gram-negative and gram-positive planktonic cultures and against climax biofilms to determine the concentration that prevented co-aggregation but did not decrease the counts of the gram-negative anaerobes.
Saliva/plaque homogeneity
To determine if the source of saliva had an effect on biofilm formation, saliva and subgingival plaque samples were collected from four individuals who were periodontally healthy and two with adult chronic periodontitis. Biofilms were grown with homogeneous combinations of saliva and subgingival plaque from the same donor and with various heterogeneous combinations of saliva and plaque from different donors. The volumes of saliva, inoculum and growth medium were the same as previously described and remained constant for each saliva/plaque combination. The biofilms were grown for 10 days under anaerobic conditions at 37°C, harvested, serially diluted and plated on TSBA-HK for total viable cell counts.
DNA isolation from planktonic and biofilm-grown cultures
All bacterial strains used as DNA probes (Table 1) were grown planktonically in pre-reduced, anaerobically-sterilized peptone-yeast-glucose broth (14) until reasonable turbidity (∼107 CFU) was present. DNA from both planktonic and biofilm-grown cells was extracted using the Wizard® Genomic DNA Purification kit (Promega, Madison, WI). All reagents used were provided in the kit unless otherwise noted. Cells, both planktonic and biofilm-grown, were centrifuged at 13,000−16,000 g for 2 min to pellet the cells and the supernatant was removed. The cells were re-suspended in 480 μl 50 mm EDTA, 60 μl 10 mg/ml lysozyme (Sigma, St Louis, MO) was added and the mixture was incubated in a 37° C water bath for 60 min. Following incubation, the samples were centrifuged for 2 min and the supernatant was removed. Then, 600 μl Nuclei Lysis Solution was added to the pellet and mixed to re-suspend the cells. The samples were incubated at 80°C for 5 min to lyse the cells and then cooled to room temperature. After this, 3 μl RNase solution was added to the cell lysate, mixed by gently inverting the tube, and incubated for 60 min at 37°C. The samples were then cooled to room temperature and 200 μl Protein Precipitation Solution was added and vortexed at a high speed for 20 s to mix the solution with the cell lysate. The samples were incubated on ice for 5 min and then centrifuged for 3 min. The supernatant containing the DNA was transferred to a clean 1.5 ml microcentrifuge tube containing 600 μl isopropanol at room temperature (Fisher). The samples were gently mixed by inversion until the thread-like strands of DNA formed a visible mass. The samples were centrifuged for 2 min, the supernatant was carefully aspirated, and the tube was drained on clean absorbent paper; 600 μl 70% ethanol at room temperature (Fisher) was added to the DNA pellet and the tube was gently inverted to wash the pellet. The samples were centrifuged for 2 min and the ethanol was carefully aspirated. The pellet was air-dried for 30−40 min and 20−50 μl of DNA Rehydration Solution was added depending on the size of the DNA pellet. The DNA was re-hydrated overnight at room temperature and total DNA quantity was measured by UV spectrum (260 nm) using a SmartSpec® Plus spectrophotometer (Bio-Rad, Hercules, CA). All DNA samples were adjusted in TE buffer to a concentration of 100 ng/μl and stored at −70° C until used.
Table 1.
Bacterial species and strain number used for construction of DNA probes
| Bacterial species | Source/strain |
|---|---|
| Actinobacillus actinomycetemcomitans | ATCC1 29523 |
| Actinomyces israelii | ATCC 10049 |
| Actinomyces naeslundii | ATCC 12102 |
| Actinomyces odontolyticus | ATCC 17929 |
| Actinomyces viscosus | ATCC 19246 |
| Bifidobacterium dentum | ATCC 27534 |
| Campylobacter rectus | ATCC 33238 |
| Campylobacter concisus | PRDC-3262 |
| Capnocytophaga gingivalis | ATCC 33624 |
| Capnocytophaga ochracea | ATCC 33596 |
| Capnocytophaga sputigena | ATCC 33612 |
| Eikenella corrodens | ATCC 43278 |
| Fusobacterium nucleatum subsp. nucleatum | ATCC 25586 |
| Fusobacterium nucleatum subsp. vincentii | ATCC 49256 |
| Fusobacterium nucleatum subsp. polymorphum | ATCC 10953 |
| Peptrostreptococcus micros | ATCC 33270 |
| Porphyromonas gingivalis | ATCC 33277 |
| Prevotella intermedia | PDRC-11 |
| Prevotella melaninogenica | ATCC 25845 |
| Prevotella nigrescens | PDRC-2B |
| Prevotella oralis | ATCC 33269 |
| Propionibacterium acnes | ATCC 11827 |
| Streptococcus gordonii | ATCC 10558 |
| Streptococcus intermedius | ATCC 27335 |
| Streptococcus oralis | ATCC 35037 |
| Streptococcus sanguis | ATCC 10556 |
| Streptococcus parasanguis | PDRC-556 |
| Streptococcus mitis | ATCC 49456 |
| Streptococcus sobrinus | ATCC 27352 |
| Streptococcus mutans | ATCC 25175 |
| Tannerella forsythensis | ATCC 43037 |
| Treponema denticola | ATCC 35405 |
| Veillonella parvula | ATCC 10790 |
| Veillonella atypica | PDRC-124 |
American Type Culture Collection.
UF Periodontal Disease Research Center laboratory strain.
Preparation of labeled DNA probes
Whole genomic DNA probes were labeled using the BrightStar® Psoralen-Biotin nonisotopic labeling kit (Ambion® , Austin, TX). All the reagents used were provided in the labeling kit. DNA samples were denatured at 99°C for 10 min and rapidly cooled in an ice/slush mixture. The following steps were performed in reduced light. A total volume of 1.5 μl of the BrightStar Psoralen-Biotin was added to 10 μl of the nucleic acid solution, mixed and transferred to a well in a clean, untreated 96-well microtiter plate on an ice bath. A 365-nm UV light source was placed on the plate directly over the samples and the samples were irradiated for 45 min. The sample was diluted to 100 μl by adding 88.5 μl TE buffer and the mixture was transferred to a clean microfuge tube. Then, 200 μl of water-saturated n-butanol was added, the sample was vortexed, and centrifuged for 1 min at 7000 g. The top n-butanol layer was removed and this step was repeated once more. Labeled DNA probes were stored at −70°C.
‘Checkerboard’ DNA–DNA hybridization
Pre-hybridization and hybridization
Although different labeling reagents and buffers were used, the basic concept of ‘Checkerboard’ DNA–DNA hybridization was performed as described by Socransky (34, 35) and Wall-Manning (40). DNA samples (500 ng in a total volume of 5 μl) were mixed with 45 μl sterile de-ionized water. The DNA samples and DNA standards, equivalent to 107,106,105 and 104 cells of the strains used as labeled probes, were boiled for 5 min to denature the DNA and cooled on ice for 5 min. The final volume for each sample was brought up to 1 ml and applied on BrightStar®-Plus positively charged nylon membrane (Ambion® ) using a Minislot® Vacuum Manifold (Immunetics® , Cambridge, MA). The DNA was fixed to the membrane using a UV stratalinker (Stratagene, La Jolla, CA) twice at the Autocrosslink setting (1200 μJ × 100). The membrane was pre-hybridized using 20 ml hybridization buffer [45% formamide (Sigma), 20X SSC (3 M NaCl, 0.3 M sodium citrate, pH 7.0), 20% sodium dodecyl sulfate, 10% dextran sulfate, 40X liquid block (Amerisham Life Science, UK)] and incubated for 2.5 h at 37° C. After incubation, the membrane was removed from the hybridization buffer. Then, 5 μl of each DNA probe was mixed with 155 μl hybridization buffer, boiled for 5 min and cooled on ice for 5 min. The membrane was oriented at a right angle to the direction the samples were applied and placed in a 45-channel Miniblotter® (Immunetics® ). The labeled DNA probes were applied to the membrane; the MiniBlotter with the membrane was sealed in a plastic bag and incubated overnight at 42°C.
Detection
After incubation, detection was performed using the BrightStar® BioDetect® nonisotopic detection kit (Ambion®). All buffers and reagents used were provided in the detection kit. The volumes of each buffer used were adjusted as needed for the membrane size of 210 cm2. The membrane was removed from the Miniblotter and washed twice for 5 min in 210 ml 1X wash buffer and twice for 5 min in 105 ml blocking buffer. The membrane was then incubated in 210 ml blocking buffer for 30 min. Diluted strep-alkaline phosphatase (Strep-AP) was prepared by mixing 20 ml blocking buffer and 2 μl Strep-AP. The membrane was incubated in the diluted Strep-AP for 30 min and then incubated for 15 min in 105 ml of blocking buffer. The membrane was then washed in 210 ml 1X wash buffer three times for 15 min. After three washes, the membrane was incubated twice in 105 ml of 1X assay buffer for 2 min. The membrane was then incubated for 5 min in 10 ml CDP-Star. After this incubation, the membrane was blotted on a piece of filter paper (Whatman International Ltd, Maidstone, UK), placed in a Kapak pouch (Kapak, Minneapolis, MN), sealed, and exposed to imaging film (X-OMAT; Eastman Kodak Co., Rochester, NY) overnight at room temperature. The resulting images were semi-quantified by digitizing the spots obtained with the four standards for each probe (on each membrane) and then comparing these values with the value obtained for the digital image of that probe for each sample, if present at detectable levels, using CHEMIDOC XRS hardware and software (Bio-Rad).
Statistical testing
Differences within and between biofilms were tested using either analysis of variance or its non-parametric version, the Kruskal–Wallis test. To determine where differences might lie the paired t-test or its non-parametric equivalents, the Wilcoxon signed rank test or the paired sign test, were performed for pairwise comparisons. Similarities between the bacterial compositions and proportions of the cultivable flora from the subgingival plaque and those of the mature biofilms grown from the plaque samples were tested using the similarity index of two multinomial distributions as described by Good (8) and as applied by Moore (26). A P ≤ 0.05 was considered as statistically significant.
Results
Effect of Tween-20 on CFU recovery
Various concentrations of Tween-20 were investigated on pure cultures and on climax (steady-state) biofilms to determine which concentration was effective in eliminating the bacterial clumping of the biofilm cells but did not result in a decrease in the viability of the more sensitive gram-negative anaerobes (Table 2). By one-way analysis of variance, there were no statistically significant differences (P = 0.875) in the recovery of viable CFUs for any of the planktonically grown cultures. There was a slight trend (P = 0.091) in CFU recovery with 2% Tween-20 relative to 1% by the paired t-test. However, highly significant differences (P < 0.001) were detected for the CFUs recovered for the climax biofilms at different Tween-20 concentrations. These differences were found to lay in the 0% and the 0.1% Tween-20 concentrations. By the paired t-test, the CFUs recovered from both the 0% and the 0.1% concentrations were significantly higher (P < 0.05) than the counts recovered at the other concentrations. There were no differences (P = 0.306) detected in the counts recovered with Tween-20 concentrations of 0.25−2.0%. Statistical testing with the equivalent non-parametric tests gave similar results. Based on these results, a Tween-20 concentration of 0.5% in Ringer solution was used for all dilution series.
Table 2.
Effect of Tween-20 concentrations on recovery of colony-forming units from climax biofilms and stationary-phase planktonic cultures
| Tween-20 concentrations (%) | ||||||
|---|---|---|---|---|---|---|
| 0.00 | 0.10 | 0.25 | 0.50 | 1.00 | 2.00 | |
| Climax biofilms1 | ||||||
| Biofilm 1 | 1.6 × 1011 | 5.8 × 1010 | 6.0 × 109 | 4.3 × 109 | 4.9 × 109 | 4.6 × 109 |
| Biofilm 2 | 1.3 × 1011 | 4.5 × 1010 | 5.0 × 109 | 6.3 × 109 | 5.7 × 109 | 5.9 × 109 |
| Biofilm 3 | 1.5 × 1011 | 1.5 × 1010 | 1.4 × 1010 | 8.9 × 109 | 8.8 × 109 | 8.6 × 109 |
| Biofilm 4 | 1.5 × 1011 | 1.5 × 1010 | 2.0 × 109 | 3.3 × 109 | 4.3 × 109 | 3.3 × 109 |
| Biofilm 5 | 4.2 × 1011 | 4.2 × 1010 | 8.2 × 109 | 6.6 × 109 | 3.3 × 109 | 4.9 × 109 |
| Biofilm 6 | 1.6 × 1011 | 7.7 × 1010 | 1.2 × 1010 | 8.9 × 109 | 6.6 × 109 | 6.9 × 109 |
| Biofilm 7 | 2.9 × 1011 | 9.3 × 1010 | 2.9 × 109 | 3.6 × 109 | 4.5 × 109 | 3.7 × 109 |
| Biofilm 8 | 4.3 × 1011 | 1.5 × 1010 | 1.2 × 1010 | 9.6 × 109 | 5.6 × 109 | 4.8 × 109 |
| Planktonic cultures2 | ||||||
| A. odontolyticus | 8.0 × 107 | 1.2 × 108 | 1.3 × 108 | 1.5 × 108 | 8.0 × 107 | 9.6 × 107 |
| S. gordonii | 1.0 × 108 | 2.0 × 108 | 2.0 × 108 | 1.2 × 108 | 2.0 × 108 | 1.4 × 108 |
| S. parasanguis | 2.2 × 108 | 2.0 × 108 | 2.1 × 108 | 2.0 × 108 | 2.4 × 108 | 2.0 × 108 |
| F. nucleatum | 4.3 × 107 | 4.5 × 107 | 3.5 × 107 | 4.3 × 107 | 3.6 × 107 | 3.9 × 106 |
| T. forsythensis | 4.3 × 104 | 4.2 × 104 | 3.5 × 104 | 4.6 × 104 | 3.6 × 104 | 8.0 × 103 |
| P. gingivalis | 3.5 × 106 | 4.2 × 106 | 2.6 × 106 | 2.9 × 106 | 5.6 × 105 | 4.6 × 105 |
| P. intermedia | 2.7 × 107 | 3.6 × 107 | 2.6 × 108 | 4.5 × 107 | 2.6 × 107 | 5.3 × 105 |
Single determination.
Mean of three determinations.
Effect of saliva/plaque homogeneity on biofilm growth
To determine if the source of the saliva used to coat the biofilm support was important to biofilm formation, biofilms were cultivated using homogeneous combinations of saliva and subgingival plaque (same subject) and heterogeneous saliva/plaque combinations. The viable counts obtained from the homogeneous combinations were expressed as 100% and that obtained for biofilms cultivated with plaque taken from a different donor to the saliva were expressed as a percentage of the homogeneous combinations (Table 3). The total CFUs of the climax biofilms cultivated with plaque and saliva from different subjects were less than 10%, and in several cases less than 1%, of that obtained when saliva and plaque were obtained from the same subject. When samples from periodontitis subjects were crossed with samples from healthy subjects, the biofilms often detached from the hydroxyapatite disks shortly after 48 h of growth.
Table 3.
Percentage of colony-forming unit (CFU) recovered with various plaque and saliva combinations
| Subject 1H1 plaque | Subject 2H plaque | Subject 3H plaque | Subject 4H plaque | Subject 1P plaque | Subject 2P plaque | |
|---|---|---|---|---|---|---|
| Subject 1H saliva | 1002 | 0.5 | 2.2 | 3.9 | 2.0 | 1.0 |
| Subject 2H saliva | 6.1 | 100 | 1.9 | 1.9 | ND3 | ND |
| Subject 3H saliva | 0.1 | 6.3 | 100 | 0.8 | ND | ND |
| Subject 4H saliva | 0.2 | 5.2 | 8.3 | 100 | 0.1 | 0.5 |
| Subject P1 saliva | 3.5 | 2.7 | ND | 0.3 | 100 | 19 |
| Subject P2 saliva | 0.3 | 0.5 | ND | 0.8 | 6 | 100 |
H, healthy subject; P, periodontally diseased subject.
CFUs recovered when plaque and saliva from the same source was used for cultivation of biofilms was arbitrarily taken as 100%.
ND, not determined, biofilm detached prematurely.
Characterization of an in vitro biofilm model of subgingival plaque
To determine reproducibility of the model, growth curves were constructed based on triplicate CFU determinations for each subgingival sample from eight periodontally healthy and eight diseased subjects. Statistical analysis showed no significant statistical difference in the CFUs obtained within or among the biofilms cultivated from healthy subjects (P ≥ 0.95). The initial inoculum contained around 104 cells/ml. After 4 h of incubation, approximately 105 viable cells were detected per disk. At 8 h post-inoculation, this number increased to ∼106. Within 48 h, the total viable counts were between 106 and 107 CFU/disk. After 5 days of incubation, the biofilm cell mass was between 107 and 108 CFU/disk and a climax biofilm was reached after 10 days with 108−109 viable cells.
CFUs obtained from biofilms cultivated from the plaque samples taken from periodontally diseased subjects showed no significant difference within each individual (P > 0.79) or among subjects (P > 0.95). Although the initial inoculum was approximately the same as that used to inoculate from the healthy subjects, slightly higher viable counts were obtained throughout. Between 105 and 106 CFU/disk were detected 4 h after inoculation and 106−107 CFU/disk after 8 h. Total viable counts reached over 108 CFU/disk within 48 h after inoculation. After 5 days of incubation, the biofilm mass was ∼109 CFU/disk. The climax biofilm, reached after 10 days of incubation, had a viable cell count of 109−1010 CFU/disk.
Biofilm development was examined using scanning electron microscopy (Fig. 1A–D) to visualize morphological structure and using predominant cultivable analysis as well as checkerboard DNA–DNA hybridization. The microbial compositions of biofilms grown from healthy and periodontally diseased sites were grouped into the microbial complexes described by Socransky (33) and are presented in Fig. 2A,B. No notable differences were detected in the morphologies seen in the scanning electron microscopys of biofilms formed from plaque from healthy subjects relative to the diseased subjects. Approximately 4−8 h after inoculation, single cells and cell clusters were adherent to the surface of the hydroxyapatite disk (Fig. 1A). The bulk (50−70%) of these cells were identified by culture as Streptococcus species and 15% or less consisted of Actinomyces, Bacteroides, and Campylobacter (Fig. 2A,B). Microcolony formation was observable 24 h after inoculation (Fig. 1B) and the composition of the biofilms began to show increased diversity from this point. The percentage of Streptococcus began to decline as the percentages of Veillonella, Actinomyces and Campylobacter increased. The scanning electron microscopy micrograph taken 5 days after inoculation (Fig. 1C) showed a thick, multi-species biofilm that contained large and small coccoid forms, rods, fusiforms and filamentous bacteria. Culture differences were noted at 5 days between the compositions of the biofilms cultivated from diseased sites relative to those from non-diseased sites (Fig. 2A,B); he compositions of the latter were approximately 25% Streptococci, 25% Veillonella and 10−15% Actinomyces. The compositions of those grown from diseased sites were roughly 35−40% Streptococci, 20% Veillonella, 6−8% Actinomyces and 10−15% Prevotella and Fusobacterium nucleatum. At 10 days of growth, the biofilms were highly diverse and showed complex structural depth and morphologies (Fig. 1D). Although not recovered culturally, a few morphological forms typical of spirochetes were detected in some of the scanning electron microscopys at the climax stage. Microbial differences were observed culturally and by DNA–DNA hybridization in the bacterial compositions present. Members of the orange complex, Campylobacter rectus, Fusobacterium nucleatum and Prevotella intermedia were 10−25% higher in biofilms cultivated from the diseased subjects (Fig. 2B). Members of the red complex, P. gingivalis, Tannerella forsythensis and Treponema denticola, were not detected in the biofilms cultivated from healthy subjects but constituted up to 5% of the biofilms cultivated from diseased subjects.
Fig. 1.
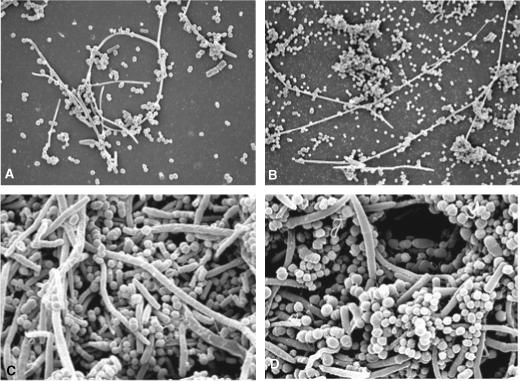
Scanning electron microscopy micrographs of (A) a 4−8-h biofilm; (B) a 24−48-h biofilm; (C) a 5-day biofilm; and (D) a 10-day (climax) biofilm.
Fig. 2.
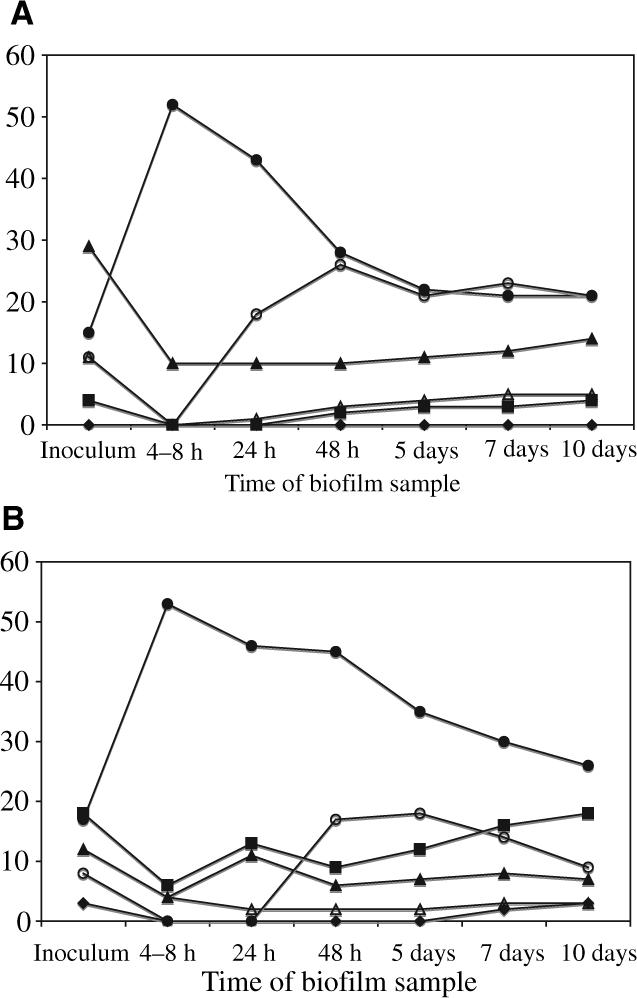
Means of the bacterial compositions of in vitro biofilms derived from healthy (A) and periodontally diseased (B) samples as determined by predominant cultivable analysis (based on triplicate determinations of biofilms cultivated from eight subjects in each category). The identified bacterial species were grouped into periodontal complexes as described by Socransky et al. (33): the yellow complex, Streptococcus species (•): the purple complex, Veillonella species (○); the blue complex, Actinomyces species (▲); the green complex, Eikenella corrodens and Campylobacter species (△); the orange complex, Prevotella species, Campylobacter rectus and Fusobacterium species (■); and the red complex, Porphyromonas gingivalis, Treponema denticola and Tannerella forsynthensis (◆).
Comparison of initial subgingival plaque with the climax biofilm
Table 4 gives a cultural comparison of the bacterial species recovered and their contribution to the total cultivable flora for the plaque samples collected from the healthy subjects relative to the climax biofilms obtained. A total of 37species was isolated and identified from eight periodontally healthy subjects. In the initial plaque samples, Actinomyces naeslundii was the most common species identified, followed by Veillonella atypica and a number of other gram-positive bacilli. In the climax biofilms cultivated from these samples, V. atypica was the most common species isolated followed by A. naeslundii.
Table 4.
Comparison of the bacterial composition of initial subgingival plaque samples collected from healthy subjects to the resulting climax biofilm grown from the samples
| Mean percentages of predominant isolates from eight subjects (±SD) | ||
|---|---|---|
| Bacterial species or taxa1 | Initial sample | Climax biofilm |
| Actinomyces naeslundii | 23.75 ± 7.19 | 11.25 ± 4.81 |
| Veillonella atypica | 6.56 ± 2.97 | 13.44 ± 4.61 |
| Streptococcus gordonii | 5.00 ± 3.28 | 6.56 ± 5.35 |
| Rothia denticariosa | 4.69 ± 2.09 | 2.81 ± 3.11 |
| Streptococcus sanguis | 4.06 ± 4.22 | 12.81 ± 9.31 |
| Actinomyces pyogenes | 3.75 ± 4.22 | 1.25 ± 1.87 |
| Bacteroides coagulans | 3.44 ± 4.22 | 2.19 ± 3.11 |
| Bifidobacterium D02A | 3.44 ± 4.22 | 3.75 ± 4.33 |
| Campylobacter concisus | 3.44 ± 3.99 | 3.44 ± 4.22 |
| Actinomyces odonotolyticus | 2.50 ± 2.66 | 2.81 ± 4.33 |
| Fusobacterium nucleatum | 2.50 ± 1.90 | 2.81 ± 4.33 |
| Leptotrichia buccalis | 2.19 ± 1.61 | 1.88 ± 2.91 |
| Streptococcus M7 | 1.88 ± 2.91 | 4.06 ± 5.66 |
| Streptococcus intermedius | 1.88 ± 2.21 | 3.13 ± 2.58 |
| Actinomyces israelii | 1.88 ± 2.21 | 1.25 ± 1.87 |
| Propionibacterium avidum | 1.56 ± 1.16 | 0.31 ± 0.88 |
| Bifidobacterium D05 | 1.56 ± 2.66 | 1.56 ± 2.29 |
| Veillonella parvula | 1.25 ± 1.87 | 3.75 ± 2.91 |
| Streptococcus anginosus | 0.94 ± 1.87 | 0.00 |
| Capnocytophaga gingivalis | 0.94 ± 1.87 | 1.88 ± 2.91 |
| Bifidobacterium breve | 0.94 ± 1.30 | 0.63 ± 1.16 |
| Capnocytophaga sputigena | 0.63 ± 1.16 | 1.25 ± 1.87 |
| Lactobacillus rogosae | 0.63 ± 1.16 | 1.56 ± 2.29 |
| Streptococcus sobrinus | 0.63 ± 1.16 | 1.25 ± 1.87 |
| Veillonella dispar | 0.63 ± 1.16 | 1.88 ± 2.91 |
| Prevotella tannerae | 0.63 ± 1.16 | 1.25 ± 1.87 |
| Lactobacillus bifermentans | 0.63 ± 1.16 | 1.25 ± 1.87 |
| Streptococcus oralis | 0.31 ± 0.88 | 0.00 |
| Streptococcus salivarius | 0.31 ± 0.88 | 0.31 ± 0.88 |
| Fusobacterium russi | 0.31 ± 0.88 | 0.00 |
| Prevotella denticola | 0.31 ± 0.88 | 0.00 |
| Prevotella intermedia | 0.31 ± 0.88 | 0.31 ± 0.88 |
| Bifidobacterium angulatum | 0.31 ± 0.88 | 0.63 ± 1.16 |
| Propionibacterium propionicum | 0.31 ± 0.88 | 0.63 ± 1.16 |
| Streptococcus parasanguis | 0.00 | 0.31 ± 0.88 |
| Actinomyces meyeri | 0.00 | 0.31 ± 0.88 |
Identified using the VPI Anaerobe database (Moore 6, Microbial ID, Inc.).
A similar comparison of samples collected from subjects with periodontitis and their climax biofilms is given in Table 5. Forty-two different species were isolated and identified from the eight periodontitis subjects. Campylobacter rectus, a putative periodontal pathogen, was the most abundant species isolated from both the initial samples of periodontitis patients and the climax biofilms. Other frequently encountered species included F. nucleatum, P. intermedia, P. gingivalis and various species of Streptococci, Veillonella and Actinomyces.
Table 5.
Comparison of the bacterial composition of initial subgingival plaque samples collected from periodontitis subjects to the resulting climax biofilm grown from the samples
| Mean percentages of predominant isolates from eight subjects (±SD) | ||
|---|---|---|
| Bacterial species or taxa1 | Initial sample | Climax biofilm |
| Campylobacter rectus | 15.94 ± 5.66 | 18.75 ± 7.08 |
| Fusobacterium nucleatum | 9.38 ± 8.65 | 7.81 ± 7.84 |
| Streptococcus sanguis | 8.13 ± 5.12 | 5.94 ± 4.22 |
| Actinomyces naeslundii | 6.88 ± 5.12 | 4.69 ± 3.11 |
| Streptococcus intermedius | 5.63 ± 4.39 | 10.94 ± 10.69 |
| Veillonella atypica | 5.00 ± 2.66 | 6.25 ± 2.66 |
| Bifidobacterium D02A | 3.75 ± 3.28 | 2.50 ± 2.66 |
| Bifidobacterium D05 | 3.75 ± 3.28 | 2.50 ± 2.66 |
| Leptotrichia buccalis | 2.50 ± 2.66 | 2.81 ± 2.09 |
| Bacteroides coagulans | 2.50 ± 1.87 | 4.38 ± 4.59 |
| Fusobacterium gonidiaformans | 2.19 ± 3.40 | 0.00 |
| Prevotella intermedia | 2.19 ± 3.65 | 2.19 ± 3.65 |
| Porphyromonas gingivalis | 2.19 ± 3.11 | 2.19 ± 3.11 |
| Actinomyces israelii | 1.88 ± 5.32 | 0.94 ± 1.30 |
| Rothia denticariosa | 1.88 ± 2.91 | 2.87 ± 3.11 |
| Prevotella tannerae | 1.88 ± 2.21 | 1.25 ± 2.29 |
| Capnocytophaga sputigena | 1.56 ± 2.29 | 1.56 ± 2.97 |
| Streptococcus gordonii | 1.25 ± 2.29 | 3.13 ± 3.99 |
| Veillonella parvula | 1.25 ± 1.87 | 2.19 ± 2.83 |
| Actinomyces odontolyticus | 0.94 ± 1.30 | 0.63 ± 1.16 |
| Propionibacterium avidum | 0.94 ± 1.30 | 0.63 ± 1.16 |
| Streptococcus M7 | 0.94 ± 1.30 | 4.38 ± 4.75 |
| Campylobacter concisus | 0.63 ± 1.16 | 0.63 ± 1.16 |
| Actinomyces pyogenes | 0.63 ± 1.16 | 0.94 ± 1.35 |
| Lactobacillus rogosae | 0.63 ± 1.16 | 1.25 ± 1.35 |
| Eikenella corrodens | 0.63 ± 1.16 | 1.56 ± 2.91 |
| Actinomyces bovis | 0.31 ± 0.88 | 0.00 |
| Actinomyces georgiae | 0.31 ± 0.88 | 0.31 ± 0.88 |
| Actinomyces gerensceriae | 0.31 ± 0.88 | 0.00 |
| Actinomyces meyeri | 0.31 ± 0.88 | 0.00 |
| Eubacterium eligens | 0.31 ± 0.88 | 0.31 ± 0.88 |
| Fusobacterium necrophorum | 0.31 ± 0.88 | 0.00 |
| Bacteroides putredins | 0.31 ± 0.88 | 0.00 |
| Bifidobacterium bifidum | 0.31 ± 0.88 | 0.00 |
| Lactobacillus brevis | 0.31 ± 0.88 | 0.00 |
| Neisseria mucosa | 0.31 ± 0.88 | 0.31 ± 0.88 |
| Streptococcus sobrinus | 0.31 ± 0.88 | 0.00 |
| Propionibacterium propionicum | 0.31 ± 0.88 | 0.00 |
| Streptococcus parasanguis | 0.00 | 1.25 ± 1.30 |
| Lactobacillus bifermentans | 0.00 | 0.31 ± 0.88 |
| Veillonella dispar | 0.00 | 0.31 ± 0.88 |
Identified using the VPI Anaerobe database (Moore 6, Microbial ID, Inc.).
Comparison of the bacterial compositions and proportions of subgingival samples from healthy subjects and the resulting mature biofilms revealed similarity indices of 81% in bacterial species and 70% in the proportions present. In the periodontitis subjects, similarity indexes were 69% and 57%, respectively, for the bacterial species recovered and the proportions present.
Discussion
In the initial development stages of the biofilm model, we attempted to create a defined model of the subgingival plaque by adding specific strains of individual species to pooled saliva-treated hydroxyapatite disks and then building on these to create a multi-species model similar to what is recovered from subgingival plaque. This proved to be difficult, was not reproducible, and, at best, yielded a biofilm more similar to supragingival than to subgingival plaque. In many instances, we were unable to obtain colonization by any species other than streptococci or else the biofilm detached prematurely. Subsequent observations led us to believe that biofilms were formed more readily when both the saliva source and the bacterial sample were obtained from the same donor.
The use of pooled saliva resulted in either no biofilm growth or the detachment of the biofilm from the surface. However, when the saliva sample and the subgingival plaque sample were collected from the same individual, biofilms formed very readily. Thus, the source of the saliva appeared to be important to the ability of the bacteria to colonize the biofilm support. To test this, saliva and plaque were collected from several subjects and various plaque/saliva combinations were used to develop biofilms. Biofilms grown using homogeneous combinations of saliva and plaque resulted in climax biofilms with 10- to 100-fold more CFUs than biofilms grown with heterogeneous combinations. Attempts to grow biofilms using saliva from healthy subjects and subgingival plaque from periodontitis subjects were unsuccessful. The source of the saliva appears to be very important in obtaining biofilm formation. Several reviews have described the specific nature of interactions between early colonizers of dental biofilms and the salivary molecules of the acquired pellicle (17, 32) as well as the genetic diversity in salivary composition that occurs from person to person (3, 7). It has been hypothesized that in a common pool of salivary molecules, a proportion of certain bacteria that bind to specific molecules within that pool could alter the diversity of the salivary molecules exposed to later colonizers through agglutination and microcolonization of those bacteria (43). It is possible that the early colonizers could influence the degree of accumulation of later colonizers. Such events might account for the differences in cell mass and bacterial composition that we have observed between homogeneous and heterogeneous saliva and biofilms.
Concurrent with the use of saliva and plaque from the same donor, we discovered that inoculation of the saliva-coated hydroxyapatite disk directly with the dispersed subgingival plaque sample routinely yielded multi-species biofilms that were similar in composition to the flora recovered from the initial plaque sample. These biofilms represented the variation in the cultivable flora observed from subject to subject as well as between periodontally healthy and diseased flora. Preliminary data indicated that the model exhibited specific, observable stages of biofilm development and yielded a climax biofilm after 10 days of growth that provided a close approximation to the cultured flora obtained, both in composition and in the proportions of the bacterial taxa present, from samples of the subgingival plaque. We are well aware that subgingival plaque is bathed by gingival crevicular fluid and not by saliva. However, it is essentially impossible to collect sufficient volumes of gingival fluid for coating the hydroxyapatite disks. For this reason, we elected to use unstimulated saliva. Since the biofilms obtained gave us a close approximation to the initial subgingival samples in both the species present and their relative proportions, we have continued to use filter-sterilized 10% saliva to coat the supports.
In our preliminary studies, the climax biofilms yielded reproducible but impossibly high counts of viable cells. These counts were around 1012−1014 CFU from a single hydroxyapatite disk (5 mm diameter, 2 mm thickness) or roughly 109−1011 viable cells per mm2. This would be equivalent to a gram or more of viable cells per disk. On examination by dark-field microscopy, clumps of bacteria were readily visible in Ringer solution following the sonication step. Our hypothesis was that the bacterial cells were either immediately co-aggregating with each other upon removal from the support by sonication or were not completely separated from the polysaccharide matrix of the biofilm. Either of these could result in the formation of clumps of bacterial cells, or mini-biofilms, which were either not disrupted by vortexing between dilutions or were readily co-aggregated following vortexing. These clumps were then transferred from dilution to subsequent dilution and were only dispersed during the plating procedure. Examination of the plate counts revealed a continuous cell number rather than a 10-fold decrease following each dilution. In an attempt to prevent this co-aggregation and the subsequent formation of micro-subunits consisting of different bacterial combinations, we tried vigorously vortexing each dilution blank, with and without glass beads, immediately before making the next dilution. When this had no effect, we tried adding various concentrations of Tween-20 to the dilution blanks. We found that a final concentration of 0.5% Tween-20 in the initial Ringer dilution blank gave us reasonable counts and a logarithm decrease in the CFUs present as the dilution series increased without exerting a detrimental effect on the recovery of gram-negative species. This phenomenon seems to be limited to this particular model because it has not been reported with other plaque biofilm models.
Biofilm development and maturation was monitored using a combination of scanning electron microscopy, predominant cultivable analysis, and checkerboard DNA–DNA hybridization. scanning electron microscopy images taken 4−8 h after exposure of the hydroxyapatite disks to the subgingival plaque inoculum demonstrated the presence of individual cocci, rods and filaments as well as small clusters of cocci dispersed across the hydroxyapatite surface. It is not known if the different morphological forms observed were all primary colonizers or if some degree of co-aggregation occurred between certain bacterial species following the plaque dispersal step. Kolenbrander et al. (17, 18) postulated that development of the supragingival plaque biofilm occurs as a series of distinct events involving primary colonizers, bridging microorganisms and secondary colonizers. Since the hydroxyapatite supports are physically moved to fresh medium at 48-h intervals, it follows that all the bacterial components found in the later biofilms must be present on the disks within the first 48 h. However, many of these were below the detectable limits of either culture methodology or DNA–DNA hybridization until the biofilms were 5 days old.
The majority of the isolates subcultured and identified from the 4- to 8-hour-old biofilms were streptococci. However, at this early stage, 35−70% of the colonies isolated for predominant cultivable analysis could not be subcultured for identification. This same phenomenon was also observed, to a lesser extent, with subcultures from biofilms aged 24−48 h but not with biofilms 5 days and older. This was puzzling. Our laboratories have successfully performed predominant cultivable analysis of subgingival plaque samples for a number of years. Thus, we do not think the failure of the subcultures to survive was a result of our technique. DNA–DNA hybridization revealed that species other than those that were successfully subcultured and identified were probably present. However, because of the number of cells present and the low sensitivity of the DNA–DNA hybridization method (approximately 105 cells are required for a definite positive reaction); the checkerboard DNA–DNA method was inconclusive for the early biofilm stages. A possible hypothesis to explain the lack of cultivability might be that a totally different phenotype is expressed and cellular metabolism is directed toward growth and the production of extracellular polysaccharides for the biofilm matrix in the earlier stages of biofilm development. Thus, it may be difficult for certain bacteria to readily convert from an accelerated biofilm mode back to the planktonic phenotype. Genomic and proteomic investigations using single-species biofilms have shown that gene expression may be up-regulated, down-regulated, or totally unique in biofilm relative to planktonically grown cells (19, 31). In certain instances, up-regulation or unique expression of certain genes is necessary for biofilm formation (4, 5, 30). It is possible, once such genes are turned on, that bacteria may not readily revert to the planktonic phenotype until a ‘steady-state’ phase is reached and metabolic activity decreases.
Overall, the developmental stages that we have observed in our subgingival biofilm model are analogous to the stages described in the current literature. Rickard et al. (32) have described the possible roles of co-aggregation in the development of multi-species oral biofilms. The first stage involves primary colonizers adhering to a conditioning film on a substratum. In the next stage, cell growth, division and extrapolysaccharide (EPS) production lead to microcolony formation. A third stage involves extensive co-aggregation of single cells, and other groups of cells to the growing biofilm. In the final stage, the biofilm matures into a complex, multi-species community. Similar stages have been described in single-species biofilms of Pseudomonas aeruginosa (36), Escherichia coli and Vibrio cholerae (41) as well as in multi-species biofilms in dental plaque (20, 28).
The bacterial species within the biofilms were grouped into six bacterial complexes as described by Socransky (33, 34). The differences between the bacterial compositions of healthy and periodontitis biofilms were assessed using these complexes. Subgingival plaque samples from healthy subjects consisted primarily of Actinomyces species (blue complex) and to a lesser extent, Streptococcus species (yellow complex) and Veillonella (purple complex). Less than 4% of the total bacteria identified were members of the orange (Fusobacterium species, Prevotella species, C. rectus) or red (P. gingivalis, T. forsythia, T. denticola) complexes, both of which are most frequently associated with periodontitis (33). The mature biofilms developed from plaque samples from healthy individuals were also composed of mostly members of the blue, yellow and purple complexes and less than 5% of the identified bacteria were from the orange and red complexes. In subgingival plaque from periodontitis patients, the dominant species were members of the orange complex, sometimes composing up to 40% of the total isolates identified. This increase in members of the orange complex was also observed in the mature biofilms grown from these samples. These results are in accordance with current literature describing a shift in bacterial composition from gram-positive cocci and rods to gram-negative rods that often accompanies the transition from a state of periodontal health to periodontal disease.
The model described here provides a method for the study of the subgingival plaque in an in vitro multi-species biofilm that closely mimics the composition of the in vivo state. The versatility of this model, combined with its simplicity and high reproducibility, makes it an effective system to study subgingival biofilm colonization and development. The model should also be useful for investigating the mechanisms of enhanced antimicrobial resistance that has been attributed to biofilm-grown bacteria.
Acknowledgments
This study was supported by grant DE014714 from the National Institute of Dental and Craniofacial Research of the National Institutes of Health. We thank Jennifer Gollwitzer for technical assistance with the checkerboard DNA–DNA hybridization experiments and Sonia Nangó-Henesy for assistance with cultures.
References
- 1.Amies CR. A modified formula for the preparation of Stuart's transport medium. Can J Public Health. 1967;58:296–300. [PubMed] [Google Scholar]
- 2.Auschill TM, Artweiler NB, Netuschil L, Brecx M, Reich ME, Sculen A. Spatial distribution of vital and dead microorganisms in dental biofilms. Arch Oral Biol. 2001;46:471–476. doi: 10.1016/s0003-9969(00)00136-9. [DOI] [PubMed] [Google Scholar]
- 3.Azen EA. Genetics of salivary protein polymorphisms. Crit Rev Oral Biol Med. 1993;4:479–485. doi: 10.1177/10454411930040033201. [DOI] [PubMed] [Google Scholar]
- 4.Beenken KE, Dunman PM, McAleese F, et al. Global gene expression in Staphylococcus aureus biofilms. J Bacteriol. 2004;186:4665–4684. doi: 10.1128/JB.186.14.4665-4684.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown TAJ, Ahn SJ, Frank RN, Chen YY, Lemos JA, Burne RA. A hypothetical protein of Streptococcus mutans is critical for biofilm formation. Infect Immun. 2005;73:3188–3191. doi: 10.1128/IAI.73.5.3147-3151.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cohen L, Rams T, Slots J, Walker C. Independent analyses of microbiological samples by three testing laboratories. J Dent Res. 2001;80:219. abstract 1465. [Google Scholar]
- 7.Dodds MWJ, Johnson A, Yeh C-K. Health benefits of saliva: a review. J Dent Res. 2004;33:223–233. doi: 10.1016/j.jdent.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 8.Good IJ. An index of separateness of clusters and a permutation test for its significance. J Stat Comput Simul. 1982;15:81–84. [Google Scholar]
- 9.Guggenheim B, Giertsen W, Schupbach P, Shapiro S. Validation of an in vitro biofilm model of supragingival plaque. J Dent Res. 2001;80:363–370. doi: 10.1177/00220345010800011201. [DOI] [PubMed] [Google Scholar]
- 10.Guggenheim B, Guggenheim M, Gmur R, Giertsen E, Thurnheer T. Application of the Zurich biofilm model to problems of cariology. Caries Res. 2004;38:212–222. doi: 10.1159/000077757. [DOI] [PubMed] [Google Scholar]
- 11.Guggenheim M, Shapiro S, Gmur R, Guggenheim B. Spatial arrangements and associative behavior of species in an in vitro oral biofilm model. Appl Environ Microbiol. 2001;67:1343–1350. doi: 10.1128/AEM.67.3.1343-1350.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Handel T, Olsen I, Walker C, Caugant DA. β-Lactamase production and antimicrobial susceptibility of subgingival bacteria from refractory periodontitis. Oral Microbiol Immunol. 2004;19:303–308. doi: 10.1111/j.1399-302x.2004.00159.x. [DOI] [PubMed] [Google Scholar]
- 13.Handel T, Olsen I, Walker C, Caugant DA. Detection and characterization of β-lactamase genes in subgingival bacteria from patients with refractory periodontitis. FEMS Microbiol Lett. 2005;242:319–324. doi: 10.1016/j.femsle.2004.11.023. [DOI] [PubMed] [Google Scholar]
- 14.Holdeman LV, Moore WEC, Cato EP. VPI anaerobic lab manual. 4th edn. VPI&SU; Blacksburg, VA: 1977. [Google Scholar]
- 15.Hope CK, Clements D, Wilson M. Determining the spatial distribution of viable and nonviable bacteria in hydrated microcosm dental plaques by viability profiling. J Appl Microbiol. 2002;93:448–455. doi: 10.1046/j.1365-2672.2002.01703.x. [DOI] [PubMed] [Google Scholar]
- 16.Hope CK, Wilson M. Biofilm structure and cell vitality in a laboratory model of subgingival plaque. J Microbiol Methods. 2006;66:390–398. doi: 10.1016/j.mimet.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 17.Kolenbrander P. Oral microbial communities: biofilms, interactions, and genetic systems. Annu Rev Microbiol. 2000;54:413–437. doi: 10.1146/annurev.micro.54.1.413. [DOI] [PubMed] [Google Scholar]
- 18.Kolenbrander PE, Andersen RN, Kazmerzak K, Wu R, Palmer RJ. Spatial organization of oral bacteria in biofilms. Methods Enzymol. 1999;310:322–332. doi: 10.1016/s0076-6879(99)10026-0. [DOI] [PubMed] [Google Scholar]
- 19.Lazazzera BA. Lessons from DNA micro-array analysis: the gene expression profile of biofilms. Curr Opin Microbiol. 2005;8:222–227. doi: 10.1016/j.mib.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 20.Marsh PD. Dental plaque as a microbial biofilm. Caries Res. 2004;38:204–211. doi: 10.1159/000077756. [DOI] [PubMed] [Google Scholar]
- 21.Moore LVH, Bourne DM, Moore WEC. Comparative distribution and taxonomic value of cellular fatty acids in thirty-three genera of anaerobic gram-negative bacilli. Int J Syst Bacteriol. 1994;44:338–347. doi: 10.1099/00207713-44-2-338. [DOI] [PubMed] [Google Scholar]
- 22.Moore WEC, Holdeman LV. Human fecal flora: the normal flora of 20 Japanese-Hawaiians. Appl Microbiol. 1974;27:961–979. doi: 10.1128/am.27.5.961-979.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moore WEC, Holdeman LV, Cato EP. Bacteriology of moderate (chronic) periodontitis in mature adult humans. Infect Immun. 1983;42:510–515. doi: 10.1128/iai.42.2.510-515.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moore WEC, Holdeman LV, Cato EP. Comparative bacteriology of juvenile periodontitis. Infect Immun. 1985;48:507–519. doi: 10.1128/iai.48.2.507-519.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moore WEC, Holdeman LV, Smibert RM, et al. Bacteriology of experimental gingivitis in young adult humans. Infect Immun. 1982;38:651–667. doi: 10.1128/iai.38.2.651-667.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moore LVH, Moore WEC, Riley C, Brooks CN, Burmeister JA, Smibert RM. Periodontal microflora of HIV positive subjects with gingivitis or adult periodontitis. J Periodontol. 1993;64:48–56. doi: 10.1902/jop.1993.64.1.48. [DOI] [PubMed] [Google Scholar]
- 27.Netuschil L, Reich E, Unteregger G, Sculean ABM. A pilot study of confocal laser scanning microscopy for the assessment of undisturbed dental plaque viability and topography. Arch Oral Biol. 1998;43:277–285. doi: 10.1016/s0003-9969(97)00121-0. [DOI] [PubMed] [Google Scholar]
- 28.Nishihara T, Koseki T. Microbial etiology of periodontitis. Periodontol 2000. 2004;36:14–26. doi: 10.1111/j.1600-0757.2004.03671.x. [DOI] [PubMed] [Google Scholar]
- 29.Pratten J, Andrews CS, Craig DQM, Wilsom M. Structural studies of microcosm dental plaques grown under different nutritional conditions. FEMS Microbiol Lett. 2000;189:215–218. doi: 10.1111/j.1574-6968.2000.tb09233.x. [DOI] [PubMed] [Google Scholar]
- 30.Rathsam C, Eaton RE, Simpson CL, et al. Up-regulation of competence- but not stress-responsive proteins accompanies an altered metabolic phenotype in Streptococcus mutans biofilms. Microbiology. 2005;151:1825–1837. doi: 10.1099/mic.0.27830-0. [DOI] [PubMed] [Google Scholar]
- 31.Resch A, Rosenstein R, Nerz C, Gotz F. Differential gene expression profiling of Staphylococcus aureus cultivated under biofilm and planktonic conditions. Appl Environ Microbiol. 2005;71:2663–2676. doi: 10.1128/AEM.71.5.2663-2676.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rickard AH, Gilbert P, High NJ, Kolenbrander PE, Handley PS. Bacterial coaggregations: an integral process in the development of multi-species biofilms. Trends Microbiol. 2003;11:94–100. doi: 10.1016/s0966-842x(02)00034-3. [DOI] [PubMed] [Google Scholar]
- 33.Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RL. Microbial complexes in subgingival plaque. J Clin Periodontol. 1998;25:134–144. doi: 10.1111/j.1600-051x.1998.tb02419.x. [DOI] [PubMed] [Google Scholar]
- 34.Socransky S, Haffajee A, Smith C, et al. Use of checkerboard DNA–DNA hybridization to study complex microbial ecosystems. Oral Microbiol Immunol. 2004;19:352–362. doi: 10.1111/j.1399-302x.2004.00168.x. [DOI] [PubMed] [Google Scholar]
- 35.Socransky SS, Smith C, Martin L, Paster BJ, Dewhirst FE, Levin AE. ‘Checkerboard’ DNA-DNA hybridization. Biotechniques. 1994;17:488–492. [PubMed] [Google Scholar]
- 36.Stoodley P, Sauer K, Davies DG, Costerton JW. Biofilms as complex differentiated communities. Annu Rev Microbiol. 2002;56:187–209. doi: 10.1146/annurev.micro.56.012302.160705. [DOI] [PubMed] [Google Scholar]
- 37.Takeuchi H, Yamanaka Y, Yamamoto K. Morphological analysis of subgingival biofilm formation on synthetic carbonate apatite inserted into human periodontal pockets. Aust Dent J. 2004;49:72–77. doi: 10.1111/j.1834-7819.2004.tb00053.x. [DOI] [PubMed] [Google Scholar]
- 38.Walker CB. The acquisition of antibiotic resistance in the periodontal microflora. Periodontol 2000. 1996;10:79–88. doi: 10.1111/j.1600-0757.1996.tb00069.x. [DOI] [PubMed] [Google Scholar]
- 39.Walker CB, Gordon JM, Socransky SS. Antibiotic susceptibility testing of subgingival plaque samples. J Clin Periodontol. 1983;10:422–432. doi: 10.1111/j.1600-051x.1983.tb01291.x. [DOI] [PubMed] [Google Scholar]
- 40.Wall-Manning GM, Sissons CH, Anderson SA, Lee M. Checkerboard DNA-DNA hybridisation technology focused on the analysis of Gram-positive cariogenic bacteria. J Microbiol Methods. 2002;51:301–311. doi: 10.1016/s0167-7012(02)00106-9. [DOI] [PubMed] [Google Scholar]
- 41.Watnick P, Kolter R. Biofilms, city of microbes. J Bacteriol. 2000;182:2675–2679. doi: 10.1128/jb.182.10.2675-2679.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wecke J, Kersten T, Madela K, et al. A novel technique for monitoring the development of bacterial biofilms in human periodontal pockets. FEMS Microbiol Lett. 2000;191:95–101. doi: 10.1111/j.1574-6968.2000.tb09324.x. [DOI] [PubMed] [Google Scholar]
- 43.Whittaker CJ, Klier CM, Kolenbrander PE. Mechanisms of adhesion by oral bacteria. Annu Rev Microbiol. 1996;50:513–552. doi: 10.1146/annurev.micro.50.1.513. [DOI] [PubMed] [Google Scholar]
- 44.Wimpenny J. Laboratory models of biofilm. In: Heman HN, Wilson N, editors. Dental plaque revisited: oral biofilms in health and disease. Bioline; Cardiff, UK: 1999. pp. 89–110. [Google Scholar]
- 45.Wood SR, Kirkham J, Manz W, Shore RC, Nattress B, Robinson C. Architecture of intact natural human plaque biofilms studied by confocal laser scanning microscopy. J Dent Res. 2000;79:21–27. doi: 10.1177/00220345000790010201. [DOI] [PubMed] [Google Scholar]
- 46.Zaura-Arite E, van Marle J, ten Cate JM. Confocal microscopy study of undisturbed and chlorhexidine-treated dental biofilm. J Dent Res. 2001;80:1436–1440. doi: 10.1177/00220345010800051001. [DOI] [PubMed] [Google Scholar]