

Abstract
The selection of tRNAs by their cognate aminoacyl-tRNA synthetases is critical for ensuring the fidelity of protein synthesis. While nucleotides that comprise tRNA identity sets have been readily identified, their specific role in the elementary steps of aminoacylation is poorly understood. By use of a rapid kinetics analysis employing mutants in tRNAHis and its cognate aminoacyl-tRNA synthetase, the role of tRNA identity in aminoacylation was investigated. While mutations in the tRNA anticodon preferentially affected the thermodynamics of initial complex formation, mutations in the acceptor stem or the conserved motif 2 loop of the tRNA synthetase imposed a specific kinetic block on aminoacyl transfer, and decreased tRNA mediated kinetic control of amino acid activation. The mechanistic basis of tRNA identity is analogous to fidelity control by DNA polymerases and the ribosome, whose reactions also demand high accuracy.
Keywords: aminoacylation, aminoacyl-tRNA synthetases, tRNA identity, asymmetric catalysis, tRNA
Biological reactions involving information transfer impose special constraints on substrate selection and mechanism, because they must exhibit speed while preserving accuracy. The low overall frequency (10-3 to 10-4) of amino acid mis-incorporation in translation reflects the cumulative fidelity of at least three principal reactions, namely the esterification of amino acids to their cognate tRNAs by aminoacyl-tRNA synthetases (aaRSs) (Ibba and Soll, 2000), the binding selectivity for cognate amino acid tRNA pairs by EF-Tu (LaRiviere et al., 2001), and the decoding process performed by the ribosome (Ogle and Ramakrishnan, 2005). By virtue of their requirement for accurate selection of both amino acid and tRNA, the aaRSs help maintain the fidelity of protein synthesis. All isoacceptors in a tRNA family possess the same RNA fold, and share common nucleotides to ensure recognition by the cognate aaRS and rejection by the non-cognate aaRS. Which of the twenty standard amino acids is attached to a given tRNA in vivo (its “identity”) reflects both its recognition by its cognate aaRS, as well as potential mischarging by near cognate aaRSs (Giege et al., 1998; Saks et al., 1994). The frequency of errors in this process has been estimated to be as low as 10-7 per aminoacylation, indicative of the surface area of the tRNA available for discrimination (Jakubowski and Goldman, 1992).
The specificity exhibited for cognate tRNA by the aaRSs arises from the summed contribution of both thermodynamic and kinetic effects. Depending on the tRNA and the particular reaction conditions, non-cognate tRNAs are rejected during binding by factors of 10-103 (Lam and Schimmel, 1975). The Vmax of the aminoacylation reaction can provide an additional 103 to 104 in specificity, such that the ratio of resulting specificity constants (kcat/Km) accounts for the low frequency of tRNAs mischarging observed experimentally (Ebel et al., 1973). Accordingly, proofreading in tRNA selection was originally thought to be unnecessary (Fersht, 1979). The identity nucleotides that define tRNA isoacceptor systems are primarily concentrated in the anticodons and acceptor stems of tRNAs, providing functional groups that can be accessed by specificity determining side chains on the enzymes (Cavarelli et al., 1993; Rould et al., 1991). However, tRNA identity also emerges from the presence of modified bases (Muramatsu et al., 1988) and more global aspects of tRNA structure in ways that are not fully understood (Hauenstein et al., 2004). Substitution of individual nucleotides within an identity set for a given tRNA can produce significant decreases in kcat, and also alter specificity of the system (Giege et al., 1998).
Despite these apparent successes in cataloging and manipulating identity determinants on tRNAs, and in localizing specificity determining side chains on aaRSs, the fundamental question of how tRNA recognition is manifested during the aminoacylation reaction itself has never been satisfactorily addressed. To address this issue, a pre-steady state kinetics approach was applied to identity mutants in tRNA and aaRS in the class II histidyl-tRNA synthetase (HisRS) system, with the goal of pinpointing the precise step(s) in the catalytic cycle where tRNA recognition exerts its greatest influence. These experiments revealed how tRNA selection arises from interactions between tRNA identity determinants and class conserved motifs in the active site that provide kinetic discrimination at discrete points throughout the catalytic cycle. The mechanisms by which the fidelity of tRNA selection is maintained by the aaRSs are highly reminiscent of the fidelity control mechanisms of DNA polymerases and the ribosome.
RESULTS
The E. coli histidine tRNA on which these experiments were based possesses as identity elements the 5’ phosphate and G-1:C73 base pair in the acceptor stem, and a GUG anticodon in which G34 is substituted by queosine (Figure 1A). Mutation of these elements diminishes aminoacylation in vitro (Fromant et al., 2000; Himeno et al., 1989; Yan et al., 1996), and histidine identity in vivo (Yan et al., 1996; Yan and Francklyn, 1994). This study employed G34U and C73U single base substitutions, and a 5’ triphosphate variant of tRNAHis. The conserved motif 2 loop of HisRS includes two important classes of residues (Figure 1B,C) whose significance was established by mutagenesis experiments (Connolly et al., 2004; Hawko and Francklyn, 2001), and phylogenetics (Ardell and Andersson, 2006). The first group, exemplified by Glu115 and Arg121, includes class II ‘universally conserved’ acidic and basic residues that interact with ATP during adenylation (Arnez et al., 1997; Belrhali et al., 1995), and with C74 during aminoacyl transfer (Cavarelli et al., 1994; Cusack et al., 1996). Another class II conserved residue is Gln127, which interacts with the α-carboxylate of the histidine substrate. The motif 2 loop also includes ‘HisRS family conserved” residues, exemplified by Gln118, Arg116, and Arg123. These have no obvious structural role in adenylation (Figure 1B), but a more significant role in providing selectivity for the G-1:C73 base pair and/or 5’ phosphate (Figure 1C). Alanine and histidine substitutions introduced at these side chains (Figure 1D) alter the sensitivity of HisRS to tRNA nucleotide substitutions, supporting their predicted role in enforcing tRNA specificity (Connolly et al., 2004; Hawko and Francklyn, 2001).
Figure 1.
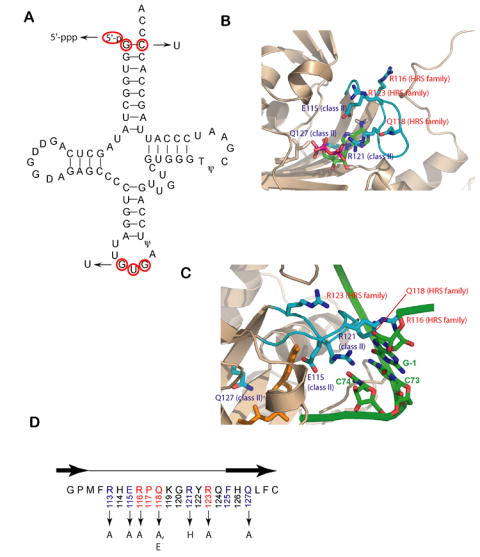
tRNA recognition elements in the HisRS/tRNAHis system.
(A) Cloverleaf diagram of tRNAHis from E. coli. Key recognition elements for tRNAHis by HisRS are shown in red. The mutant substitutions used in this work are denoted by arrows.
(B) Close up of the active site of the E. coli HisRS histidinol-ATP complex, showing residues of the motif 2 loop in contact (PDB id: 1KMN). The motif 2 loop and residues thereof are colored in cyan, while the surrounding active site secondary structures are rendered in tan. Class II conserved residues are labeled in blue, while the HisRS-family conserved residues are labeled in red.
(C) Interactions between HisRS and tRNAHis predicted by homology modeling with the AspRS-tRNAAsp crystal structure (Connolly et al., 2004). The coloring of the enzyme is as noted in panel A, while the nucleotides of tRNAHis are colored green. The aminoacyl-adenylate is shown in orange. The model has not been manipulated to reflect conformational rearrangements likely associated with tRNA binding. The configuration of the motif 2 loop represents the “T state” seen in one of the two monomers of the histidyl adenylate complex (PDB: 1KMM).
(D) Primary sequence of the E.coli HisRS motif 2 loop. The β strands flanking loop are depicted above the sequence as black arrows. Class II conserved amino acids are highlighted in light blue, while HisRS conserved sequences are highlighted in red. Amino acid substitutions in the motif 2 loop examined in this study are indicated by arrows.
Single turnover and pre-steady state assays developed previously (Guth et al., 2005) were employed to study the tRNA and HisRS mutants described above. In the single turnover aminoacylation assay, histidyl-adenylate (labeled with [14C] in the amino acid) is pre-formed on the enzyme, and then rapidly mixed in excess over the tRNA in a rapid quench device. The resulting first order progress curves provide the rate of aminoacyl transfer. In the second assay, pre-incubated HisRS:tRNAHis complex is rapidly mixed with a large excess of histidine and Mg2+-ATP, and then sampled under pre-steady conditions (Fersht and Kaethner, 1976). The slope of the multiple turnover plot provides the limiting rate for the overall aminoacylation cycle, equivalent to kcat measured under steady state conditions. Significantly, the use of [14C] labeled amino acid or [α-32P] labeled ATP in parallel reactions provides information about the two different half reactions, and the stoichiometry of ATP consumption relative to aminoacyl-tRNA product formation.
Mutations at identity positions in tRNA and HisRS significantly reduce the rate of aminoacyl transfer
The first group of single turnover assays clearly revealed a difference in the aminoacyl transfer kinetics of the G34U anticodon mutant, versus the C73U discriminator base and 5’ triphosphate mutants. While the former had virtually no effect on the rate of transfer (12.5 s-1, versus 18.8 s-1 for wild type), the latter decreased transfer by 940-fold (Figure 2, summarized in Table 1). As measured by the apparent K1/2 for maximal transfer rate, the G34U mutant tRNAHis required an eight-fold higher concentration than the wild type to reach a saturating rate of transfer, while C73U tRNAHis required only a three-fold increase (Table 1). In this system, tRNA binding is not in rapid equilibrium (E.G. and C.F., unpublished), so KD cannot be readily extracted from plots of the apparent ktrans versus [tRNA]. Nevertheless, the elevated K1/2 is consistent with the increased Km previously reported for anticodon mutants (Himeno et al., 1989; Yan et al., 1996). A significantly decreased (134-fold) rate of transfer and elevated K1/2 was also seen for 5’ triphosphate tRNAHis (Table 1). Thus, while identity-determining mutations in tRNAHis could be seen to affect both the thermodynamics of initial complex formation (as estimated from K1/2) and the kinetics of transfer, deleterious mutations preferentially affected the latter.
Figure 2.
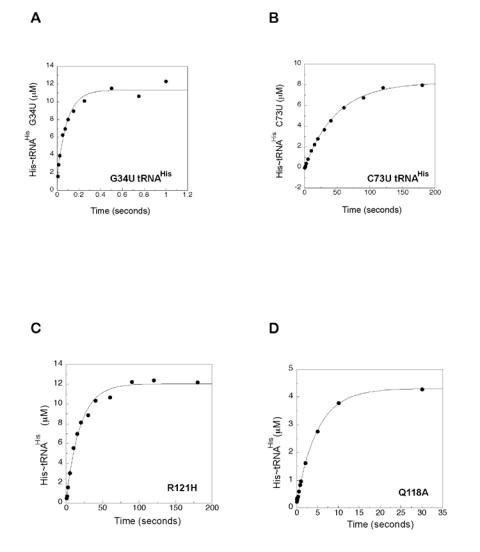
Representative single turnover progress curves for aminoacyl transfer to tRNAHis mutants catalyzed by wild type E.coli HisRS, and mutant versions of HisRS. HisRS with preformed adenylate was rapidly mixed with tRNAHis mutants at 37°C, pH 7.5 in the RQF-3 quench flow apparatus as described in “Experimental Procedures”.
(A) G34U-tRNAHis and wild type HisRS.
(B) C73U-tRNAHis and wild type HisRS.
(C) Wild type tRNAHis and R121H HisRS.
(D) Wild type tRNAHis and Q118A HisRS.
Table 1.
Transient Kinetics analysis of tRNAHis Variants
| tRNA | Single Turnover rate of aminoacyl transfer (sec-1) | Apparent K1/2 for tRNA in single turnover (μM)* | Multiple Turnover rate (sec-1) | [32P] AMP burst amplitude, mol AMP/mol active sites | Burst phase rate of [32P] AMP formation (sec-1) | Linear Turnover Number, [14C]- His-tRNAHis subsat. [ATP] (sec-1) | Linear Turnover Number, [32P]-AMP subsat. [ATP] (sec-1) |
|---|---|---|---|---|---|---|---|
| Wt | 18.8 | 2.5 | 2.01 | No burst | No burst | 2.31 | 1.74 |
| G34U | 12.5 | 20 | 0.37 | No burst | No burst | 0.41 | 0.67 |
| 5’ppp | 0.141 | 12.5 | 0.195 | 0.669 | 0.349 | 0.0969 | 0.0869 |
| C73U | 0.020 | 8 | 0.0063 | 0.578 | 7.920 | 0.00610 | 0.0134 |
This value is calculated as one half the concentration of tRNA required to obtain a limiting (highest) rate of aminoacyl transfer, which can be assumed to reflect saturation for tRNA binding. The apparent K1/2 is higher than reported Km for wild type tRNAs in steady state kinetics, as the latter value reflects the effect of two tRNA molecules binding to dimeric enzyme during the catalytic cycle, whereas single turnover experiments reflect one tRNA binding to the dimer in the first turnover. All values represent the mean of two to three independent determinations. The standard deviations on these values were typically 5-15%.
When single turnover experiments were performed on the two classes of HisRS motif 2 loop mutants (Figure 2, Supplementary Figure 1, and Table 2), the class II conserved E115A mutation had a relatively modest effect (2.41 sec-1, 7.8-fold decreased), whereas R121H was more severely decreased (0.051 sec-1, a 368-fold decrease). Based on the structural homology between the E. coli AspRS and HisRS active sites, Arg121 of HisRS is predicted to make two hydrogen bonds to C74, while Glu115 makes one (Figure 1C). Differences in transfer rates therefore correlate with the predicted interaction strengths to C74. The Q127A mutant, which alters a contact to the substrate histidine, was only two-fold decreased in aminoacyl transfer (Guth et al., 2005), indicating that, within the HisRS active site, adenylation and amino transfer are functionally separable. Substitution mutants of HisRS family conserved motif 2 residues were generally more compromised than the class II conserved mutants. R116A, which alters a putative contact to G-1, was 20-fold decreased in transfer, while Q118A and Q118E exhibited 69- and 8000-fold decreases in the transfer rate. Substitution of Arg123, a putative contact to the 5’ phosphate, produced a 200-fold decrease in aminoacyl-transfer. Thus, mutations of identity elements on either the tRNA or the aaRS had quantitatively similar effects on the rate of aminoacyl transfer, and these were much more severe than the effect of an anticodon substitution.
Table 2.
Transient Kinetics of HisRS mutants
| Enzyme | Single Turnover rate of aminoacyl transfer (sec-1) | Apparent K1/2 for tRNA in single turnover (μM)* | Multiple Turnover rate, (sec-1) | [32P] AMP burst amplitude, mol AMP/mol active sites | Burst phase rate of [32P] AMP formation (sec-1) | Linear Turnover, [14C]-His-tRNAHis (sec-1) | Linear Turnover Number, [32P]-AMP (sec-1) |
|---|---|---|---|---|---|---|---|
| Wt HRS | 18.8 | 2.5 | 2.012 | No burst | No burst | 2.31 | 1.74 |
| E115A | 2.41 | 2.5 | 0.696 | No burst | No burst | 0.190 | 0.216 |
| Q127A | 9.20 | 2.5 | 0.123 | No burst | No burst | 0.0195 | 0.0140 |
| R116A | 0.346 | 15 | 0.291 | 0.509 | 10.7 | 0.199 | 0.149 |
| Q118A | 0.270 | 25 | 0.228 | 0.470 | 20.4 | 0.056 | 0.0523 |
| Q118E | 0.00230 | 5 | 0.00166 | 0.408 | 4.36 | 0.000774 | 0.0155 |
| R121H | 0.051 | 50 | 0.0150 | 0.617 | 0.103 | 0.0120 | 0.016 |
| R123A | 0.092 | 25 | 0.0995 | 0.427 | 7.79 | 0.074 | 0.087 |
All values represent the mean of two to three independent determinations. The standard deviations on these values were typically 5-15%, with larger variance observed for severely kinetically compromised mutants (e.g., Q118E) due to high background values relative to product formation. Deviation in values did not affect the relative rates of product formation.
Comparison of the rates of aminoacyl transfer (ktrans) to multiple turnover (kcat) conditions
The single turnover rate of aminoacyl transfer (ktrans) catalyzed by HisRS is nearly ten-fold faster than kcat, an observation previously rationalized by the inhibitory effect of the cognate tRNA on the rate of adenylation (Guth et al., 2005). To probe whether tRNA and HisRS identity mutants change the rate determining step, the rapid quench assay was performed under multiple turnover conditions, employing excess histidine, ATP and tRNA substrates. The resulting linear progress curves all extrapolated back to the origin, indicating that neither the wild type nor any of the mutants is limited by product release (data not shown). Some tRNA and HisRS mutants exhibited a rate of aminoacyl transfer that was faster than the overall kcat, while for others, the two rates were nearly equal (Table 1 and 2). Mutants that retained the wild type relationship (ktrans > kcat) included G34U tRNAHis, E115A, Q127A and, to a somewhat lesser extent, R116A. The faster rate of transfer for G34U tRNAHIs relative to kcat suggests that the mutation principally affects the initial encounter of tRNA with the enzyme, rather than the subsequent aminoacyl transfer step. Both E115A and Q127A mutations alter side chains participating in the adenylation reaction rather than transfer (Figure 1B), so their elevated ktrans/kcat ratios likely reflect preferential effects on adenylation. While R121H exhibited significant decreases in both ktrans and kcat, the overall ktrans was three fold faster than kcat. This result is consistent with the predicted role of this side chain in the binding of both ATP during the first reaction, and of C74 during the second (Figure 1B,C). In contrast, those mutants in both tRNA and aaRS with a direct role in enforcing the tRNA specificity of the interaction (including C73U tRNAHis, 5’ppp-tRNAHis, Q118A, Q118E, and R123A) exhibited essentially equivalence between ktrans and kcat. For all these mutants, the simplest explanation for equivalence of the two rates is that aminoacyl transfer is rate limiting for the overall reaction.
Measuring the rate of adenylation under pre-steady state conditions and the stoichiometry of ATP utilization
Previously, pyrophosphate exchange (Calender and Berg, 1966) and stopped flow fluorescence assays (Fersht et al., 1975) have been used to measure the rate of amino acid activation, but neither takes into account the influence of tRNA. The multiple turnover pre-steady state rapid quench assay was modified to allow the rate of [α-32P] ATP consumption to be monitored, and thereby overcome this limitation. These reactions were performed in parallel with reactions (under otherwise identical enzyme and substrate concentrations) featuring cold ATP and [14C]-histidine to follow [14C]-His~tRNAHis production, allowing the ratio of [α-32P] AMP/[14C] His~tRNAHis production to be followed temporally. As a technical constraint, these experiments were performed at a sub-saturating concentration of [ATP] (100 M) to achieve an acceptable signal to noise ratio.
The resulting progress curves for these experiments employing wild type and mutant tRNAHis and HisRS are depicted in Figure 3 and in Supplementary Figure 2. Distinct curve patterns were observed that served to segregate the mutants into three different classes. For the wild type tRNA, G34U tRNAHis, and wild type, E115A, and Q127A HisRS, the progress curves for [α-32P] AMP and [14C] His~tRNAHis formation were linear and essentially parallel. These are the same mutants for which ktrans was greater than kcat. For these tRNAs and HisRS enzymes, the rate of [α-32P] AMP synthesis was constant over the course of the experiment, and was identical to the rate of His-tRNAHis formation. Adenylation is therefore likely to remain the rate limiting step for the overall aminoacylation for these tRNAs and enzymes, despite apparent decreases in the overall rate as measured by kcat (Table 1, 2). The equivalence in slopes of the two progress curves indicates that each molecule of histidyl-adenylate formed is quantitatively converted to [14C] His~tRNAHis product. This complete conversion of adenylate to product argues that the interactions perturbed in these mutants have little or no impact on the overall fidelity of aminoacyl transfer.
Figure 3.
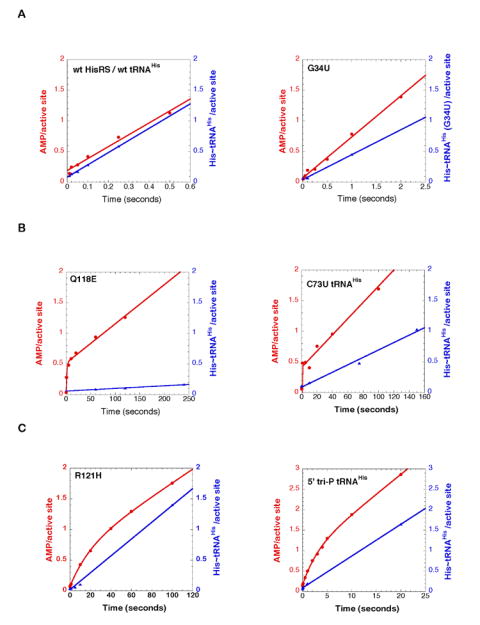
Representative multiple turnover progress curves for wild type HisRS and mutant tRNAHis comparing the production of [α-32P]-AMP (filled red circles) and [14C]-His~tRNAHis (filled blue triangles) at pH 7.5 and 37°C.
(A) Representative enzyme-tRNA combinations in which ktrans was greater than kcat, and progress curves for the two products were linear. The plots for wt tRNAHis/wt HisRS, and G34U tRNAHis/wt HisRS are depicted here. The remaining plots for other mutants are presented in Supplementary Figure 2.
(B) Representative enzyme-tRNA combinations in which there was a burst of AMP formation in the first turnover. The plots for wt tRNAHis/Q118E HisRS, and C73U tRNAHis/wt HisRS are depicted here. The remaining plots for other mutants are presented in Supplementary Figure 2.
(C) Enzyme-tRNA combinations where slow burst kinetics was observed, owing to effects on both adenylation and aminoacyl transfer. The wt tRNAHis/ R121H HisRS and 5’-ppp tRNAHis/wt HisRS were the only combinations that exhibited this behavior.
For the second class of mutants, which included C73U tRNAHis, as well as R116A, Q118A, Q118E, and R123A HisRS, there was a linear progress curve for His~tRNAHis production, but the curves for [α-32P] AMP formation were more accurately described by burst kinetics. The amplitudes of these bursts were uniformly equal to 0.5 mol AMP/mol active site, and, with two exceptions, the linear portions of the [α-32P] AMP formation curves were parallel to the [14C] His~tRNAHis product curves. The burst kinetics indicates that, in the first but not subsequent turnovers, the rates of [α-32P] AMP formation in these mutants are considerably faster (ranging from 4.3 sec-1 for Q118E to 20 sec-1 for Q118A HisRS) than both the rate of aminoacyl transfer (ktrans) and the overall rate of aminoacylation (kcat). As concluded above, these mutants are limited by the rate of transfer, and not adenylation. The data also underscore a linkage between the decrease in aminoacyl transfer rate and tRNA mediated attenuation of histidyl-adenylate formation.
For the Q118E mutant and C73U tRNAHis, multiple determinations of the [α-32P] AMP /[14C] His~tRNAHis formation ratio indicated that the rate of consumption of ATP was faster (14-fold and two-fold, respectively) than the rate of aminoacyl transfer. These particular HisRS:tRNAHis combinations may therefore bring about an active proofreading process leading to excess consumption of ATP, or, alternatively, reflect a rate of aminoacyl transfer that is inherently slower than the endogenous rate of adenylate synthesis and breakdown in the absence of tRNA. These alternatives were investigated by measuring the rate of AMP formation in the presence and absence of a non-cognate tRNA. The linear rates of adenylate in the presence or absence of a non-cognate tRNA in the steady state phase of the experiments (0.025 and 0.017 sec-1, respectively, Figure 4) were much faster than the rate of decomposition of the adenylate when pre-formed on the enzyme (1.8 × 10-4 sec-1, Supplementary Figure S3). The former rates are comparable to the rates of ATP consumption for Q118E and C73U tRNAHis, which were 0.015 sec-1 (Table 2), and 0.0134 sec-1 (Table 1). Thus, neither non-cognate tRNA, nor cognate tRNAs highly compromised with respect to identity, nor corresponding HisRS identity mutants, accelerated ATP consumption/amino acid activation over a basal tRNA-independent rate of 0.015- 0.02 sec-1. Additional control experiments (Figure 5) indicated that this basal rate of AMP formation is enzyme dependent, and reflects kinetic partitioning between solvolysis of the adenylate on the enzyme, and its release and subsequent hydrolysis in solution. The release of free adenylate into solution from a fractional subset of HisRS active sites occurred with a rate constant of 11.1 ± 4.7 × 10-2 min-1, while the enzyme independent decomposition of adenylate in solution occurred at a rate of 4.9 ± 1.2 × 10-2 min-1 (Figure 5). These values are 8.7- and 19.5-fold slower than tRNA independent basal rate of AMP production described above, arguing that the majority of the adenylate is subject to solvolysis on the enzyme when transfer is highly compromised.
Figure 4.
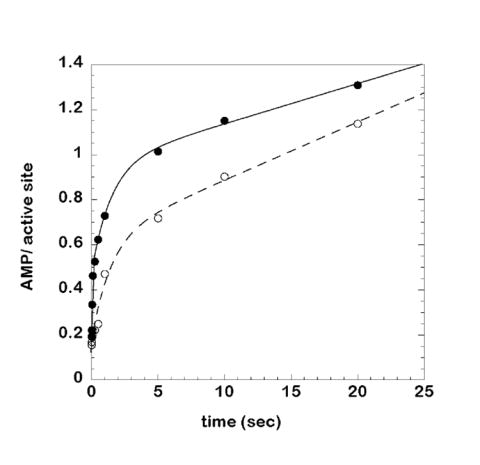
Adenylate turnover by wild type HisRS in the presence and absence of non-cognate tRNATrp at 37 °C and pH 7.5. [α-32P]-AMP formation was measured in the presence (open circles and broken line) and absence (solid circles and solid line) of non-cognate tRNATrp. The experiment was performed as described in “Experimental Procedures.” The progress curves were fit to a bi-exponential followed by a linear phase. The exponentials represent the rates of formation of adenylate in active sites one and two, followed by a slow linear rate that is likely represents reiterative cycles of adenylate formation, hydrolysis, and product dissociation in site two.
Figure 5.
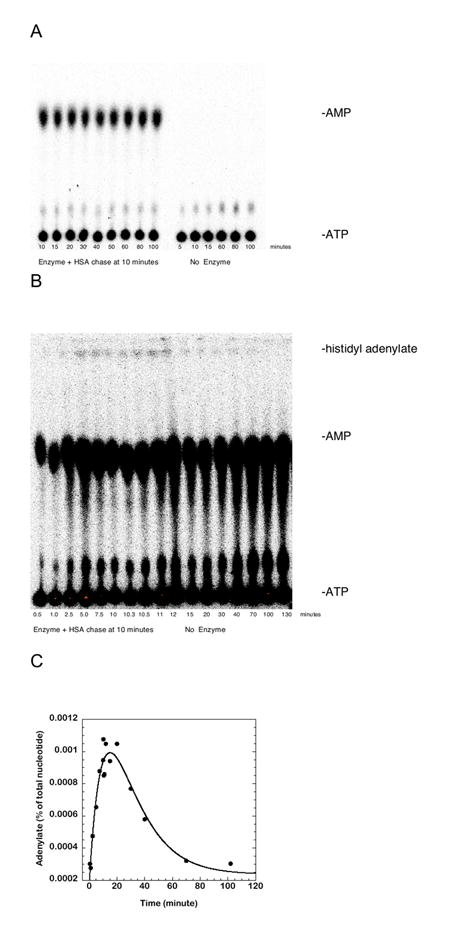
Enzyme dependent production of AMP and non-enzyme dependent hydrolysis of histidyl adenylate at pH 7.5 and 37°C.
(A) Thin layer chromatography experiment showing that the production of AMP is strictly dependent on the presence of enzyme over the relevant time course of experimentation. The left side of the panel is the complete reaction including enzyme, while the right panel is the complete reaction in the absence of enzyme. The reactions were prepared, imaged, and analyzed as described in “Experimental Procedures”.
(B) TLC showing the accumulation and destruction of the adenylate fraction during the experimental time course in the absence of tRNA. The observed adenylate is only a small fraction of the total AMP production.
(C) Graphical re-plot of data in (B), showing accumulation of adenylate in solution, and its subsequent decomposition in solution.
A third class of identity defects, described by R121H HisRS and 5’ppp tRNAHis, demonstrated intermediate effects with respect to the first two classes. These mutants demonstrated burst phase kinetics when adenylation was monitored, indicating aminoacyl transfer is rate limiting. However, the burst phase rate was considerably slower than that observed for mutants in the second class, ranging from 0.103 sec-1 for R121H HisRS to 0.349 sec-1 for 5’ppp tRNAHis. Previous studies implicated R121 as a participant in both halves of the reaction (Hawko and Francklyn, 2001), so a decrease in adenylation in addition to aminoacyl transfer is not unexpected. In the case of 5’ppp tRNAHis, the single turnover rate of aminoacyl transfer was essentially equivalent to the steady state rate, as well as equivalent to the linear rate of adenylation observed when accumulation of AMP was monitored. The observed burst of adenylate formation in the first turnover, coupled with the relatively slow rate at which that occurs, suggests that the 5’ppp tRNAHis mutant has lost some (but not all) of the capacity to regulate the rate of the first amino acid activation step. As noted above, only wild type HisRS and the mutants in the first class were unaffected in this function.
DISCUSSION
Previously, investigations into tRNA selection have been framed by the concepts of identity (Normanly and Abelson, 1989), determined through in vivo suppression, and tRNA recognition, determined by in vitro kinetic analysis (Sampson et al., 1992). In agreement with previous steady state work (Himeno et al., 1989; Yan et al., 1996), substitutions of the anticodon of tRNAHis increased the K1/2 for transfer, but had minimal effect on the rate of transfer (Table 1). Thus, anticodon mutations imposed a defect associated with formation of the initial encounter complex, but not on subsequent events associated with aminoacyl transfer, nor on the kinetics of adenylation. By contrast, the 5’ppp and C73U tRNAHis acceptor stem mutants significantly decreased the saturating rate of transfer, increased the concentration of tRNA necessary to achieve saturation, and lost the ability to attenuate the rate of the first amino acid activation step. The parallel between these kinetic effects and those observed with HisRS variants mutated at key tRNA recognition residues (e.g., Q118A, E and R123A) argues that mutations in both partners affect the same process. In view of the use of saturating tRNA concentrations, and the absence of burst kinetics under multiple turnover conditions, these step(s) must occur subsequent to tRNA binding, but prior to product release. Consistent with these observations, an earlier pre-steady state study of tRNATrp recognition found that a G73C discriminator mutant preferentially decreased the apparent transfer rate, while an A36G anticodon mutation did not (Ibba et al., 1999). This earlier study, recent work by Uter and Perona (Uter and Perona, 2004), and the work reported here all underscore the roles of different ends of the tRNA in the various phases of aminoacylation.
These diverse effects can be rationalized by proposing that tRNA aminoacylation is a multistep process initiated by formation of the “encounter complex,” the stability of which is dependent on interactions with the anticodon arm (or, for type II tRNAs, the variable arm). Conformational changes at the anticodon may facilitate direct readout by aaRSs, providing a thermodynamic basis for non-cognate tRNA rejection (Briand et al., 2000; Cusack, 1998; Eiler et al., 1999). This thermodynamic selection is significant in vivo, as tRNA identity can be readily manipulated by altering tRNA/aaRS ratios (Swanson et al., 1988). Following encounter complex formation, further conformational changes in the tRNA and enzyme likely constitute a mutual “accommodation” process that positions the CCA end in the active site with the appropriate geometry for aminoacyl transfer. The analysis reported here suggests that the kinetics of accommodation is controlled by interactions between recognition determinants on the tRNA and the enzyme. Comparative structural analysis from the aspartyl-, seryl-, and prolyl-systems argues for the existence of at least two distinct conformational states in the class II active site, namely an adenylate-bound “A-state”, and a tRNA-bound “T-state” (Cavarelli et al., 1994; Cusack et al., 1996; Moulinier et al., 2001; Yaremchuk et al., 2001). For the motif 2 loop, the switch between the A- and T-state involves an exchange of interactions between the β.γ phosphates in the adenylation reaction, and the CCA end in aminoacyl transfer (Cusack et al., 1996; Yaremchuk et al., 2001). In the HisRS system, the corresponding motif 2 loop residues E115 and R121 are predicted to be particularly important in this transition. On the tRNA, the accommodation process is likely to include both the proper juxtaposition of the acceptor helix against the catalytic domain and, potentially, a switch of the CCA end from a catalytically non-productive orientation to an orientation suitable for chemistry (Moulinier et al., 2001).
Consistent with this structural model, our kinetic experiments provided evidence for functional specialization of the motif 2 loop. Notably, mutation of residues with preferential roles in adenylation and thus the A state (i.e., Glu115 and Gln127) had only minimal impact on transfer rates, while mutation of side chains conferring specificity for the G-1:C73 identity determinant (i.e., R116, Q118, and R123) preferentially decreased transfer. An important caveat is that the single turnover measurements reported here did not allow the specific rates associated with accommodation and chemistry to be determined individually. Rather, the rates of both steps are likely to blend to give an overall composite ktrans. Identity mutations may render the equilibrium associated with the accommodation process unfavorable, and/or specifically decrease the actual rate of chemistry. By virtue of its specific interactions with the γ phosphate in the A-state and C74 in the T-state, R121 is likely to be particularly important for A-and T-site switching, and the positioning of the CCA end in the active site. The qualitatively similar kinetic consequences of R121H relative to Q118A and R123A provides important evidence that specificity determining contacts in the acceptor stem may be linked to the disposition of the CCA end. Future studies will be necessary to determine the magnitude of the equilibrium constant associated with accommodation, and the elementary rate constant associated with aminoacyl transfer chemistry.
This view of how aaRS achieve selectivity for cognate tRNA is reminiscent of models proposed to explain the fidelity of nucleotide selection by DNA polymerases, as well as selection of aminoacyl-tRNA by the elongating ribosome. Recently, Johnson and colleagues proposed a new paradigm for DNA polymerase specificity based on the T7 DNA polymerase (Tsai and Johnson, 2006). For this highly accurate DNA polymerase, binding of a correct nucleotide is followed by a favorable isomerization (Kaccom ~410) and a fast (360 sec-1) polymerization rate. Similar to the mutant tRNAs described in the study here, selection against a mismatched nucleotide involves an initial thermodynamic selection (~ 10-fold increased KD), followed by kinetic control resulting from a slightly unfavorable isomerization step (Kaccom ~0.5), and a drastically (~2 × 103 -fold) reduced rate of the chemical step. This supports the original postulate that specificity can be expressed as the ratio (kcat/Km) for correct and mismatched substrates (Fersht, 1999), but different elementary steps in the cognate and near-cognate reactions contribute to these steady state terms (Tsai and Johnson, 2006). Fluorescence data from the T7 DNA polymerase system further suggests that complexes with a cognate nucleotide are structurally distinct from those with a mis-matched nucleotide. This is highly reminiscent of the mispositioned CCA end observed in the near cognate E. coli AspRS: yeast tRNAAsp complex (Moulinier et al., 2001). If the above comparison is valid, then identity defects should specifically compromise the rate of isomerization of the aaRS: tRNA complex, a model theoretically testable by stopped flow fluorescence assays.
By virtue of the induced fit that occurs in the decoding center following codon recognition (Ogle et al., 2002), and the differential rate of GTPase activation for true and near-cognates (Gromadski and Rodnina, 2004), near cognate tRNA selection by the ribosome during decoding shares some important fidelity preservation features with the model proposed here. In the ribosome, the GTPase activation step provides the kinetic basis of selection, while subsequent hydrolysis steps provide irreversibility. The ribosome, DNA polymerases, and many aaRSs also exhibit hydrolytic proofreading, a form of editing that overcomes the energetic limitations of kinetic proofreading (Fersht, 1999). In kinetic proofreading, a branched reaction scheme allows non-cognate intermediates to be preferentially dissociated and then decomposed (Hopfield, 1974). Kinetic directionality is provided by NTP hydrolysis, such that kinetic proofreading schemes can be operationally distinguished by excess consumption of ATP, or its equivalent. In agreement with the prediction of Fersht (Fersht, 1979), but in disagreement with Hoppfield (Yamane and Hopfield, 1977), we observed that neither mutants of tRNAHis nor non-cognate tRNAs elicited kinetic proofreading by HisRS (Figures 4 and 5). Thus, tRNA selection in this system must principally rely on the kinetic discrimination mechanisms described above that are not dependent on tRNA mediated stimulation of ATP hydrolysis. In the cases of both Q118E HisRS and C73U tRNAHis, an apparent failure to correctly orient the CCA acceptor end of the tRNA in the active site influences the kinetics of adenylate formation in the opposite active site. This may facilitate entry of adventitious solvent into the first active site, where it brings about solvolysis of the adenylate. Alternatively, active rounds of adenylate synthesis and decomposition may be stimulated in the second active site, a possibility currently being tested in ongoing experiments (E. Guth and C. Francklyn, unpublished). A similar process has been described as a ‘selective release’ model for pre-transfer editing in the proline system, although it is not strictly dependent on the presence of tRNA (Hati et al., 2006). The ‘selective release’ model may be less likely for HisRS, as the apparent rate of AMP production was significantly faster than the rate of adenylate hydrolysis in solution (Figure 5). Our observations more closely parallel the glutamine system, where the presence of tRNA is required for adenylate formation. Here, a 2’ deoxy variant of tRNAGln was found to elicit multiple rounds glutaminyl adenylate formation and hydrolysis, at rates which were significantly faster than the spontaneous rate of glutaminyl adenylate hydrolysis in solution (Gruic-Sovulj et al., 2005).
A unified model for the class II aaRS catalytic cycle
The rapid kinetics analysis of HisRS provides additional insights into the class II aaRS catalytic cycle that were not apparent from earlier steady state kinetic studies. As noted in our earlier work (Guth et al., 2005), a stoichiometry of one mole of adenylate was formed per mol of dimeric enzyme in single turnover experiments. In addition, the presence of transfer RNA equalizes the rate of amino acid activation in the two active sites, such that it apparently constitutes the rate determining step for aminoacylation under physiological conditions. A notable feature of the identity mutants reported here in both tRNA and enzyme is their burst of one [32P] AMP per dimer of synthesis under multiple turnover conditions (Figure 3). This burst is equivalent in amplitude to the synthesis of adenylate in the first subunit of wild type enzyme in the absence of tRNA. These observations suggest a fundamental difference between the first turnover of aminoacylation and subsequent turnovers. Class II aaRSs are, with only rare exceptions, dimers or multiples of dimers, and the coordination of catalysis in the two active sites remains a largely unexplored issue.
These findings and other observations can be incorporated into a unified model for the class IIa aaRS family reaction cycle that features explicit priming and multiple turnover phases (Figure 6). The cycle starts with the preferential synthesis of adenylate in one of the two active sites on the dimer, consistent with the stoichiometry of the adenylate complex isolated by size exclusion spin chromatography, and the burst amplitudes of ATP consumption of mutants under pre-steady state conditions (Figures 3). Next, tRNA preferentially binds to the subunit containing the adenylate (Bovee et al., 1999), and then undergoes a single round of aminoacyl transfer to produce a “primed complex” of one aminoacylated tRNAHis per dimer (notated in green in Figure 6). While this species could, in principle, undergo dissociation from the complex, the model that it remains bound better agrees with the results presented here and in prior work. Were the aminoacylated tRNA to dissociate at this step, the steady state rate of adenylate synthesis would not be subject to the tRNA’s influence, and complex stoichiometry would never exceed one tRNA bound per dimer. Both possibilities are discounted by prior kinetic and structural work. Accordingly, we propose that amino acid and ATP are recruited into site two (notated in red in Figure 6) while aminoacylated tRNA remains bound to site one. Following formation of adenylate in site two, tRNA is recruited to site two, constituting the first of two intervals in the overall cycle where two tRNAs are bound simultaneously. The next and critical step is the release of aminoacyated tRNA from site one, which is accompanied by the aminoacyl transfer of amino acid to the tRNA in site two. This step produces aminoacylated tRNA in site two, and the converse set of steps serves to regenerate a single aminoacylated tRNA in site one.
Figure 6.
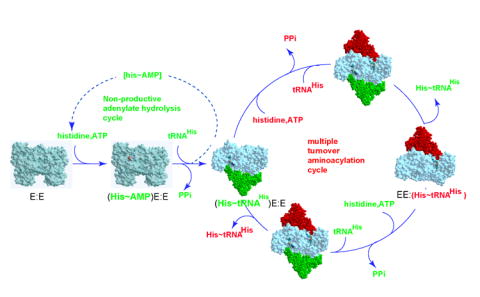
Alternating site catalysis model for the HisRS catalytic cycle. The HisRS dimer is depicted in surface representation, and the modeled, bound tRNA in CPK rendering. The reaction sequence presents initial formation of adenylate prior to tRNA binding, but there is no evidence that this order of substrate binding is obligatory. Interactions with monomer one are depicted in green, and substrate interactions with monomer two are depicted in red. Following formation of the first equivalent of aminoacylated tRNA (green), the model is rotated 90° to allow the occupancy of the ‘red’ and ‘green’ sites to be depicted separately. The step at which AMP is dissociated is not shown.
The specific order of substrate addition and product release for a number of steps remains to be determined. The rejection of near and non-cognate tRNAs, and the influence of identity, may be manifested in at least two different ways. Some tRNAs will lack the determinants for initial complex formation, and will be rejected rapidly owing to high dissociation rates. Other tRNAs, which can be considered as near cognates, will readily form initial encounter complexes, but will not be able to progress through accommodation and subsequent aminoacyl transfer. If this picture of tRNA identity is accurate, then similar kinetic blocks to transfer will be characteristic of strong identity mutations in other aaRS systems, regardless of their location on the tRNA or enzyme.
The unified model described above was developed primary by consideration of data from the histidine system, so its generality with respect to other class II enzymes is untested. In addition to HisRS, however, at least three other class II enzymes (AlaRS, ProRS, and ThrRS), exhibit a slower overall catalytic cycle than the rate of aminoacyl transfer, implying coupling across the dimeric interface (Zhang et al., 2006). Crystallographic analysis of a number of class II systems (including SerRS, ProRS, and ThrRS) also provides evidence for structural asymmetry in their active sites; some readily form complexes of one tRNA bound per dimmer (Cusack et al., 1996; Yaremchuk et al., 2000). Finally, the heterologous E. coli AspRS: yeast tRNAAsp complex provides an example of an apparently blocked complex in which the dramatically slowed aminoacylation of the first tRNA has apparently prevented accommodation of the second tRNA (Moulinier et al., 2001). The kinetic regimen described here provides an approach to examine these features dynamically, allowing the generality of structural asymmetry to be tested more broadly in other class II aaRS systems.
EXPERIMENTAL PROCEDURES
Preparation of enzyme and transfer RNA reagents
All variants of tRNAHis were produced by in vitro transcription, using T7 RNA polymerase (Connolly et al., 2004; Hawko and Francklyn, 2001). Transcription templates were produced by Fok I digestion of plasmid pJA15 harboring wild type tRNAHis or mutants generated by the Stratagene Quikchange mutagenesis kit (Stratagene, USA). The procedures employed for in vitro transcription reactions have been described previously (Hawko and Francklyn, 2001), and the gel fractionated transcripts were purified by electroelution. The concentrations of active tRNA were determined from plateau charging. The wild type and mutant HisRSs were expressed in E.coli, and purified utilizing a His6 tag and nickel-NTA affinity chromatography as described previously (Yan et al., 1996). The construction of the mutant enzymes is described previously (Connolly et al., 2004; Hawko and Francklyn, 2001) The concentrations of enzyme active sites were determined by use of the extinction coefficient for HisRS (127,097 M-1 cm-1) and active site titration. The suppliers of chemicals, general molecular biology enzymes, and other reagents were as cited previously.
Pre-steady state and single turnover transient kinetics
The single turnover kinetics of HisRS and tRNAHis mutants were determined under conditions of five-fold enzyme excess, as described in Guth et al. (Guth et al., 2005). Reactions with half lives on the millisecond time scale were sampled by use of a Kin-Tek RQF-3 chemical quench flow apparatus (Kin-Tek, Austin, Texas), while reactions operating on the multi-second time scale were manually sampled. The standard reaction buffer consisted of 50mM HEPES pH 7.5, 100mM KCl, 10mM MgCl2, and 1mM DTT. All single turnover reactions were performed by generating enzyme:histidyl-adenylate complex, followed by subsequent mixing with tRNA. Prior to analysis of the transfer reaction, the enzyme:adenylate complex was typically pre-formed by the incubation of HisRS (35.5 μM), ATP (2.5 mM), [U-14C] Histidine (100 μM) and 0.2U/mL inorganic pyrophosphatase at 37°C for 30 minutes. Some 90-95% of the enzyme-adenylate complex was routinely recovered by G-25 Sephadex size exclusion spin chromatography (Roche, Switzerland). The resulting enzyme-adenylate was rapidly mixed with tRNA in a Kin-Tek RQF-3 Rapid Quench Apparatus, quenching reactions with 3M NaOAc (pH 4.5). The collection tubes also contained 10 μL of 20% SDS to ensure dissociation of protein. The His-tRNAHis formed during the reaction was precipitated with trichloroacetic acid on Whatmann 3MM filter pads as described previously (Guth et al., 2005). For single turnover reactions, the aliquots of the reaction were typically collected out to at least 10-half lives of the reaction. Bench scale experiments allowed the determination of reaction progress from 10 seconds to two hours. In these experiments, aliquots of the reaction mixture were spotted directly on to 5% TCA soaked Whatmann 3 mm filter pads (Guth et al, 2005).
To observe the transition from the pre-steady state to steady state, enzyme (10 μM) and tRNA (100 μM) were pre-incubated in syringe one of the RQF-3, with ATP (200 μM or 6 mM), histidine (400 μM), and inorganic pyrophosphatase in the second syringe. The MgCl2 concentration in the buffer was increased to 18 mM in conjunction with 6 mM ATP. To monitor the accumulation of aminoacylated tRNA, 400 μM [U-14C] was included in syringe 2 along with saturating concentrations of ATP (6 mM), with the subsequent work up of product as for the single turnover assay. As subsequent washing steps remove unbound radiolabeled amino acid, the initial concentration of amino acid can be held well above saturation in the assay.
Alternatively, progress of the first reaction under multiple turnover conditions was monitored by the inclusion of [α-32P] labeled ATP in syringe two. In order to gain the sensitivity necessary to accurately determine the events occurring during the initial turnover of the enzyme, sub-saturating concentrations of 100 μM ATP were required. By virtue of the instability of the histidyl-adenylate during quenching and work-up, any [α-32P] AMP produced reflects the sum of adenylate remaining on the enzyme and AMP released into solution as product. The amount of AMP produced was measured by spotting 0.5 μL of the reaction on polyethyleneimmine thin layer chromatography (PEI-TLC) plates, using 0.75M NaH3PO4 as mobile phase to separate AMP and ATP. The fractional conversion of ATP to AMP on the TLC plates was determined by densitometry using a Bio-Rad K-screen and Bio-Rad FX personal molecular imaging system (Bio-Rad, USA). The resulting images were analyzed using the Bio-Rad Quantity One software package. To ensure that product formation rates of [U-14C] His~tRNAHis and [α-32P] AMP could be compared with confidence, the multiple turnover [U-14C] His~tRNAHis experiments were repeated at sub saturating concentrations of ATP (100 μM), and these rates are reported in column 7 of Tables 1 and 2.
Adenylate stability and decomposition assays were adapted from previously published studies (Gruic-Sovulj et al., 2005; Hati et al., 2006). The 50 μL adenylate kinetic stability reactions were composed of 50mM HEPES pH 7.5, 10mM MgCl2, 100mM KCl, 1mM DTT, 2.5 mM histidine, 10 μM ATP, 0.06 M α-P32 ATP, 0.02U/mL PPiase and 1 μM HisRS. Reactions were carried out at 37°C and initiated by addition of enzyme. 2 μL reaction aliquots were then removed from reactions containing no chase, or chase solutions consisting of 10 mM HSA (5’-O-[N-(L-histidyl)sulfamoyl]adenosine) or 10 mM cold ATP, and then quenched by addition to 8 μL of 0.2M NaOAc (pH 5) at various time points. Alternatively, the reactions were terminated by mixing with an equal volume of phenol:chloroform: isoamyl alcohol 25:24:1 (pH 6.7). To allow resolution of the adenylate, 1 μL aliquots of the quenched reaction were spotted onto PEI-TLC plates, resolved in 0.1M ammonium acetate and 5% acetic acid, and then imaged as described above. The position of adenylate on the thin layer chromatography was inferred from the mobility of HSA in the same solvent system. In order to assure the enzyme independent nature of adenylate hydrolysis, control reaction mixtures lacking enzyme were brought to 10mM in HSA, or added to an equal volume of phenol:chloroform:isoamyl alcohol pH 6.7 (25:24:1) after 10 minutes of incubation at 37°C and monitored as above.
Data Analysis
For single turnover experiments, progress curves were fit to a single exponential equation
| (1.1) |
where a is the y intercept offset to account for background radioactivity, b is the amplitude, k is the observed rate constant, and t is time. tRNA titrations were performed to ensure saturation of enzyme-adenylate during the first turnover. The progress curves for pre-steady state reactions were fit to either a linear or burst equation, where appropriate:
| (1.2) |
where a, b, k and t are as defined in eq (1.1) with the additional term c is the post-burst steady state rate and additional term d is the corresponding linear y intercept. Data acquired in experiments monitoring the accumulation and destruction of the adenylate were fitted to the equation:
| (1.3) |
where a is the y intercept offset, b and c are amplitudes of the respective exponential functions, k1 is the production of adenylate (more accurately the release of free adenylate into solution from a fractional subset of HisRS active sites) and k2 is the enzyme independent decomposition of adenylate in solution.
Supplementary Material
Acknowledgments
The authors thank Charles Carter Jr., for the plasmid encoding tRNATrp and insightful comments on the manuscript, and anonymous reviewers are acknowledged for suggesting the adenylate stability experiments. This work was supported by grants from NIGMS (GM54899) and NIEHS (T32-ES07122).
References
- Ardell DH, Andersson SG. TFAM detects co-evolution of tRNA identity rules with lateral transfer of histidyl-tRNA synthetase. Nucleic Acids Res. 2006;34:893–904. doi: 10.1093/nar/gkj449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnez JG, Augustine JG, Moras D, Francklyn CS. The first step of aminoacylation at the atomic level in histidyl-tRNA synthetase. Proc Natl Acad Sci U S A. 1997;94:7144–7149. doi: 10.1073/pnas.94.14.7144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belrhali H, Yaremchuk A, Tukalo M, Berthet-Colominas C, Rasmussen B, Bösecke P, Diat O, Cusack S. The structural basis for seryl-adenylate and Ap4A synthesis by seryl-tRNA synthetase. Structure. 1995;3:341–352. doi: 10.1016/s0969-2126(01)00166-6. [DOI] [PubMed] [Google Scholar]
- Bovee ML, Yan W, Sproat BS, Francklyn CS. tRNA discrimination at the binding step by a class II aminoacyl-tRNA synthetase. Biochemistry. 1999;38:13725–13735. doi: 10.1021/bi991182g. [DOI] [PubMed] [Google Scholar]
- Briand C, Poterszman A, Eiler S, Webster G, Thierry J-C, Moras D. An Intermediate Step in the Recognition of tRNAAsp by Aspartyl-tRNA synthetase. J Mol Biol. 2000;299:1051–1060. doi: 10.1006/jmbi.2000.3819. [DOI] [PubMed] [Google Scholar]
- Calender R, Berg P. Purification and physical characterization of tyrosyl-ribonucleic acid synthetases from Escherichia coli and Bacillus subtilis. Biochemistry. 1966;5:1681–1690. doi: 10.1021/bi00869a033. [DOI] [PubMed] [Google Scholar]
- Cavarelli J, Eriani G, Rees B, Ruff M, Boeglin M, Mitschler A, Martin F, Gangloff J, Thierry JC, Moras D. The active site of yeast aspartyl-tRNA synthetase: structural and functional aspects of the aminoacylation reaction. EMBO J. 1994;13:327–337. doi: 10.1002/j.1460-2075.1994.tb06265.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavarelli J, Rees B, Ruff M, Thierry JC, Moras D. Yeast tRNAAsp recognition by its cognate class II aminoacyl-tRNA synthetase. Nature. 1993;362:181–184. doi: 10.1038/362181a0. [DOI] [PubMed] [Google Scholar]
- Connolly SA, Rosen AE, Musier-Forsyth K, Francklyn CS. G-1:C73 recognition by an arginine cluster in the active site of Escherichia coli histidyl-tRNA synthetase. Biochemistry. 2004;43:962–969. doi: 10.1021/bi035708f. [DOI] [PubMed] [Google Scholar]
- Cusack S. Aminoacyl-tRNA synthetases. Current Opinions in Structural biology. 1998;7:881–889. doi: 10.1016/s0959-440x(97)80161-3. [DOI] [PubMed] [Google Scholar]
- Cusack S, Yaremchuk A, Tukalo M. The crystal structure of the ternary complex of T. thermophilus seryl-tRNA synthetase with tRNASer and a seryl-adenylate analogue reveals a conformational switch in the active site. EMBO J. 1996;15:2834–2842. [PMC free article] [PubMed] [Google Scholar]
- Ebel JP, Giegé R, Bonnet J, Kern D, Befort N, Bollack C, Fasiolo F, Gangloff J, Dirheimer G. Factors determining the specificity of the tRNA aminoacylation reaction. Non-absolute specificity of tRNA-aminoacyl-tRNA synthetase recognition and particular importance of the maximal velocity. Biochimie. 1973;55:547–557. doi: 10.1016/s0300-9084(73)80415-8. [DOI] [PubMed] [Google Scholar]
- Eiler S, Dock-Bregeon A, Moulinier L, Thierry JC, Moras D. Synthesis of aspartyl-tRNA(Asp) in Escherichia coli--a snapshot of the second step. Embo J. 1999;18:6532–6541. doi: 10.1093/emboj/18.22.6532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fersht A. Editing Mechanisms in the Aminoacylation of tRNA. Cold Spring Harbor Lab; 1979. [Google Scholar]
- Fersht AR. Specificity and Editing Mechanisms. New York: WH Freeman; 1999. [Google Scholar]
- Fersht AR, Kaethner MM. Mechanism of aminoacylation of tRNA. Proof of the aminoacyl adenylate pathway for the isoleucyl- and tyrosyl-tRNA synthetases from Escherichia coli K12. Biochemistry. 1976;15:818–823. doi: 10.1021/bi00649a014. [DOI] [PubMed] [Google Scholar]
- Fersht AR, Mulvey RS, Koch GLE. Ligand binding and enzymic catalysis coupled through subunits in tyrosyl-tRNA synthetase. Biochemistry. 1975;14:13–18. doi: 10.1021/bi00672a003. [DOI] [PubMed] [Google Scholar]
- Fromant M, Plateau P, Blanquet S. Function of the extra 5′-phosphate carried by histidine tRNA. Biochemistry. 2000;39:4062–4067. doi: 10.1021/bi9923297. [DOI] [PubMed] [Google Scholar]
- Giege R, Sissler M, Florentz C. Universal rules and idiosyncratic features in tRNA identity. Nucleic Acids Res. 1998;26:5017–5035. doi: 10.1093/nar/26.22.5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromadski KB, Rodnina MV. Kinetic determinants of high-fidelity tRNA discrimination on the ribosome. Mol Cell. 2004;13:191–200. doi: 10.1016/s1097-2765(04)00005-x. [DOI] [PubMed] [Google Scholar]
- Gruic-Sovulj I, Uter N, Bullock T, Perona JJ. tRNA-dependent aminoacyl-adenylate hydrolysis by a nonediting class I aminoacyl-tRNA synthetase. J Biol Chem. 2005;280:23978–23986. doi: 10.1074/jbc.M414260200. [DOI] [PubMed] [Google Scholar]
- Guth E, Connolly SH, Bovee M, Francklyn CS. A substrate-assisted concerted mechanism for aminoacylation by a class II aminoacyl-tRNA synthetase. Biochemistry. 2005;44:3785–3794. doi: 10.1021/bi047923h. [DOI] [PubMed] [Google Scholar]
- Hati S, Ziervogel B, Sternjohn J, Wong FC, Nagan MC, Rosen AE, Siliciano PG, Chihade JW, Musier-Forsyth K. Pre-transfer editing by class II prolyl-tRNA synthetase: Role of aminoacylation active site in “selective release” of noncognate amino acids. J Biol Chem. 2006 doi: 10.1074/jbc.M605856200. [DOI] [PubMed] [Google Scholar]
- Hauenstein S, Zhang CM, Hou YM, Perona JJ. Shape-selective RNA recognition by cysteinyl-tRNA synthetase. Nat Struct Mol Biol. 2004;11:1134–1141. doi: 10.1038/nsmb849. [DOI] [PubMed] [Google Scholar]
- Hawko SA, Francklyn CS. Covariation of a specificity-determining structural motif in an aminoacyl-tRNA synthetase and a tRNA identity element. Biochemistry. 2001;40:1930–1936. doi: 10.1021/bi0025316. [DOI] [PubMed] [Google Scholar]
- Himeno H, Hasegawa T, Ueda T, Watanabe K, Miura K, Shimizu M. Role of the extra G-C pair at the end of the acceptor stem of tRNA(His) in aminoacylation. Nucleic Acids Res. 1989;17:7855–7863. doi: 10.1093/nar/17.19.7855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopfield JJ. Kinetic proofreading : a new mechanism for reducing errors in biosynthesis processes requiring high specificity. PNAS. 1974;71:4135–4139. doi: 10.1073/pnas.71.10.4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibba M, Sever S, Praetorius-Ibba M, Soll D. Transfer RNA identity contributes to transition state stabilization during aminoacyl-tRNA synthesis. Nucleic Acids Res. 1999;27:3631–3637. doi: 10.1093/nar/27.18.3631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibba M, Soll D. Aminoacyl-tRNA synthesis. Annu Rev Biochem. 2000;69:617–650. doi: 10.1146/annurev.biochem.69.1.617. [DOI] [PubMed] [Google Scholar]
- Jakubowski H, Goldman E. Editing of errors in selection of amino acids for protein synthesis. Microbiol Rev. 1992;56:412–429. doi: 10.1128/mr.56.3.412-429.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam SSM, Schimmel PR. Equilibrium measurements of cognate and noncognate interactions between aminoacyl transfer RNA synthetases and transfer RNA. Biochemistry. 1975;14:2775–2780. doi: 10.1021/bi00683a034. [DOI] [PubMed] [Google Scholar]
- LaRiviere FJ, Wolfson AD, Uhlenbeck OC. Uniform binding of aminoacyl-tRNAs to elongation factor Tu by thermodynamic compensation. Science. 2001;294:165–168. doi: 10.1126/science.1064242. [DOI] [PubMed] [Google Scholar]
- Moulinier L, Eiler S, Eriani G, Gangloff J, Thierry JC, Gabriel K, McClain WH, Moras D. The structure of an AspRS-tRNA(Asp) complex reveals a tRNA-dependent control mechanism. Embo J. 2001;20:5290–5301. doi: 10.1093/emboj/20.18.5290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramatsu T, Nishikawa K, Nemoto F, Kuchino Y, Nishimura S, Miyazawa T, Yokoyama S. Codon and amino-acid specificities of a transfer RNA are both converted by a single post-transcriptional modification. Nature. 1988;336:179–181. doi: 10.1038/336179a0. [DOI] [PubMed] [Google Scholar]
- Normanly J, Abelson J. Transfer RNA Identity. Annual Review of Biochemistry. 1989;58:1029–1049. doi: 10.1146/annurev.bi.58.070189.005121. [DOI] [PubMed] [Google Scholar]
- Ogle JM, Murphy FV, Tarry MJ, Ramakrishnan V. Selection of tRNA by the ribosome requires a transition from an open to a closed form. Cell. 2002;111:721–732. doi: 10.1016/s0092-8674(02)01086-3. [DOI] [PubMed] [Google Scholar]
- Ogle JM, Ramakrishnan V. Structural insights into translational fidelity. Annu Rev Biochem. 2005;74:129–177. doi: 10.1146/annurev.biochem.74.061903.155440. [DOI] [PubMed] [Google Scholar]
- Rould MA, Perona JJ, D S. Structural basis of anticodon loop recognition by glutaminyl-tRNA synthetase. Nature. 1991;352:213–218. doi: 10.1038/352213a0. [DOI] [PubMed] [Google Scholar]
- Saks ME, Sampson JR, Abelson JN. The Transfer RNA Identity Problem: A Search for Rules. Science. 1994;263:191–197. doi: 10.1126/science.7506844. [DOI] [PubMed] [Google Scholar]
- Sampson JR, Behlen LS, DiRenzo AB, Uhlenbeck OC. Recognition of Yeast tRNAPhe by Its Cognate Yeast Phenylalanyl-tRNA Synthetase: An Analysis of Specificity. Biochemistry. 1992;31:4164–4167. doi: 10.1021/bi00132a002. [DOI] [PubMed] [Google Scholar]
- Swanson R, Hoben P, Sumner-Smith M, Uemura H, Watson L, Soll D. Accuracy of in vivo aminoacylation requires proper balance of tRNA and aminoacyl-tRNA synthetase. Science. 1988;242:1548–1551. doi: 10.1126/science.3144042. [DOI] [PubMed] [Google Scholar]
- Tsai YC, Johnson KA. A new paradigm for DNA polymerase specificity. Biochemistry. 2006;45:9675–9687. doi: 10.1021/bi060993z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uter NT, Perona JJ. Long-range intramolecular signaling in a tRNA synthetase complex revealed by pre-steady-state kinetics. Proc Natl Acad Sci U S A. 2004;101:14396–14401. doi: 10.1073/pnas.0404017101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamane T, Hopfield JJ. Experimental evidence for kinetic proofreading in the aminoacylation of tRNA by synthetase. Proc Natl Acad Sci U S A. 1977;74:2246–2250. doi: 10.1073/pnas.74.6.2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan W, Augustine J, Francklyn C. A tRNA identity switch mediated by the binding interaction between a tRNA anticodon and the accessory domain of a class II aminoacyl-tRNA synthetase. Biochemistry. 1996;35:6559–6568. doi: 10.1021/bi952889f. [DOI] [PubMed] [Google Scholar]
- Yan W, Francklyn C. Cytosine 73 is a discriminator nucleotide in vivo for histidyl-tRNA in Escherichia coli. J Biol Chem. 1994;269:10022–10027. [PubMed] [Google Scholar]
- Yaremchuk A, Cusack S, Tukalo M. Crystal structure of a eukaryote/archaeon-like protyl-tRNA synthetase and its complex with tRNAPro(CGG) Embo J. 2000;19:4745–4758. doi: 10.1093/emboj/19.17.4745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaremchuk A, Tukalo M, Grotli M, Cusack S. A succession of substrate induced conformational changes ensures the amino acid specificity of Thermus thermophilus prolyl-tRNA synthetase: comparison with histidyl-tRNA synthetase. J Mol Biol. 2001;309:989–1002. doi: 10.1006/jmbi.2001.4712. [DOI] [PubMed] [Google Scholar]
- Zhang CM, Perona JJ, Ryu K, Francklyn C, Hou YM. Distinct kinetic mechanisms of the two classes of Aminoacyl-tRNA synthetases. Journal of Molecular Biology. 2006;361:300–311. doi: 10.1016/j.jmb.2006.06.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.