

Abstract
A serine proteinase pathway in insect hemolymph leads to prophenoloxidase activation, an innate immune response against pathogen infection. In the tobacco hornworm Manduca sexta, recombinant hemolymph proteinase 14 precursor (pro-HP14) interacts with peptidoglycan, autoactivates, and initiates the proteinase cascade (Ji, C., Wang, Y., Guo, X., Hartson, S., and Jiang, H. (2004) J. Biol. Chem. 279, 34101–34106). Here, we report the purification and characterization of pro-HP14 from the hemolymph of bacteria-injected M. sexta larvae. The zymogen, consisting of a single polypeptide with a molecular mass of 68.5 kDa, is truncated at the amino terminus. It is converted to a two-chain active form in the presence of β-1,3-glucan (a fungal cell wall component) and β-1,3-glucan recognition protein-2. The 45-kDa heavy chain contains four low-density lipoprotein receptor A repeats, one Sushi domain, and one unique cysteine-rich region, whereas the 30-kDa light chain contains a serine proteinase domain, which was labeled by [³H]diisopropyl fluorophosphate. Pro-HP14 in the plasma strongly binds curdlan, zymosan, and yeast and interacts with peptidoglycan and Micrococcus luteus. Addition of autoactivated HP14 elevated phenoloxidase activity level in the larval plasma. Recombinant M. sexta serpin-1I reduced prophenoloxidase activation by inhibiting HP14. These data are consistent with the current model on initiation and regulation of the prophenoloxidase activation cascade upon recognition of pathogen-associated molecular patterns by specific pattern recognition proteins.
Innate immunity is essential for the wellbeing of vertebrates and invertebrates. Key features of this defense system include pattern recognition of infectious agents, stimulation of extra- and intracellular signaling pathways, immediate cellular/humoral responses, and induced synthesis of immune factors (e.g. antimicrobial peptides) (1–3). Serine proteinase cascades in plasma, such as the human complement system and horseshoe crab hemolymph coagulation pathway, mediate many of the defense responses (4, 5). In insects, extracellular serine proteinases generate spätzle, phenoloxidase (PO),2 and plasmatocyte spreading peptide via limited proteolysis (4, 6). In Drosophila, a proteinase cascade activates spätzle to induce antimicrobial peptide synthesis by the Toll pathway. While Persephone (a clip-domain serine proteinase) is a component of the cascade (7), its activating proteinase and protein substrate remain unclear. Neither is it known how Gram-positive bacteria trigger the cascade activation (8) or how melanization often associates with malfunctioning of this proteinase pathway (9–11).
Microbial infection or tissue damage often leads to proteolytic activation of prophenoloxidase (pro-PO) in insects. Active PO catalyzes the formation of quinones, reactive intermediates for melanization (12–14). Quinones may also participate in wound healing and sequestration of parasites or pathogens. To minimize the cytotoxicity of quinones to host tissues/cells, conversion of pro-PO to PO occurs as a local reaction involving a system of interacting proteins (e.g. pattern recognition receptors, serine proteinases, serine proteinase homologs, serpins, and pro-PO). Although some of these molecules have been characterized at the molecular level, the constituents, order, initiation, and regulation of the proteinase pathway are still poorly understood. Our current knowledge is mainly limited to pathogen recognition and pro-PO activation, the first and last steps of the cascade (6, 15).
Recently, we reported the cDNA cloning, recombinant expression, and functional elucidation of Manduca sexta HP14, an immune-responsive hemolymph proteinase (16). This Sushi-domain enzyme contains seven Cys-rich regulatory domains and a serine proteinase domain. Incubation with peptidoglycan leads to autoprocessing of the recombinant pro-HP14 and generation of an amidase activity. Supplementation of hemolymph with the zymogen enhanced pro-PO activation in response to Micrococcus luteus. These data suggest that pro-HP14 binds to the Gram-positive bacteria, autoactivates, and triggers the immune proteinase pathway in M. sexta. To further explore the physiological function of HP14, we purified and characterized pro-HP14 from the hemolymph of M. sexta larvae injected with microbes. In the presence of a fungal cell wall component and its recognition protein, autoproteolysis of pro-HP14 yielded active HP14 that enhanced pro-PO activation in the plasma. Its binding to microbial surface components and inhibition by M. sexta serpin-1I were also investigated.
EXPERIMENTAL PROCEDURES
Insect Rearing, Bacterial Challenge, and Hemolymph Collection
M. sexta eggs were purchased from Carolina Biological Supply. The larvae were reared on an artificial diet (17). Day 2, fifth instar larvae were injected with a mixture of formaldehyde-killed Escherichia coli (3 ✕ 107 cells), M. luteus (30 µg), and curdlan (30 µg) in 50 µl of H2O. Hemolymph was collected from cut prolegs of the larvae at 24 h after the immune challenge. Individual hemolymph samples were immediately mixed with equal volume of pH 7.0, 100% saturated (NH4)2SO4 to prevent melanization.
Proteins, Microorganisms, and Microbial Polysaccharides
M. sexta β-1,3-glucan recognition protein-2 (βGRP2), immulectin-2, and serpin-1I were isolated as described previously (18–20). M. luteus (2 ✕ 109 cells/ml), E. coli (2 ✕ 109 cells/ml), and Saccharomyces cerevisiae (5 ✕ 108 cells/ml) were treated with 0.4% formaldehyde at room temperature for 2 days and thoroughly washed with sterilized 0.85% NaCl. Curdlan (from Alcaligenes faecalis), laminarin (from Laminaria digitata), lipopolysaccharide (from E. coli O127:B8), lipoteichoic acid (from Streptococcus faecalis), peptidoglycan (from Staphylococcus aureus), and zymosan A (from S. cerevisiae) were purchased from Sigma. These microbial cells and cell-wall components were heated at 100 °C for 5 min to inactivate possible contaminating proteinases.
Purification of Pro-HP14 from Induced M. sexta Hemolymph
All procedures for pro-HP14 purification were carried out at 4 °C using buffers filtered through a 0.22-µm membrane. The induced hemolymph sample (55 ml, stored at −80 °C) was thawed as 50% (NH4)2SO4 suspension. After centrifugation at 17,400 ✕ g for 20 min, the protein precipitate was collected and dissolved in 50 ml of buffer A (1 mm benzamidine, 0.01% 1-phenyl-2-thiourea, 0.5 m NaCl, 10mm potassium phosphate, pH 6.8). The solution was centrifuged at 17,400 ✕ g for 30 min to remove flocculent materials. Saturated (NH4)2SO4 solution was slowly added to the supernatant to a final saturation of 35%. After centrifugation, the precipitate was collected and dissolved in 15 ml of buffer A.
For rapid removal of (NH4)2SO4, the protein sample was passed through a Sephadex G25 column (2.5 cm i.d. ✕ 16 cm) containing buffer A. Protein fractions were pooled (~40 ml) and applied to a hydroxylapatite column (2.5 cm inner diameter ✕ 7 cm, Bio-Rad) equilibrated with the same buffer. After washing with 100 ml of buffer A, bound proteins were eluted with a linear gradient of 10–150 mm potassium phosphate in buffer A at 0.5 ml/min for 6 h. Fractions were analyzed by SDS-PAGE and immunoblotting using HP14 antibody. The flow-through fractions were combined and precipitated with 50% saturated (NH4)2SO4. Following centrifugation, the pellet was dissolved in 3 ml of buffer B (pH 7.5, 20 mm Tris-HCl, 0.5 m NaCl, 0.01% Tween 20, and 1 mm benzamidine) and separated on a Sephacryl S100-HR column (2.5 cm inner diameter ✕ 120 cm) equilibrated with buffer B.
The pro-HP14 fractions were combined, supplemented with 1 mm CaCl2 and MgCl2, and loaded onto a concanavalin A-Sepharose column (5 ml). After washing, pro-HP14 was eluted from the lectin column with buffer B containing 1 mm CaCl2, 1 mm MgCl2, and 0.4 m methyl-α-D-mannopyranoside. Eluted proteins were diluted with 10 volumes of buffer C (0.01% Tween 20, 2 mm benzamidine, 10 mm potassium phosphate, pH 6.4) and applied to a dextran sulfate-Sepharose CL-6B column (5 ml) (21). After washing with 30 ml of buffer C, a linear gradient of 0–1.0 m NaCl in buffer C was employed to elute the bound proteins at 1 ml/min for 30 min. Twenty fractions of 1.5 ml were collected and analyzed by SDS-PAGE. The purified pro-HP14 was stored at −80 °C before use. Prior to functional analyses, the purified protein was passed through a Sephadex G25 column (1.5 cm inner diameter ✕ 15 cm) equilibrated with pH 7.5, 20 mm Tris-HCl, 20 mm NaCl to remove benzamidine.
Characterization of M. sexta Pro-HP14
MALDI-TOF mass spectrometry of pro-HP14 was carried out at Nevada Proteomics Center. Briefly, the purified protein (2 µg) was separated by 10% SDS-PAGE under reducing condition and stained with Coomassie Blue. Individual protein bands were excised from the gel, treated with 100 mm iodoacetamide, and digested with sequencing-grade trypsin (25 µl and 5 ng/µl) (Promega) in 25 mm ammonium bicarbonate at 37 °C for 16 h (22). In a parallel experiment, gel slices containing 2 µg of pro-HP14 were reacted with iodoacetamide and then 100 ng of Glu-C (Princeton Separation) at 30 °C for 16 h. After desalting with a Zip Tip C18 (Millipore), the peptides were analyzed on an ABI4700 MALDI-TOF/TOF mass spectrometer (Applied Biosystems). Mass data were acquired in reflector mode within the range of 800–4,000 Da, and 2500 laser shots were averaged for each mass spectrum. Molecular masses of the peptides from pro-HP14 were calculated (prospector.ucsf.edu/) and compared with those of observed monoisotopic peaks to locate their positions. The isoelectric point of pro-HP14 was measured on a precast PhastGel IEF-9 (Amersham Biosciences) (23).
Cleavage Activation of Pro-HP14 Induced by Recognition of Microorganisms
Purified pro-HP14 (200 ng, 10 µl), microbial cells, or microbial cell wall components (10 µg, 1 µl), corresponding recognition proteins (40 ng, 2 µl), CaCl2 (100 mm, 1 µl), and buffer D (20 mm Tris-HCl, 20 mm NaCl, 5 mm CaCl2, pH 8.0, 2 µl) were incubated at 37 °C for 1 h. The reaction mixtures (8 µl) were analyzed by 12% SDS-PAGE followed by silver staining or immunoblotting to reveal possible proteolytic cleavage.
[³H]Diisopropyl Fluorophosphate Labeling of the Active Site Serine Residue in HP14
Pro-HP14 (200 ng, 10 µl) was incubated with curdlan (10 µg, 1 µl), βGRP2 (40 ng, 2 µl), [³H]DFP (1 µl, 0.1 mm, 10mCi/mmol, PerkinElmer Life Sciences), CaCl2 (100 mm, 1 µl), and buffer D (2 µl) at 37 °C for 1 h. The reaction mixture (17 µl), along with 14C-labeled Mr standards, was resolved by 12% SDS-PAGE under reducing condition. After 2,5-diphenyloxazole treatment (24), the gel was dried and exposed to an x-ray film for 30 days at −80 °C prior to development.
Optimization of Conditions for Pro-HP14 Autoactivation
The general conditions for curdlan-induced pro-HP14 activation were: purified pro-HP14 (200 ng, 10 µl), curdlan (10 µg, 1 µl), βGRP2 (40 ng, 2 µl), CaCl2 (100 mm, 1 µl), and buffer D (2 µl) incubated at 37 °C for 1 h. One of these conditions was changed as specified in Fig. 3 to test the effects of βGRP2/curdlan amount, incubation time/temperature/pH, and NaCl concentration. To assess cleavage extent, the reaction mixtures (8 µl) were separated by 10% SDS-PAGE under reducing condition and visualized by silver staining. The effect of pH was investigated by replacing buffer D with 3 µl, 1:5 diluted Polybuffer 96 (Amersham Biosciences) adjusted to various pH.
FIGURE 3. Optimization of the reaction conditions for pro-HP14 autoprocessing.

The general conditions for pro-HP14 autoactivation were: pro-HP14 (10 µl and 20 ng/µl), curdlan (1 µl and 10 µg/µl), and βGRP2 (2 µl and 20 ng/µl) incubated with 20 mm Tris-HCl, pH 7.8, 20 mm NaCl, 5 mm CaCl2 at 37 °C for 90 min. Incubation time (A), βGRP2 amount (B), NaCl concentration (C), or curdlan amount (D) was changed in each experiment. The reaction mixtures were separated by 12% SDS-PAGE under reducing condition and visualized by silver staining. a–d, see Fig. 2 legend.
Determination of the Proteolytic Cleavage Site in HP14
HP14 generated under the optimal conditions was resolved by SDS-PAGE and transferred to a polyvinylidene difluoride membrane. After staining with Ponceau S (Sigma), the catalytic chain of HP14 was subjected to automated Edman degradation at Harvard Microchemistry Facility.
Binding of Plasma Pro-HP14 to Microorganisms
Following 0–35% ammonium sulfate fractionation and centrifugation, hemolymph (0.5 ml) from the injected larvae was dissolved in 200 µl of 20 mm Tris-HCl, 0.5 m NaCl, 1 mm CaCl2, 1 mm MgCl2, 0.01% 1-phenyl-2-thiourea, pH 7.5. The plasma fraction was incubated with M. luteus (2 ✕ 109 cells), E. coli (2 ✕ 109 cells), S. cerevisiae (5 ✕ 107 cells), or microbial cell wall components (1 mg and 100 µl) at room temperature for 30 min. After centrifugation at 13,000 ✕ g for 1 min, the supernatants were removed. The pellets were washed five times with 50 mm Tris-HCl, 5 mm CaCl2, pH 7.8. Bound proteins were eluted with 40 µl of 2 ✕ SDS sample buffer at 100 °C for 5 min. As controls, the microbial cells/polysaccharides only were treated similarly. The supernatants, bound proteins, and control samples were subjected to 10% SDS-PAGE and immunoblot analysis using 1:2000 diluted HP14 antiserum as the first antibody. Distribution of pro-HP14 in the supernatant and pellet was estimated by comparing relative intensities of the immunoreactive bands.
Binding of Purified Pro-HP14 to Microorganisms
Purified pro-HP14 (1 µg of 50 µl) and CaCl2 (100 mm of 5 µl) were incubated with microbial cells or cell-surface components in the presence or absence of their binding proteins (200 ng and 10 µl) at 37 °C for 60 min. The total volume was adjusted to 65 µl with buffer D. As described above, the supernatants and bound proteins were obtained and subjected to SDS-PAGE followed by silver staining or immunoblot analysis. Microbial cells or cell-wall components, pro-HP14, and recognition proteins were included as controls.
Stimulation of Pro-PO Activation System by HP14
Pro-HP14 (1 µg and 50 µl), curdlan (1-mg pellet), βGRP2 (200 ng and 10 µl), and CaCl2 (100 mm and 5 µl) were incubated at 37 °C for 90 min. After centrifugation, 2 µl of the supernatant was incubated with plasma (1:10 diluted in buffer D, 5 µl) and buffer D(10 µl) on ice for 20 min. PO activity in the reaction mixture was assayed using dopamine as a substrate (25). The preincubation mixture without pro-HP14 or curdlan-βGRP2 was used as negative controls.
Inhibition of HP14-induced PO Activation by Serpin-1I
To detect the formation of an SDS-stable complex of HP14 and serpin-1I, purified pro-HP14 (200 ng and 10 µl), serpin-1I (3 µg and 1 µl), curdlan (10 µg and 1 µl), βGRP2 (40 ng and 2 µl), and CaCl2 (100 mm and 1 µl) were incubated at 37 °C for 90 min. In the controls, pro-HP14 or serpin-1I was replaced by the same volumes of buffer D. The reaction mixture and negative controls (10 µl) were separated by 7.5% SDS-PAGE and subjected to immunoblot analysis using 1:1000 diluted serpin-1 antiserum. To examine the effect of serpin-1I on pro-PO activation induced by HP14, the active proteinase was generated under the optimal conditions. After centrifugation, supernatant (2 µl), serpin-1I (3 µg and 1 µl) and buffer D (9 µl) were incubated on ice for 10 min. Induced plasma (5 µl and 1:10 diluted) was added to the HP14-serpin-1I mixture and further incubated on ice for 20 min prior to PO activity assay. A mixture of the supernatant (2 µl), buffer D (10 µl), and induced plasma (5 µl and 1:10 diluted) was included as a positive control, whereas plasma only was a negative control.
RESULTS
Purification and Characterization of M. sexta Pro-HP14 from the Induced Larval Hemolymph
Using polyclonal antiserum against HP14, we detected a protein doublet in the hemolymph, whose level increased after an immune challenge (16). The immunoreactive proteins with apparent molecular masses of 75 and 67 kDa were mostly found in 0 –35% ammonium sulfate fraction of the induced plasma. After desalting and buffer exchange, we resolved the fractionated hemolymph by hydroxylapatite chromatography. The flow-through contained the large protein, whereas the bound part included the small one (Fig. 1, A and B). Because the secreted recombinant pro-HP14 had an apparent Mr of ~75,000 (16), we decided to isolate pro-HP14 from the unbound fractions, representing ~20% of the total proteins loaded onto the hydroxylapatite column. Following a concentration step, the pooled flow-through fractions were separated by gel-filtration chromatography on a Sephacryl S100-HR column. About 85% of the proteins were removed in this step. The combined pro-HP14 fractions were loaded onto a concanavalin A-Sepharose column. After nearly half of the loaded proteins were removed, pro-HP14 and other glycoproteins were eluted from the lectin column. The pooled pro-HP14 fractions were resolved by ion-exchange chromatography onto a dextran sulfate column. Pro-HP14 eluted from the column at ~0.25 m NaCl. With ~7 ✕ 10³-fold purification, 179 µg of pro-HP14 was isolated from 55 ml of induced hemolymph. As judged by SDS-PAGE analysis, the proenzyme was essentially pure, and it migrated to the 75-kDa position under reducing condition (Fig. 1B).
FIGURE 1. SDS-PAGE analysis of pro-HP14 isolated from the hemolymph of M. sexta larvae injected with bacteria.
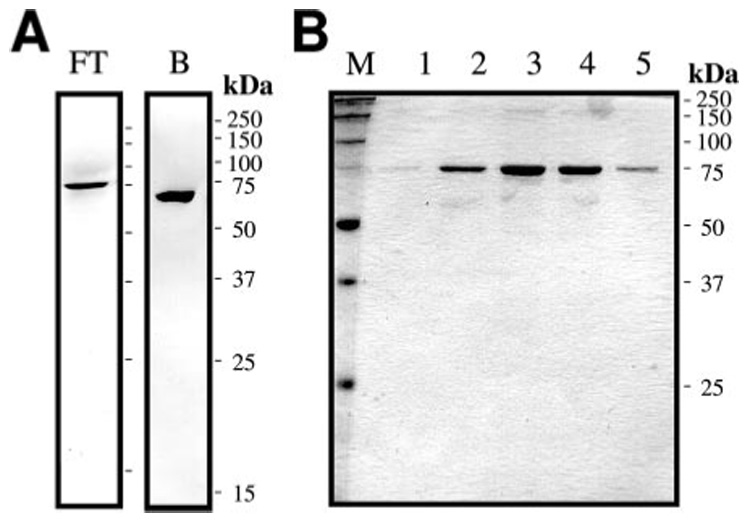
A, electrophoretic separation and immunoblot detection of pro-HP14 in 12 µl of flow-through fraction 18 (left) and bound fraction 54 (right) from the hydroxylapatite column. B, SDS-PAGE and silver staining of the 75-kDa pro-HP14 eluted from the dextran sulfate column. The fractions 9–13 (12 µl each, lanes 1–5) were treated with SDS sample buffer containing dithiothreitol, separated on a 12% SDS-PAGE gel, and stained with silver.
We determined by MALDI-TOF mass spectrometry that the molecular mass of “75-kDa” pro-HP14 from the induced plasma was actually 68,545 ± 69 Da, smaller than the theoretical value of the mature proenzyme (residues 1–649, 71,774 Da) (16). Peptide mass fingerprint analysis indicated that 28 predicted trypsinolytic fragments (800–4,000 Da) were apparent on the mass spectrum, covering 59% of the sequence from residue 68 to the carboxyl terminus (Table 1). The identification of 644FWTDEY649 demonstrated that the proenzyme was intact at the carboxyl terminus and, therefore, truncated at the amino terminus to account for the mass reduction. Due to amino-terminal blocking, automated Edman degradation failed to yield any sequence from pro-HP14. However, we detected two peptides (760.34 and 949.46 Da) that are identical in mass to pyroGlu62LSNCR67 (760.337 Da, carboxy-amidomethyl Cys (CAM-C)) and 68ISQWQCK74 (949.457 Da, CAM-C). Right before Gln62, there is a recognition site of intracellular processing enzymes: Arg58-Ser-Arg-Arg61 (26).
TABLE 1.
Trypsinolytic peptides from 75-kDa pro-HP14 matched with the deduced sequence
| Calculated mass | Observed massa | Δ | Start | End | Missed cleavage | Sequence |
|---|---|---|---|---|---|---|
| 949.4566 | 949.4631 | −0.0065 | 68 | 74 | 0 | (R) ISQWQCK (D) |
| 1112.4682 | 1112.4788 | −0.0106 | 75 | 84 | 0 | (K) DGSCINFDGK (C) |
| 3284.3353 | 3284.2832 | 0.0521 | 75 | 103 | 1 | (K) DGSCINFDGKCDGIVDCPDASDETHALCR (E) |
| 2190.8854 | 2190.8821 | 0.0033 | 85 | 103 | 0 | (K) CDGIVDCPDASDETHALCR (E) |
| 1486.6650 | 1486.6813 | −0.0163 | 104 | 113 | 1 | (R) ERQCQYNWFR (C) |
| 1201.5213 | 1201.5363 | −0.0150 | 106 | 113 | 0 | (R) QCQYNWFR (C) |
| 2987.2280 | 2987.2517 | −0.0237 | 115 | 143 | 1 | (R) CTYGACVDGTAPCNGVQDCADNSDELLPR (R) |
| 1877.7394 | 1877.7474 | −0.0080 | 167 | 183 | 0 | (K) HCDGVADCADGSDETLR (S) |
| 2973.2640 | 2973.2065 | 0.0575 | 189 | 213 | 1 | (K) TCLSYLFQCAYGACVDKDSDCNGIR (E) |
| 3148.5093 | 3148.4790 | 0.0303 | 230 | 258 | 1 | (R)NTSVQPVKEGACVLPEYPEHGGYVVSGMOXK(N) |
| 2279.0477 | 2279.0491 | −0.0014 | 238 | 258 | 0 | (K) EGACVLPEYPEHGGYVVSGMK (N) |
| 2295.0426 | 2295.0591 | −0.0165 | 238 | 258 | 0 | (K)EGACVLPEYPEHGGYVVSGMOXK(N) |
| 2049.9493 | 2049.9507 | −0.0014 | 285 | 301 | 0 | (R) NDVTCTNGWWNFTPLPK (C) |
| 990.4896 | 990.4926 | −0.0030 | 310 | 317 | 0 | (K) EHVSVEYK (C) |
| 1197.5686 | 1197.5807 | −0.0121 | 318 | 328 | 0 | (K) CVLSGSSQGFR (T) |
| 1405.6752 | 1405.6886 | −0.0134 | 346 | 356 | 0 | (R) KPNYYSETTFR (Y) |
| 1789.8406 | 1789.8472 | −0.0066 | 357 | 372 | 0 | (R) YMSCIGAVGAWNYVAK (C) |
| 1708.8699 | 1708.8761 | −0.0062 | 394 | 408 | 0 | (R) AQFGELPWQAGIYTK (N) |
| 979.5100 | 979.5131 | −0.0031 | 444 | 452 | 0 | (K) EEYAVALGK (L) |
| 1087.6053 | 1087.6149 | −0.0096 | 453 | 460 | 0 | (K) LYRPWQPK (M) |
| 1802.9553 | 1802.9653 | −0.0100 | 469 | 483 | 1 | (K) SEIRDIHISPYFLGR (T) |
| 1317.6955 | 1317.7108 | −0.0153 | 473 | 483 | 0 | (R) DIHISPYFLGR (T) |
| 1093.5893 | 1093.5852 | 0.0041 | 520 | 529 | 0 | (K) EQLYVGSLGK (V) |
| 1868.9870 | 1868.9882 | −0.0012 | 530 | 547 | 1 | (K) VAGWGIKDEAGNPSQVLK (V) |
| 1157.5802 | 1157.5829 | −0.0027 | 537 | 547 | 0 | (K) DEAGNPSQVLK (V) |
| 2895.4715 | 2895.4326 | 0.0389 | 551 | 575 | 0 | (K) LPYVDVLQCISQSPQAFRPYITGDK (I) |
| 1333.6501 | 1333.6622 | −0.0121 | 589 | 602 | 0 | (K) GDSGGGLSFPAVNR (L) |
| 860.3467 | 860.3444 | 0.0023 | 644 | 649 | 0 | (R) FWTDEY (−) |
Monoisotopic mass in daltons. The starting and ending numbers for individual tryptic peptides were based on the deduced amino acid sequence of mature pro-HP14 (16).
Was pro-HP14 truncated somewhere before Gln62, and, after trypsin cleavage at Arg61, the exposed Gln62 spontaneously became pyroGlu62? Although it is possible, evidence suggests this is not the case: 1) there is no other known processing site in the first LDLr A repeat or in the following linker (residues 44–65); 2) the first Gln in 106QCQYNWFR113 did not become pyroGlu106 under the same condition (Table 1); 3) Glu-C digestion generated a peptide (1805.835 Da) with amass identical to that of pyroGlu62LSNCRISQWQCKD (1805.802 Da, 2 CAM-C). Therefore, the 75-kDa pro-HP14 starts at pyroGlu62 and Gln62-pyro-Glu62 conversion occurred before trypsin digestion.
The calculated mass of pro-HP14 (residues 62–649) is 65,183 Da, smaller than the experimental value (68,545 Da). Because pro-HP14 binds concanavalin A, glycosylation appears to be responsible for the mass difference of 3,362 Da. We predict that the post-translational modification occurs at N145ET, N383GT, and/or N582GT, because peptide masses containing these were not found (Table 1). The isoelectric point of pro-HP14 was determined to be 5.7, slightly higher than the calculated value (5.5) from its amino acid sequence (residues 62–649).
Cleavage Activation of Pro-HP14 Induced by Microorganisms and Their Binding Proteins
To test whether or not microorganisms could activate pro-HP14, we incubated purified pro-HP14 with various microbial cells or cell-surface molecules in the presence/absence of their binding proteins. SDS-PAGE analysis indicated that pro-HP14 was cleaved after incubating with yeast, curdlan, or zymosan A in the presence of βGRP2 (Table 2). However, laminarin (a soluble form of β-1,3-glucan) did not lead to pro-HP14 cleavage activation. E. coli did not cause such a change with or without immulectin-2, and neither did M. luteus. Whereas incubation of pro-HP14 with peptidoglycan did lead to pro-HP14 cleavage, the efficiency was lower than with β-1,3-glucan and βGRP2.
TABLE 2.
Effect of microbial cells or cell wall components on autoactivation of pro-HP14
| Microbial cells or surface molecules | Binding protein | Proteolysis of pro-HP14 |
|---|---|---|
| E. coli | Immulectin-2 | − |
| Lipopolysaccharide | Immulectin-2 | − |
| Peptidoglycan | − | + |
| M. luteus | βGRP2 | − |
| Lipoteichoic acid | βGRP2 | − |
| S. cerevisiae | βGRP2 | + |
| Laminarin | βGRP2 | − |
| Curdlan | βGRP2 | + |
| Zymosan | βGRP2 | + |
Proteolytic processing of pro-HP14 occurred when insoluble β-1,3-glucan and βGRP2 were present at the same time (Fig. 2A). Because pro-HP14 cleavage did not happen after incubation with curdlan or βGRP-2 alone, binding-induced autoactivation (rather than a contaminating proteinase) is responsible for the limited proteolysis. Cleaved HP14 was separated into 45- and 30-kDa bands by SDS-PAGE under reducing condition, whereas, under nonreducing condition, HP14 migrated as a single band at ~70 kDa (Fig. 2B). This agrees with our prediction based on the deduced sequence and domain structure of HP14: an interchain disulfide bond links the 30- and 45-kDa polypeptides (16). Although the 45-kDa heavy chain was blocked at the amino terminus, we determined the first ten amino acid residues of the light chain as: Val-Leu-Gly-Gly-Glu-Arg-Ala-Gln-Phe-Gly. This perfectly matched the sequence after the predicted proteolytic activation site (Gly-Thr-Glu-Leu*Val-Leu-Gly-Gly). Affinity labeling by [³H]DFP demonstrated that the light chain was catalytically active (Fig. 2C). In other words, the 30-kDa band represents the proteinase domain at the carboxyl terminus.
FIGURE 2. Autoactivation of pro-HP14 upon exposure to β-1,3-glucan and βGRP2.

A, the purified pro-HP14 (200 ng) was incubated with 10 µg of curdlan and 40 ng of βGRP2 at 37 °C for 2 h. After being treated with SDS sample buffer containing dithiothreitol, the reaction mixture and controls were subjected to 12% SDS-PAGE and silver staining. B, a duplicate reaction mixture separated by SDS-PAGE under nonreducing condition. C, to label the active site Ser residue, the reaction mixture was incubated at 37 °C in the presence of [³H]DFP (1 µl, 0.1 mm, and 10 mCi/mmol). The labeling mixture was resolved by 12% SDS-PAGE under reducing condition and subjected to fluorography (24). a, 75-kDa pro-HP14; b, βGRP2;, c, 45-kDa HP14 heavy chain; and d, 30-kDa HP14 light chain.
We optimized the reaction conditions for pro-HP14 activation. At 15 min after 200 ng of pro-HP14 was incubated with 10 µg of curdlan and 10 ng of βGRP2, nearly half of the proenzyme was converted to the two-chain active HP14 (Fig. 3). The cleavage reaction was >90% complete at 45 min. Further increase in βGRP2 or curdlan level had little effect on the proteolytic activation. Ionic strength of the reaction mixture negatively impacted the molecular interactions: 80% of the cleavage was blocked in the presence of 200 mm NaCl. KCl, Na2SO4, and NaAc had a similar effect (data not shown). The optimum pH for pro-HP14 autoactivation was ~7.5–8.5, and the cleavage was more complete at 37 °C than 25 °C (data not shown). At the physiological pH (6.7) and ionic strength (equivalent of 200 mm NaCl) (27), the proteolysis in the larval plasma is estimated to occur at 15% of the optimal rate determined in vitro.
Binding of Pro-HP14 to Microorganisms in the Presence or Absence of Plasma Proteins
To better understand the role of pro-HP14 in immune responses, we tested its binding with various microbial cells or cell-surface molecules using 0–35% ammonium sulfate fraction of the plasma. In the presence of high salt and 1-phenyl-2-thiourea, pro-HP14 activation and protein cross-linking were prevented. Pro-HP14 in the fractionated hemolymph bound to the Gram-positive bacteria and yeast but not E. coli (Fig. 4B). Peptidoglycan and β-1,3-glucan appeared to be responsible for the binding. Although the reduction of pro-HP14 in M. luteus- and peptidoglycan-treated supernatants were relatively small, strong interactions between pro-HP14 and curdlan/zymosan completely depleted the proenzyme in the solution phase (Fig. 4A). The binding of pro-HP14 to S. cerevisae greatly reduced the proenzyme in the supernatant.
FIGURE 4. Association of microbes and their cell wall components with pro-HP14 in the fractionated plasma.

Aliquots of 0–35% ammonium sulfate fraction of the induced hemolymph were incubated with M. luteus (lane 1), peptidoglycan (lane 2), E. coli (lane 3), zymosan (lane 4), yeast (lane 5), or curdlan (lane 6) at room temperature for 30 min. After centrifugation at 13,000 ✕ g for 1 min, the supernatants (A) were removed for SDS-PAGE under reducing condition. The pellets were washed five times with 50 mm Tris-HCl, 5mm CaCl2, pH 7.8. Bound proteins (B) were eluted from the pellet by SDS sample buffer at 100 °C for 5 min and resolved by SDS-PAGE. Electrotransfer and immunoblot analysis were performed to detect pro-HP14 using its antibodies. Some of the signal (panel B, lane 1) came from the cross-reactivity with a co-migrating molecule from M. luteus.
We further examined whether the purified pro-HP14 can directly associate with microbes in the presence or absence of their recognition proteins. Immunoblot analysis showed that most pro-HP14 remained in supernatants treated with E. coli, M. luteus, or S. cerevisae (Fig. 5A and Table 3). Because supplementing the mixtures with immulectin-2 or βGRP2 did not increase the amount of binding, other plasma proteins must have contributed to the binding of pro-HP14 to M. luteus or yeast (Fig. 4). In the presence of βGRP2, zymosan or curdlan caused larger reduction of pro-HP14 in the supernatants than yeast did (Fig. 5A). This reduction was partly due to the proteolytic processing of pro-HP14. Although βGRP2 did not seem to enhance the binding of pro-HP14 to S. cerevisae, its coexistence with curdlan induced the proteolytic processing of pro-HP14 (Fig. 5B).
FIGURE 5. Binding of purified pro-HP14 to microbes and their surface molecules.
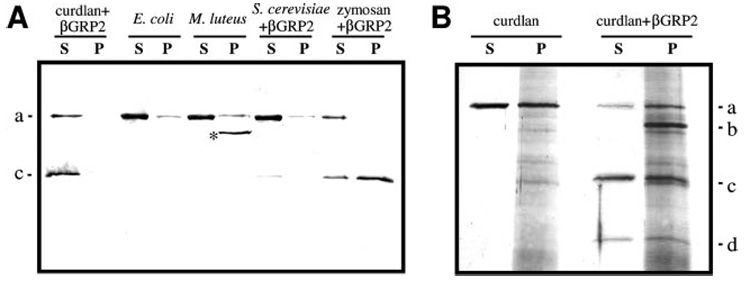
Purified pro-HP14 (1 µg) was incubated with 1 mg of heat-treated microbial cells in the presence or absence of their binding proteins (200 ng) in 70 µl, 20 mm Tris-HCl, 20 mm NaCl, 5 mm CaCl2, pH 7.8, at 37 °C for 60 min. After centrifugation at 13,000 ✕ g for 1 min, the supernatants (S) were recovered for SDS-PAGE analysis. The pellets were washed five times with 20 mm Tris-HCl, 5 mm CaCl2, pH 7.8, and the bound proteins (P) were eluted by treating the pellets with SDS sample buffer at 100 °C for 5 min. A, detection of pro-HP14 or HP14 binding by immunoblot analysis. For background reduction, 1:1000 diluted HP14 antiserum was preincubated with lysates of E. coli, M. luteus, and S. cerevisae.*, unknown; note that the HP14 polyclonal antibodies hardly recognize the 30-kDa catalytic domain (16). B, association of pro-HP14 with curdlan examined by SDS-PAGE and silver staining. a–d, see Fig. 2 legend.
TABLE 3.
Binding of pro-HP14 to microbes or their surface polysaccharides
| Curdlan | S. cerevisiae | Zymosan | M. luteus | E. coli | Peptidoglycan | ||
|---|---|---|---|---|---|---|---|
| % | |||||||
| Pro-HP14 in plasmaa | 0 | 25 | 0 | 66 | 100 | 33 | |
| Purified pro-HP14b | 82 | 97 | 97 | 94 | 95 | Not determined |
Binding of pro-HP14 in plasma is represented as % of the total proenzyme in the supernatant. The relative levels of pro-HP14 were determined by immunoblotting and densitometry of the protein band.
Binding of pro-HP14 to microbial cells or cell-wall components is represented as % of the total purified zymogen in the unbound fraction.
Initiation of Pro-PO Activation System in the Larval Hemolymph by Autocleaved HP14
To test whether or not HP14 triggers the pro-PO activation pathway in the plasma, we first used curdlan and βGRP2 to completely activate pro-HP14 under the optimal conditions (Fig. 3). After removing the elicitor and associated βGRP2 by centrifugation, the supernatant containing the two-chain HP14 was added to plasma from the naïve or bacteria-injected larvae. After incubation, high levels of PO activity developed in the control and induced hemolymph samples (Fig. 6). The enzyme activities (0.06 and 1.21 units) were low in the plasma controls and became slightly higher when pro-HP14 was added (0.32 and 1.59 units), thus, the proteinase zymogen had only a small effect on pro-PO activation. In another control, after the supernatant of curdlan and βGRP2 mixture was incubated with the two plasma samples, higher PO activities (0.52 and 3.18 units) were detected, suggesting that the supernatant may contain trace amounts of curdlan and βGRP2 to activate the proteinase cascade. Nevertheless, such activity increases were much lower than those in HP14-treated plasma samples (20.5 and 43.4 units). These results are consistent with the conclusion that HP14 initiated the pro-PO activation system in the hemolymph.
FIGURE 6. Activation of pro-PO by autocleaved HP14.
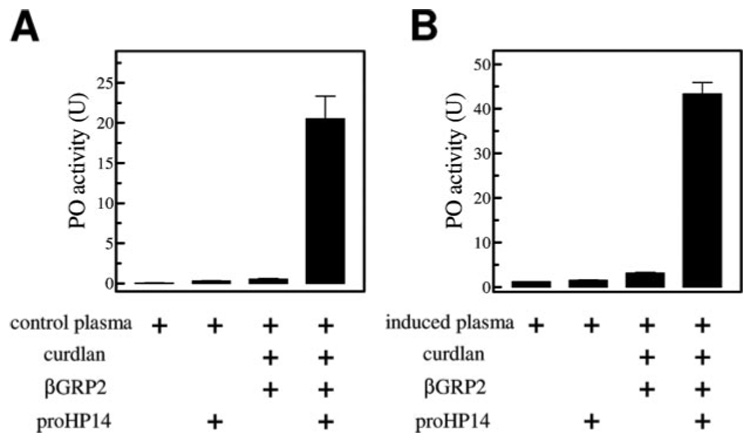
Purified pro-HP14 (1 µg), 1 mg of curdlan, and 200 ng of βGRP2 were incubated at 37 °C for 90 min in 20 mm Tris-HCl, 20 mm NaCl, 5 mm CaCl2, pH 8.0. After centrifugation, 2 µl of the supernatant was incubated on ice with 17.5 µl of buffer and 0.5 µl of plasma samples from naïve (A) or bacteria-injected (B) larvae. Plasma alone, plasma and pro-HP14, as well as a mixture of plasma, curdlan, and βGRP2 were included as controls. After 20 min, PO activity was determined using dopamine as a substrate (25) and plotted as mean ± S.E. (n = 3) in the bar graph.
Inhibition of HP14 by Serpin-1I
Although [³H]DFP labeling, pro-HP14 auto-processing, and pro-PO activation demonstrated HP14 is an active serine proteinase, we failed to detect its amidase activity using Ala-Ala-Pro-Leu-p-nitroanilide, as we did with peptidoglycan-activated recombinant HP14 (16). To further demonstrate that HP14 is an active proteinase, we decided to test if HP14 could form an SDS-stable complex with serpin-1I. Among the 12 variants of M. sexta serpin-1, serpin-1I has a reactive site sequence (AL*SL) most similar to the proteolytic activation site for HP14 (EL*VL). After curdlan-activated HP14 was incubated with recombinant serpin-1I, we detected a complex using serpin-1 antibodies (Fig. 7A). The immunoreactive band, absent in the controls of serpin-1I and HP14 only, had an apparent Mr (~75 kDa) close to that predicted for the complex formed between the HP14 catalytic domain and serpin-1I. No such complex was formed with serpin-1J, a negative control with an Arg at the P1 position (data not shown). We further examined the effect of serpin-1I on pro-PO activation induced by active HP14. As shown in Fig. 6, supplementation of active HP14 greatly enhanced pro-PO activation in the induced hemolymph. Preincubation of activated HP14 with serpin-1I significantly suppressed pro-PO activation (Fig. 7B).
FIGURE 7. Down-regulation of pro-PO activation by serpin-1I, an HP14 inhibitor.

A, to detect the covalent complex of HP14 and serpin-1I, 200 ng of purified pro-HP14, 10 µg of curdlan, and 40 ng of βGRP2 were incubated with 3 µg of serpin-1I in pH 8.0, 20 mm Tris-HCl, 5 mm CaCl2 at 37 °C for 90 min. The reaction mixture (lane 3) was analyzed by immunoblot analysis using 1:1000 diluted serpin-1 antiserum as the first antibody. The activation mixture (lane 1) and inhibitor alone (lane 2) were run as negative controls. B, to examine the effect of serpin-1I on pro-PO activation triggered by HP14, 1 µg of pro-HP14 was first activated by 1 mg of curdlan, and 200 ng of βGRP2 in the presence of 5 mm CaCl2 at 37 °C for 90 min. After centrifugation, the supernatant (2 µl) and serpin-1I (3 µg and 1 µl) were reacted on ice for 10 min in 20 mm Tris-HCl, 5 mm CaCl2, pH 8.0. Plasma (0.5 µl) from naïve larvae was then added to the HP14-serpin-1I mixture or HP14 (positive control). Plasma alone and a mixture of plasma and serpin-1I were used as negative controls. After 20 min on ice, PO activities in the reaction and controls were measured and plotted as mean ± S.E. (n = 3).
DISCUSSION
Innate immunity in vertebrates and invertebrates is activated by host proteins recognizing conserved surface determinants of pathogens (28). Upon binding, these proteins stimulate plasma factors and blood cells to immobilize and kill the invading microorganisms. Although many defense reactions are mediated by extracellular serine proteinase pathways (4, 5), limited information is available on how pathogen recognition leads to proteinase activation. A current model suggests that the binding of recognition proteins to a microbial surface induces a conformational change required for interacting with the first proteinase zymogen, leading to its autoactivation and pathway initiation (4, 12, 29). The lectin-mediated pathway for complement activation in mammals is an example for this model: mannose-binding protein and mannose-binding protein-associated serine proteinase (MASP) trigger the serine proteinase cascade (30). In this report, we provide biochemical evidence that the pathway for pro-PO activation in a lepidopteran insect is initiated in similar manner: β-1,3-glucan and βGRP2 together induced pro-HP14 autoactivation (Fig. 2), and active HP14 by itself started the pro-PO activation cascade (Fig. 6). These similarities in the pro-PO activation pathway in arthropods and the complement system in vertebrates suggest that proteinase autoactivation to trigger innate immune responses is an ancient evolutionary adaptation for defense against infection (4, 14).
A few known arthropod initiation serine proteinases greatly differ in their domain and subunit organizations. The β-1,3-glucan-responsive proteinases, M. sexta HP14 and Tachypleus tridentatus factor G, trigger pro-PO activation and hemolymph coagulation cascades, respectively (4, 5). In the insect, recognition of β-1,3-glucan is performed by a separate protein (βGRP2), whereas in the horseshoe crab, a subunit of factor G executes the same function. This subunit, which associates with the proteinase subunit, is encoded by a separate gene. In contrast, the horseshoe crab factor C consists of a single polypeptide. This lipopolysaccharide-responsive enzyme, by strictest definition, is a pattern-recognition proteinase. Although T. tridentatus factor C and M. sexta HP14 both contain a Sushi domain and a catalytic domain, the types of other structural modules (e.g. LDLr A repeat, C-type lectin) are strikingly different, and, unlike Factor C, pro-HP14 does not autotivate in the presence of lipopolysaccharide.
The HP14 zymogen produced by baculovirus-infected insect cells had two forms: the 87-kDa pro-HP14 was primarily intracellular, whereas the 75-kDa one was mostly in the medium (16). With an apparent Mr similar to the purified hemolymph pro-HP14, the latter may also start with pyroGlu62. Indeed, the 75-kDa recombinant protein was blocked at its amino terminus and intact at the carboxyl terminus. In light of these findings, we further speculate that the 87-kDa protein represents the full-length pro-HP14 with five LDLr A repeats. Because the 87-kDa protein was not detected in M. sexta larval plasma, the function of its first LDLr A repeat seems dispensable. Orthologs of HP14 in D. melanogaster, A. gambiae, and Apis mellifera contain four instead of five LDLr A repeats at the amino terminus. On the other hand, the 67-kDa protein in plasma may represent pro-HP14 with the first two LDLr A repeats removed, antibodies specific for LDLr, Sushi, 7C, or proteinase domain all recognized this band.3 Structural and functional comparisons of this form of pro-HP14 with the 75-kDa pro-HP14 are under active investigation.
The structure-function relations of human MASPs have shed light on our analysis of M. sexta HP14. Corresponding to the amino-terminal LDLr A repeats, the CUB-EGF-CUB region in MASP-1 and MASP-2 is responsible for interacting with mannose-binding protein and for forming homodimer (31). The following two Sushi domains in MASP-2 stabilize the structure of the serine proteinase domain at the carboxyl terminus and are directly involved in the enzyme autoactivation (32). In HP14, the second Sushi domain does not exist: between the Sushi and proteinase domains, there is another cysteine-rich region (16). Perhaps, the last (or seventh) Cys in this region forms the interchain disulfide bond with Cys510 in the catalytic domain. If proven true, the part that contains the first six cysteine residues may represent the prototype of a unique domain structure stabilized by three disulfide bonds. We tentatively call that “Wonton domain.” Structural and functional analyses are required to test if the Sushi and Wonton domains take part in the auto-activation of HP14 and interaction with its substrate.
One important feature of the pro-HP14 autoproteolysis induced by curdlan and βGRP2 is the high specificity: a single cleavage at the predicted activation site yielded two polypeptides (Fig. 3). In contrast, after recombinant pro-HP14 had been incubated with peptidoglycan, a series of low Mr bands appeared in a time-dependent manner (16). This apparent low specificity could be caused by the lack of particular plasma factors in the activation reaction. In fact, formation of a macromolecular complex containing HP14 and other immune proteins was observed in M. sexta larval plasma incubated with microbial surface components (33, 34). Consequently, further studies are needed to test if other proteins (e.g. peptidoglycan recognition proteins) are required for specific cleavage activation of pro-HP14 by peptidoglycan. Such experiments would further establish the physiological role of HP14 as a common entry point for responses against Gram-positive bacteria and fungi.
We demonstrated that HP14 is an active proteinase that can be trapped in a covalent complex with a suicidal inhibitor (Fig. 7). Although this is not to say that HP14 is the physiological target of serpin-1I (because, even at a high concentration, serpin-1I failed to completely block pro-PO activation), it did suggest a possible regulatory mechanism for pathway initiation. Moreover, the Leu residue preceding the cleavage site in serpin-1I (and HP14) provided important clues for elucidating the protein substrate of HP14. Because HP14 is a chymotrypsin-like enzyme preferring to cleave after Leu, we could test the ability of HP14 to activate several M. sexta hemolymph proteinase precursors (pro-HP2, pro-HP16, pro-HP19, pro-HP21, and pro-HP22) that have a Leu at the putative activation site (35). This will be our next step toward elucidating the pro-PO activation system in M. sexta.
Acknowledgments
We thank Drs. M. R. Kanost, M. Gorman, J. Dillwith, and A. Mort for critical review of the manuscript.
Footnotes
This work was supported by National Institutes of Health Grant GM58634. The article was approved for publication by the Director of Oklahoma Agricultural Experimental Station and supported in part under project OKLO2450.
The abbreviations used are: PO and pro-PO, phenoloxidase and its precursor; βGRP2, β-1,3-glucan recognition protein-2; HP14 and pro-HP14, hemolymph proteinase 14 and its precursor; MALDI-TOF, matrix-assisted laser desorption ionization-time of flight; DFP, diisopropyl fluorophosphate; CAM-C, carboxyamidomethyl cysteine; LDLr, low-density lipoprotein receptor; MASP, mannose-binding protein-associated serine proteinase.
Y. Wang and H. Jiang, unpublished data.
REFERENCES
- 1.Gillespie JP, Kanost MR, Trenczek T. Annu. Rev. Entomol. 1997;42:611–643. doi: 10.1146/annurev.ento.42.1.611. [DOI] [PubMed] [Google Scholar]
- 2.Hoffmann JA. Nature. 2003;426:33–38. doi: 10.1038/nature02021. [DOI] [PubMed] [Google Scholar]
- 3.Lavine MD, Strand MR. Insect Biochem. Mol. Biol. 2002;32:1295–1309. doi: 10.1016/s0965-1748(02)00092-9. [DOI] [PubMed] [Google Scholar]
- 4.Jiang H, Kanost MR. Insect Biochem. Mol. Biol. 2000;30:95–105. doi: 10.1016/s0965-1748(99)00113-7. [DOI] [PubMed] [Google Scholar]
- 5.Iwanaga S, Kawabata S, Muta T. J. Biochem. 1998;123:1–15. doi: 10.1093/oxfordjournals.jbchem.a021894. [DOI] [PubMed] [Google Scholar]
- 6.Kanost MR, Jiang H, Yu X. Immunol. Rev. 2004;198:97–105. doi: 10.1111/j.0105-2896.2004.0121.x. [DOI] [PubMed] [Google Scholar]
- 7.Ligoxygakis P, Pelte N, Hoffmann JA, Reichhart JM. Science. 2002;297:114–116. doi: 10.1126/science.1072391. [DOI] [PubMed] [Google Scholar]
- 8.Michel T, Reichhart JM, Hoffmann JA, Royet J. Nature. 2001;414:756–759. doi: 10.1038/414756a. [DOI] [PubMed] [Google Scholar]
- 9.De Gregorio E, Han SJ, Lee WJ, Baek MJ, Osaki T, Kawabata S, Lee BL, Iwanaga S, Lemaitre B, Brey PT. Dev. Cell. 2002;3:581–592. doi: 10.1016/s1534-5807(02)00267-8. [DOI] [PubMed] [Google Scholar]
- 10.Ligoxygakis P, Pelte N, Ji C, Leclerc V, Duvic B, Belvin M, Jiang H, Hoffmann JA, Reichhart JM. EMBO J. 2002;21:6330–6337. doi: 10.1093/emboj/cdf661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Levashina EA, Langley E, Green C, Gubb D, Ashburner M, Hoffmann JA, Reichhart JM. Science. 1999;285:1917–1919. doi: 10.1126/science.285.5435.1917. [DOI] [PubMed] [Google Scholar]
- 12.Ashida M, Brey PT. In: Molecular Mechanisms of Immune Responses in Insects. Brey PT, Hultmark D, editors. London: Chapman & Hall; 1998. pp. 135–172. [Google Scholar]
- 13.Nappi AJ, Vass E. Adv. Exp. Med. Biol. 2001;484:329–348. doi: 10.1007/978-1-4615-1291-2_33. [DOI] [PubMed] [Google Scholar]
- 14.Nappi AJ, Christensen BM. Insect Biochem. Mol. Biol. 2005;35:443–459. doi: 10.1016/j.ibmb.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 15.Cerenius L, Söderhäll K. Immunol. Rev. 2004;198:116–126. doi: 10.1111/j.0105-2896.2004.00116.x. [DOI] [PubMed] [Google Scholar]
- 16.Ji C, Wang Y, Guo X, Hartson S, Jiang H. J. Biol. Chem. 2004;279:34101–34106. doi: 10.1074/jbc.M404584200. [DOI] [PubMed] [Google Scholar]
- 17.Dunn P, Drake D. J. Invert. Pathol. 1983;41:77–85. [Google Scholar]
- 18.Jiang H, Kanost MR. J. Biol. Chem. 1997;272:1082–1087. doi: 10.1074/jbc.272.2.1082. [DOI] [PubMed] [Google Scholar]
- 19.Yu X, Kanost MR. J. Biol. Chem. 2000;275:37373–37381. doi: 10.1074/jbc.M003021200. [DOI] [PubMed] [Google Scholar]
- 20.Jiang H, Ma C, Lu Z, Kanost MR. Insect Biochem. Mol. Biol. 2004;34:89–100. doi: 10.1016/j.ibmb.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 21.Wang Y, Jiang H. Insect Biochem. Mol. Biol. 2004;34:731–742. doi: 10.1016/j.ibmb.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 22.Finhout E, Lee K. Electrophoresis. 2003;24:3508–3516. doi: 10.1002/elps.200305615. [DOI] [PubMed] [Google Scholar]
- 23.Ji C, Wang Y, Ross J, Jiang H. Protein Exp. Purif. 2003;23:328–337. [Google Scholar]
- 24.Skinner MK, Griswold MD. Biochem. J. 1983;209:281–284. doi: 10.1042/bj2090281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang H, Wang Y, Yu XQ, Kanost MR. J. Biol. Chem. 2003;278:3552–3561. doi: 10.1074/jbc.M205743200. [DOI] [PubMed] [Google Scholar]
- 26.Bergeron F, Leduc R, Day R. J. Mol. Endocrinol. 2000;24:1–22. doi: 10.1677/jme.0.0240001. [DOI] [PubMed] [Google Scholar]
- 27.Jungreis AM, Jatlow P, Wyatt GR. J. Insect Physiol. 1973;19:225–233. doi: 10.1016/0022-1910(73)90235-7. [DOI] [PubMed] [Google Scholar]
- 28.Medzhitov R, Janeway CA., Jr Science. 2002;296:298–300. doi: 10.1126/science.1068883. [DOI] [PubMed] [Google Scholar]
- 29.Krem MM, Di Cera E. Trends Biochem. Sci. 2002;27:67–74. doi: 10.1016/s0968-0004(01)02007-2. [DOI] [PubMed] [Google Scholar]
- 30.Matsushita M, Fujita T. Immunol. Rev. 2001;180:78–85. doi: 10.1034/j.1600-065x.2001.1800107.x. [DOI] [PubMed] [Google Scholar]
- 31.Thielens NM, Cseh S, Thiel S, Vorup-Jensen T, Rossi V, Jensenius JC, Arlaud GJ. J. Immunol. 2001;166:5068–5077. doi: 10.4049/jimmunol.166.8.5068. [DOI] [PubMed] [Google Scholar]
- 32.Gal P, Harmat V, Kocsis A, Bian T, Barna L, Ambrus G, Vegh B, Balczer J, Sim RB, Naray-Szabo G, Zavodszky P. J. Biol. Chem. 2005;280:33435–33444. doi: 10.1074/jbc.M506051200. [DOI] [PubMed] [Google Scholar]
- 33.Tong Y, Jiang H, Kanost MR. J. Biol. Chem. 2005;280:14932–14942. doi: 10.1074/jbc.M500532200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zou Z, Jiang H. J. Biol. Chem. 2005;280:14341–14348. doi: 10.1074/jbc.M500570200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang H, Wang Y, Gu Y, Guo X, Zou Z, Scholz F, Trenczek TE, Kanost MR. Insect Biochem. Mol. Biol. 2005;35:931–943. doi: 10.1016/j.ibmb.2005.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]