

Abstract
Pyramidal relay neurons in limbic cortex are vulnerable to denervation lesions, i.e. pyramidal neurons in layer IIα of piriform cortex undergo transsynaptic apoptosis after lesions that interrupt their inputs from the olfactory bulb. We have previously established the role of inhibitory interneurons in elaborating signals that lead to the apoptosis of projection neurons in these lesion models, i.e. the upregulation of neuronal NOS and release of nitric oxide. Thus, we have proposed that cortical interneurons play an essential role in transducing injury to degenerative effects for nearby pyramidal neurons. In the present study, we extend the previous findings to a toxic model of degeneration of pyramidal neurons in the adult paralimbic cortex, i.e. after exposure to the NMDA channel blocker MK801. Our findings indicate that treatment of adult rats with MK801 in doses previously found to cause alterations in pyramidal neurons of the retrosplenial cortex (5 mg/kg) results in an active caspase 3 (+), ultrastructurally apoptotic type of cell death involving the same projection neurons of layer IIα that are also vulnerable to bulbotomy lesions. Interneurons of layer I are induced by MK801 treatment to higher levels of nNOS expression and the selective nNOS inhibitor BRNI ameliorates pyramidal cell apoptosis caused by MK801. Our results indicate that certain pyramidal neurons in piriform cortex are very sensitive to NMDA blockade as they are to disconnection from modality-specific afferents and that inhibitory interneurons play significant roles in mediating various types of pro-apoptotic insults to cortical projection neurons via nNOS/NO signaling.
Keywords: glutamate, excitotoxicity, limbic system, neuronal cell death, olfactory, GABA
INTRODUCTION
We have previously demonstrated the vulnerability of a population of layer II neurons in piriform cortex to lesions that disconnect this cortical area from modality-specific afferents originating in the olfactory bulb (Capurso et al., 1997). These neurons are modified pyramidal cells that are located superficially in layer II and are often described as semilunar neurons based on their shape after Golgi impregnation. We have also shown that transsynaptic apoptosis of these pyramidal neurons is triggered, at least in part, by the upregulation of neuronal nitric oxide synthase (nNOS) in small GABAergic interneurons located superficially in piriform cortex and the subsequent release of nitric oxide (Koliatsos et al., 2004). In addition, we have found that one of the upstream steps of nNOS induction is glutamate binding to AMPA-kainate receptors that are enriched in these nNOS (+) interneurons (Zhou et al., 2004). In separate studies, Olney and colleagues have shown that pyramidal neurons in paralimbic cortex, primarily retrosplenial cortex, undergo degenerative changes consisting of widespread argyrophilia and some ultrastructural evidence of cytoplasmic vacuolization and necrosis after treatment with NMDA antagonists. The effects of the prototypical non-competitive N-methyl-D-aspartate (NMDA) channel blocker MK801, a compound with broad anticonvulsant, sympathomimetic, and anxiolytic properties as well as protective effects in models of anoxia-ischemia have been a major focus of attention (Wozniak et al., 1998; Fix et al., 1993). A closer examination of data published on the effects of MK801 shows that piriform cortex is also part of the degenerative response and that argyrophilic neurons appear throughout the antero-posterior extent of this paralimbic cortical area (Wozniak et al., 1998), although their ultrastructural pathology is unclear.
The present study was designed to bridge concepts from the above two experimental lines and, specifically, to examine the morphology of degenerating neurons in piriform cortex after treatment with MK801 and to explore the hypothesis that nNOS induction and activation of layer I interneurons may be a common denominator linking lesion- and NMDA antagonist-induced death of pyramidal neurons in limbic cortex. Our findings indicate that treatment of adult rats with the NMDA antagonist MK801 replicates an active caspase 3 (+), ultrastructurally apoptotic type of cell death involving the same projection neurons of layer IIα that are also vulnerable to bulbotomy lesions. Within the MK801 dose regimen used in our experiments (5 mg/kg), we found that piriform cortex is selectively vulnerable to the apoptotic effects of the drug. We also found that MK801 treatment results in higher levels of nNOS expression in layer I interneurons and that selective nNOS inhibitors block MK801-induced apoptosis of pyramidal neurons.
MATERIALS AND METHODS
Animals and Experimental Design
Experimental subjects were adult female (250-275g) Sprague–Dawley rats obtained from Charles River Laboratories (Wilmington, MA) and handled according to protocols approved by the Institutional Animal Care and Use Committee of the Johns Hopkins Medical Institutions. Female rats were chosen because they are markedly more sensitive to the effects of MK801 as reported by others (Wozniak et al., 1998) and also confirmed by our own pilot investigations. This report is based on five complementary experiments. The first experiment explores the effects of MK801 on layer II pyramidal neurons in piriform cortex 24 or 72 hours after treatment; control animals are treated with saline vehicle (n=5 per group, total n=20); the single outcome here is the magnitude of apoptotic cell death in layer II pyramidal neurons assessed by stereology. The second experiment involves the morphological confirmation of apoptosis by electron microscopic analysis or active caspase-3 immunocytochemistry (ICC) and utilizes MK801 or vehicle-treated subjects (n=3 per group) allowed to survive 24 or 72 hours after treatment. The third experiment explores the expression, by layer I interneurons, of the NR1 and NR2 NMDA receptor subunits and uses banked coronal sections through piriform cortex kept in an antifreeze solution in -20 °C for no more than 2-3 months. The fourth experiment addresses the induction, by MK801, of nNOS/NADPHd (+) neurons in layer I of piriform cortex by ICC and histochemistry and uses tissues obtained from animals in the first and fifth experiments. The fifth experiment investigates the ameliorating effect of the selective nNOS inhibitor 3-Bromo-7-Nitroindazole (BRNI) on MK801-induced degeneration of piriform pyramidal neurons 24 hours after MK801 treatment and involves two main treatment groups, the one treated with MK801 and the other with MK801-BRNI and three control groups treated with saline, DMSO and BRNI alone (n=5 per group, total n=25); as in the first experiment, the outcome is the magnitude of apoptotic cell death in layer II pyramidal neurons. All aspects of animal care and handling, including pharmacological treatments described here were carried out according to protocols approved by the Animal Care and Use Committee of the Johns Hopkins Medical Institutions.
Pharmacological Treatment with MK801 and BRNI
MK801 was given in a single dose of 5mg/kg in saline by i.p. injection. Higher doses had more than 60% mortality in our hands and were not further considered as practical experimental options. Animals were allowed to survive for 24 or 72 hours post treatment. Immediately after treatment and throughout the survival period, animals were closely supervised by a dedicated caretaker because of their profound paucity of movement and inability to eat or drink as well as loss of body heat. Rats were injected every few hours with 3 ml saline s.c. to avoid dehydration, were gently massaged to increase blood flow, and a heat lamp was used at regular intervals to maintain body temperature.
The selective nNOS inhibitor 3-Bromo-7-Nitroindazole (BRNI) (A. G. Scientific Co, San Diego, California) was given in a group of animals also treated with MK-801 in three doses of 20mg/kg i.p. in DMSO (n=10). BRNI was first given immediately after the MK801 injection and then again at 6 and 12 hours later. The main comparison group was treated with MK801. Control animals were treated with DMSO i.p. (n=5), BRNI (n=5) and saline (n=5). BRNI was kept in -20 °C and dissolved in DMSO immediately before use.
Histology, Histochemistry and Immunocytochemistry (ICC)
Twenty four or 72 hours after MK-801 treatment, rats were deeply anesthetized with sodium pentobarbital (50 mg/kg) and perfused through the ascending aorta with ice-cold PBS for 2 min, followed by neutral-buffered 4% paraformaldehyde/0.1M phosphate buffer for 20 minutes. Brain blocks containing the forebrain were postfixed overnight, treated with 30% sucrose till they sunk and sectioned coronally (40 μm) on a freezing microtome. Sections were stained in series for: cresyl violet; NADPH diaphorase histochemistry using the method of Vincent with minor modifications (Vincent et al., 1983); neuronal nitric oxide synthase (nNOS) ICC; and active caspase-3 ICC. All ICC reactions were based on the avidin-biotin-peroxidase (ABC) labeling protocol using commercially available kits (Vector Labs, Burlingame, CA) and goat anti-rabbit IgG as a linker (Vector Labs). Neuronal NOS and caspase-3 ICC each used a rabbit primary antiserum (1:10000, Santa Cruz, CA, and 1:500, Cell Signaling Technology, MA, respectively). Details on solvents, including buffers and detergents have been described elsewhere (Koliatsos et al., 1994). Sections were studied with a Zeiss Axiophot microscope and images were captured with a Spot RT Slider digital camera (Diagnostic Instruments Inc., Sterling Heights, MI) using software SPOT Advanced (version 3.5.7 for Windows) provided by the manufacturer.
NMDA glutamate receptor (NR) ICC utilized banked sections from normal brains sectioned at either the coronal or the saggital plane and stained with NR1 as well as NR2A/B antibodies for the NMDA receptor complex (AB1516 and AB1548 rabbit antisera, respectively; Chemicon). Primary antibody reaction was linked to avidin-biotin-peroxidase with goat anti-rabbit IgG (Vector Labs, Burlingame, CA) essentially as described (Koliatsos et al., 1994). A dual immunofluorescence protocol was also used to detect immunoreactivity for reelin, a marker of inducible nitrinergic interneurons in piriform cortex (Koliatsos et al., 2004) in conjunction with NR1 and NR2A/B immunoreactivities. Reelin immunoreactivity utilized the well-characterized monoclonal antibody CR-50 (1:500; a gift from Dr. Kazunori Nakajima, Jikei University, Tokyo, Japan) and was directly visualized with Cy2-conjugated goat anti-mouse IgG (1:100, Jackson ImmunoResearch Laboratories, West Grove, PA) emitting at the green range. NR1 and NR2A/B immunoreactivities (with primary antibodies diluted 1:500) were visualized with Cy3-conjugated goat anti-rabbit IgG (1:100; Jackson ImmunoResearch) emitting at the red range. Secondary incubations were performed for 2-4 hours at RT. Sections were counterstained with the nuclear dye DAPI, dehydrated, coversliped with DPX, and studied with a Zeiss Axiophot microscope equipped for epifluorescence. Images were captured with a Spot RT Slider digital camera as above. Because of a partial cytoplasmic overlap of NR1 and NR2 immunoreactivity with reelin, we performed confocal verification of the cytoplasmic localization of NR on a Zeiss LSM 410 unit. Rates of colocalization of NR within reelin (+) neurons were studied in three sections through the anterior piriform cortex corresponding to horizontal planes passing through the major island of Calleja caudally and through two additional levels one and two mm rostral to the previous level. All these sections were counterstained with DAPI for the appreciation of cytoarchitectonics.
Stereological estimates of apoptotic neurons in piriform cortex
The total number of apoptotic neurons in layer II of piriform cortex was estimated 24 and 72 hours after MK801 or vehicle treatment or 24 hours after MK801-BRNI or MK801-DMSO treatment, along with the appropriate control groups as laid out in the Animals and Experimental design section. Apoptotic neuron number was estimated with the optical fractionator probe as proposed by West and colleagues (1991). Nine serial 40 μm-thick, cresyl violet stained-sections were taken from each animal and studied on a Zeiss Axioplan photomicroscope equipped with a LEP motorized stage (Ludl Electronic Products, LTD, Hawthorne, NY) and connected to a Hitachi video camera and a computer fitted to operate the Stereo Investigator 5.0 software (Microbrightfield Inc. Williston, VT). Sections were collected starting at a random level in the beginning of the piriform cortex rostrally and then proceeding systematically (every 12th from the standard coronal series from each animal) to the level of the decussation of the anterior commissure caudally. Piriform cortex was outlined at 5x from the olfactory tubercle medially to just below the rhinal fissure laterally based on the characteristic cell density of layer II. Apoptotic profiles were identified at 100x in layer II by an investigator blinded to experimental record on the basis of cytological (nuclear and cytoplasmic condensation and repackaging of chromatin into single or multiple spheres) and histochemical (blue metachromasia) features of apoptosis on cresyl violet-stained material. Numbers of apoptotic profiles were estimated with a 40×35 mm optical fractionator (West et al., 1991). Stereologically estimated apoptotic profiles per individual subject were grouped as per experimental animal group and studied as outlined in the Experimental Design. Differences between groups were analyzed either with a two-tailed, unpaired Students’ t test when two groups were compared (as in the first experiment) or with one-way ANOVA followed by Bonferroni’s multiple-comparison testing when many groups were evaluated (in the fifth experiment).
The total number of nNOS cells in layer I 16 and 24 hours after MK801 or vehicle treatment were also counted using the optical fractionator, except that in this case counts were limited to layer I. Numbers of nNOS (+) layer I neurons were grouped per experimental history and differences between MK801 and vehicle groups were analyzed separately in the two time points with a two-tailed, unpaired Students’ t test.
Electron Microscopy
Ultrastructural details of degenerating neurons in piriform cortex were studied with electron microscopy as described (Liu et al., 2001). Briefly, Animals for EM were perfused with 30 ml of 0.1M PB (pH 7.4) at 37°C (1 min), followed by 2% glutaraldehyde and 2% paraformaldehyde in 0.1M PB at 37°C (10 min) and at 4°C (20 min). Brains were post-fixed in the glutaraldehyde/paraformaldehyde solution at room temperature for 2 hours. Piriform area was subdissected and post-fixed further at 4°C overnight. Tissue blocks were then treated with 1% osmium tetroxide and then 1% uranyl acetate before dehydration and embedding in Epon. Blocks were cut into semithin and thin sections. Semithin sections (1μm thick) were counterstained with Mallory’s trichrome (Multiple Stain Polyscience, Warrenton, PA). Thin sections were collected on grids without further treatment with uranyl acetate or lead citrate and examined with a Phillips CM120 transmission electron microscope.
RESULTS
MK801 causes apoptotic death of pyramidal neurons in piriform cortex
Treatment with MK-801 induces histological apoptosis in neurons located superficially in layer II of piriform cortex that is evident both at 1 and 3 days post-treatment (Fig. 1). Consistent with a progressive course of degeneration, apoptotic profiles in day 1 are featured primarily by nuclear and cytoplasmic condensation and chromatin repackaging, whereas day 3 profiles show extensive fragmentation into basophilic apoptotic bodies. Based on fractionator counts, we found 5,311 apoptotic neurons 1 day after MK-801 treatment, with no significant further increases by day 3. The corresponding apoptotic cell density data (apoptotic cell number/ total contour area) from the nine serial sections are consistent with our fractionator counts. In the control group, average apoptotic cell density is 0.35 ± 0.13 cells/mm2. One and three days after MK801 treatment, apoptotic cell density increases to 14.80 ± 2.60 and 10.91 ± 5.89 cells/mm2, respectively. The examination of semithin and thin sections further confirms the apoptotic morphology of neurons that degenerate in layer IIα after MK801 treatment (Fig. 2). All salient morphological features of apoptosis, including cytoplasmic and nuclear condensation, fragmentation of chromatin and cytoplasmic vacuolation, first in the periphery and then in the center of the cytoplasm, are present.
Figure 1. Histological demonstration of MK-801-induced apoptosis in the adult piriform cortex after MK801 treatment. Apoptotic profiles are concentrated in layer IIα, as per anatomical designations laid out in (A).

A. This coronal section is taken through the anterior septum from a normal rat and is used to show the boundaries (arrows) and layers of piriform cortex as applied in the present study.
B-C. Typical apoptotic profiles encountered in layer II at 24 (B) and 72 (C) hours after MK801 treatment. Chromatin condensation within shrunken, but intact perikarya is the hallmark presentation at 24 hours (B, arrows). In 72 hours, chromatin is fragmented and nuclear/perikaryal boundaries not always clear (C, arrows).
D. Bar diagram of differences in numbers of apoptotic neurons between MK801 and saline groups 24 and 72 hours post-treatment. Differences are significant at p <0.01 Size bars: A, 100μm; B, C, 20 μm
Abbreviations: aic, agranular insular cortex; ot, olfactory tubercle
Figure 2. Morphological features of degenerating neurons in piriform cortex 24 hours after MK801 treatment.
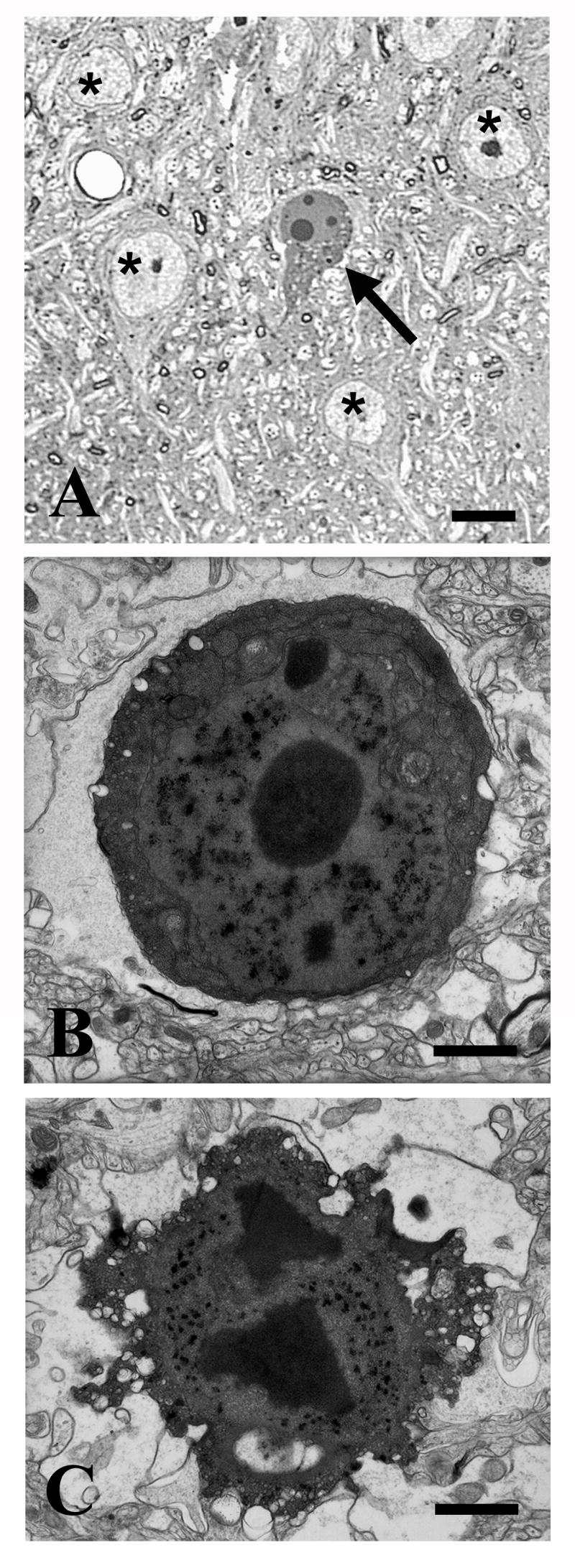
A-B. A representative semithin section (A) and an electron micrograph (B) illustrating early apoptotic changes in layer IIα pyramidal neurons including cytoplasmic and nuclear condensation, chromatin rearrangement in large and smaller spherical structures and peripheral cytoplasmic vacuolation (blebbing, B). Profiles indicated with asterisks in (A) represent healthy pyramidal neurons.
C. This electron micrograph illustrates a late apoptotic neuron with advanced condensation and some dissolution of structure, including both central and peripheral cytoplasmic vacuolation (bubbling).
Size bars: A, 10 μm; B, C, 1μm
Caspase-3 ICC using antibodies specific for the active form of the enzyme serves to further confirm the presence of apoptotic cell death but also to clarify the identity of dying neurons as the semilunar pyramids of superficial layer II. These nerve cells receive relatively exclusive inputs from the olfactory bulb and relay olfactory signals for further processing in other parts of the piriform cortex and the limbic system (Fig. 3). In our hands, specific active caspase-3 immunoreactivity is featured by cytoplasmic and dendritic staining, as contrasted with the nuclear background staining present in normal neurons and in vehicle-treated animals.
Figure 3. Activation of caspase-3 in layer IIa neurons 24 hours after MK801 treatment.

Note the cytoplasmic and dendritic immunoreactivity of layer IIα (semilunar) pyramids that is distinct from the nuclear background staining (arrows in lower panel; see more details in right-hand inset). Upper panel shows a plane through the piriform cortex of a vehicle-treated animal; only non-specific nuclear immunoreactivity in present.
Size bars: 100 μm
MK801 induces layer I interneurons in layer I of piriform cortex to higher levels of nNOS expression
The remarkable similarity in the morphology and anatomical specification of dying neurons between MK801-treated subjects used here and bulbotomized animals from previous experiments (Koliatsos et al., 2004) raises the issue of potential similar mechanisms signaling transsynaptic and neurotoxic apoptosis, including the activation of layer I small GABAergic interneurons that are induced by lesions to higher levels of expression of nNOS. The glycoprotein reelin is a sensitive and selective marker for these superficially located nerve cells (Koliatsos et al., 2004). To respond to MK801 treatment, these reelin (+) interneurons must be shown to express functional NMDA receptors comprised of NR1 and NR2 subunits (Kemp and McKernan, 2002). In addition, we must demonstrate that these neurons are induced 24-72 hours after MK801 treatment by either increased NADPHd histochemistry or nNOS immunoreactivity.
The examination of sections from normal animals stained with NR antibodies in single and dual ICC protocols demonstrates the enrichment of piriform cortex in NR immunoreactivity (Fig. 4). Antibodies against the NR1 subunit of the NMDA receptor complex reveal NR1 immunoreactivity in both layers I and II of piriform cortex (Fig. 4A). NR2A/B immunoreactivity has a very similar distribution (Fig. 4B). Over 80% of reelin (+) interneurons in layer I are also NR1 and NR2A/B (+)[reelin-NR1: 85±13% of reelin (+) neurons (n=3); reelin-NR2A/B: 82±22% of reelin (+) neurons(n=3)](Fig. C-D). The localization of these receptor proteins inside the cytoplasm of reelin (+) neurons was confirmed with confocal microscopy (Fig. 4E). The presence, in reelin (+) neurons, of both the NR1 and NR2 receptor subunits enables these cells to respond to pharmacological manipulations of the NMDA receptor.
Figure 4. Layer I interneurons are enriched in NMDA glutamate receptors based on ICC for the NR epitopes 1 and 2.

A. Immunoperoxidase ICC for the NMDA subunit NR1 reveals widespread expression of NR1 immunoreactivity in layers I and II of piriform cortex, including expression in layer I interneurons (arrows).
B. Immunoperoxidase staining of NR2A/B protein in piriform cortex shows that both layer II pyramidal neurons and layer I interneurons (arrows) express high level of immunoreactivity for this NR subunit.
C-D. Immunofluorescent microscopy of sections dually labeled for CR-50 --a reelin epitope that is a selective marker of layer I interneurons-- and NR1 (C) or NR2A/B (D) reveals colocalization of reelin and these NMDA receptor epitopes in over 80% of reelin (+) layer I interneurons.
E. Confocal microscopy of sections dually labeled for CR-50 and NR1 (left-hand panel) or NR2A/B (right-hand panel). NMDA receptor immunoreactivity in the cytoplasm is classically punctate.
Abbreviations: ot, olfactory tract.
Size bars: A-B, 50μm; C-D, 25 μm; E, 10μm
To examine the induction, by MK801, of layer I interneurons to increased levels of nNOS expression, we counted the numbers of these neurons expressing nNOS immunoreactivity 16 and 24 hours after treatment with MK801 or DMSO vehicle (Fig. 5A-B). By fractionator counts, there are 1643±128 neurons expressing immunoreactive nNOS in vehicle-treated and 2606±275 neurons in MK801-treated animals at 16 hours post-treatment; at 24 hours, there are 1827±79 nNOS (+) neurons in vehicle- and 2817±105 neurons in MK801- treated subjects (Fig. 5D). In both cases, the magnitude of difference is more than 1/3 of the baseline vehicle population and is statistically significant (Fig. 5D). This increase in nNOS expression correlates with apparent increases in NADPHd histochemical activity, primarily within the processes of layer I interneurons (Fig. 5C).
Figure 5. MK801 increases nNOS expression in layer I interneurons based on nNOS ICC (A-B, D) and NADPHd histochemical activity (C).

A-B. These images are taken from representative coronal sections through anterior piriform cortex that were stained for nNOS immunoperoxidase. No significant nNOS reactivity is seen in layer II. There is more immunostaining in superficial layer I (layer Iα) than layer Iβ. Numbers of nNOS-immunoreactive neurons increase (arrows in panel B) with MK801 treatment.
C. NADPHd histochemical activity, shown to be identical to nNOS enzymatic activity, in especially enriched in the processes of layer Iα neurons (all panels) and increases at 1 and 3 days after MK801 treatment (compare middle and right-hand panel with left-hand panel).
D. Bar diagram summarizing data on numbers of nNOS (+) neurons at 16 (left-hand bars) and 24 (right-hand bars) hours after vehicle (blue) or MK801 (red) treatment. For both time points, difference between the two groups is significant by Students’ t test (p<0.01).
Size bars: 50 μm
Selective nNOS blockade with BRNI abrogates MK801-induced apoptosis in piriform cortex
The ability of layer I interneurons to respond to NMDA manipulations by increasing their expression of nNOS at or before the time of MK801-induced apoptosis suggests that blocking nNOS activity with selective inhibitors may abrogate MK801- induced apoptosis in piriform cortex. The selective nNOS inhibitor BRNI was used to block the nNOS-inductive effects of MK801 and the effect was studied in a multiple-group comparison design including animals treated with MK801 vehicle (saline), BRNI vehicle (DMSO), MK801, MK801 plus BRNI and BRNI alone (Fig. 6). Variance in numbers of apoptotic neurons among groups was very significant by one-way ANOVA followed by post-hoc testing and was likely caused by both by the large apoptotic effect of MK801 treatment and the substantial ameliorating effect of BRNI. The latter reduces the population of apoptotic neurons to 2/5 of the size observed with MK801 treatment.
Figure 6.
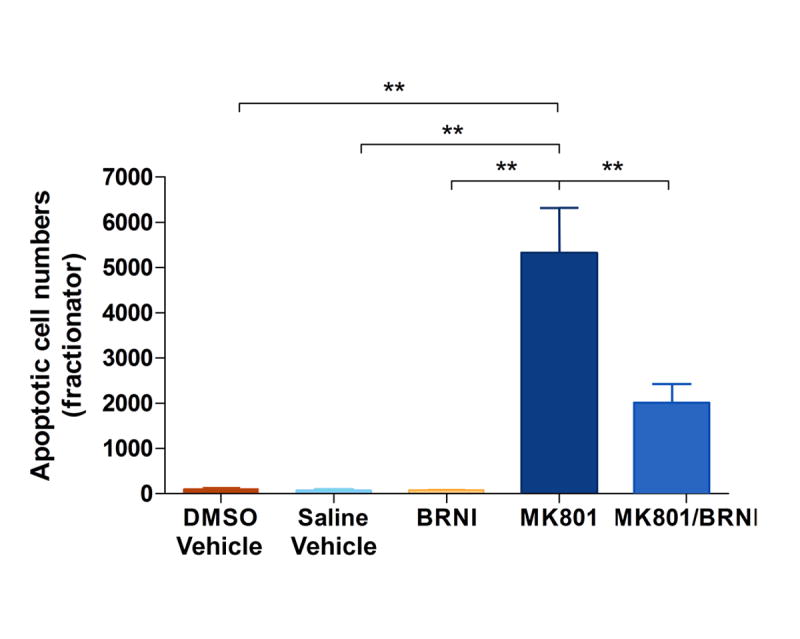
Bar diagram showing the variance in apoptotic neuron numbers in piriform cortex after NMDA blockade with MK801 and both NMDA blockade and nNOS inhibition with MK801 and BRNI treatment. Three control groups, treated with DMSO vehicle, saline vehicle and with BRNI alone were also introduced in the ANOVA design. Overall significance is very high at p<0.0001. Post-hoc analysis with Tukey’s multiple comparisons test shows differences between saline or DMSO or BRNI groups and the MK801 group at p<0.001 and between MK801 and MK801-BRNI at p<0.001.
DISCUSSION
The findings presented here show that pyramidal neurons in layer IIα are vulnerable to apoptotic degeneration when exposed to the selective non-competitive NMDA channel blocker MK801 in adult rats. These are the same nerve cells that are also vulnerable to apoptotic degeneration after deprivation of their modality-specific afferents from the olfactory bulb (Capurso et al., 1997;Koliatsos et al., 2004). In addition, MK801 induces nNOS/NADPHd expression in layer I interneurons, an effect also seen with bulbotomy lesions. The concomitant treatment of animals exposed to MK801 with the selective nNOS inhibitor BRNI prevents one-third to one-half of the size of the pro-apoptotic effect of MK801. Together, these data demonstrate that pyramidal neurons located superficially in layer II of piriform cortex have a constitutive vulnerability to apoptosis with both traumatic and toxic insults and that inhibitory interneurons may play a prominent role in signaling the apoptotic death of these pyramidal neurons. The apparently indirect degenerative effect of MK801 via induction of nNOS in inhibitory interneurons adds to arguments on the importance of these local circuit neurons in cortical degeneration raised by us (Koliatsos et al., 2004) as well as by Olney and colleagues (Corso et al., 1997) and invites more work on cortical inhibitory interneurons as mediators of traumatic and toxicological plasticity in mammalian cortex.
The ability of inhibitory interneurons to mediate the effects of MK801 is founded on the expression, by these cells, of both the NR1 and NR2 NMDA receptor components (Kemp and McKernan, 2002). This understanding is based on ICC data presented here that focus on layer I interneurons and is consistent with earlier broader accounts on NR1 (Petralia et al., 1994b) and NR2A/NR2B (Petralia et al., 1994a) immunoreactivity in rat brain. Importantly, these reelin-and-GAD67 (+) interneurons (Koliatsos et al., 2004) also express various constituents of the AMPA/kainate complex, including Glur1 and Glur2/3 (Zhou et al., 2006), a pattern also in keeping with the original anatomical studies on the distributions of AMPA/kainate receptors in the rat forebrain (Martin et al., 1993;Martin et al., 1992). As might be predicted by their enrichment in AMPA/kainate receptors, these interneurons also respond to AMPA/kainate manipulations. For example, AMPA-kainate antagonists like NBQX prevent the upregulation of nNOS expression in layer I interneurons after bulbotomy lesions (Zhou et al., 2006). Thus, the blockade of the two major types of ionotropic glutamate receptors in layer I interneurons in piriform cortex appears to have opposing effects on nNOS regulation, suggesting that activation of AMPA/kainate receptors eventually increases and the activation of NMDA receptors decreases the expression of nNOS in these inhibitory interneurons (Fig. 7). The apparent involvement of these interneurons, via nNOS/NO, in signaling of apoptosis for adjacent pyramidal neurons in layer IIα (Koliatsos et al., 2004) may explain why AMPA/kainate receptor blockade ameliorates the transsynaptic apoptosis of pyramidal neurons after bulbotomy lesions whereas NMDA receptor blockade appears to have a direct pro-apoptotic effect for these pyramidal cells.
Figure 7.
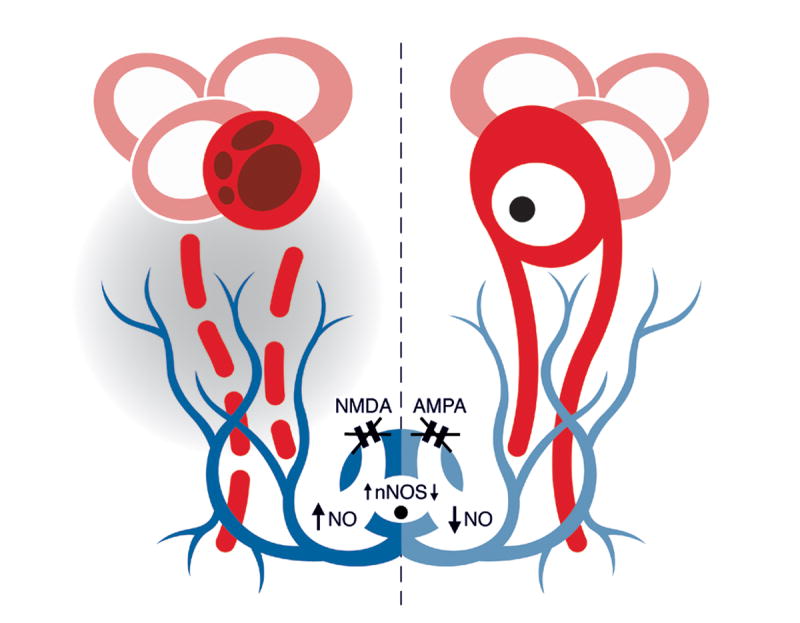
A summary sketch laying out our hypothesis on the role of inhibitory interneurons (blue) as regulators of pro-apoptotic signals for pyramidal neurons (red) of piriform cortex. The pro-apoptotic signal proposed here is extracellular nitric oxide (NO). This hypothesis is deduced from data on NMDA (this study) and AMPA (Zhou et al., 2006) antagonism. NMDA blockade (left) is associated with upregulation of nNOS in these interneurons followed by nitric oxide release, via cell bodies and axon branches, in the extracellular space (shaded area). Free-radical accumulation within the dendrites and, perhaps, cell bodies of nearby pyramidal neurons increases risk for apoptotic cell death. In contrast, AMPA blockade (right) is associated with reductions in lesion-induced nNOS and, thus, decrements in extracellular nitric oxide, thereby lowering the apoptotic risk for pyramidal neurons. Glutamate receptor events and changes in nNOS/NO levels are deduced from pharmacological studies in this and previous studies and, although depicted here in a logical temporal sequence, they should not be viewed as directly linked steps along a single biochemical cascade. In view of the substantial complexity of glutamate signaling in neurons, the effects of pharmacological blocking of NMDA and AMPA receptors do not necessarily imply the opposite effects when these receptors are activated.
Our finding that NMDA antagonism has a pro-apoptotic effect, especially via an increase in nNOS expression, is somewhat surprising. The conventional thinking originating primarily in models of anoxia-ischemia is that NMDA activation induces rather than suppresses nNOS expression, a step that releases nitric oxide intracellularly and initiates the cell death cascade (Garthwaite et al., 1989;Dawson et al., 1991). Recent data on hippocampal slices have disputed the general applicability of this concept (Keynes et al., 2004). In addition, the previous hypothesis is based on in vitro preparations and involves single-cell concepts, i.e. concepts in which a single cell is the host of both the NMDA receptor activation and downstream induction of nNOS. It is now clear that, at least in some complex models such as rat cerebellar slices, nitric oxide can effectively serve as a chemical signal between neurons (Southam and Garthwaite, 1991). Our proposed mechanism for MK801-mediated apoptosis of pyramidal neurons in piriform cortex is based on a cell-interactive model that requires the partnership of two nerve cells. The first cell, i.e. the inhibitory nNOS (+) interneuron in layer I is the initial sensor of the MK801 effect and the releaser of nitric oxide to whom it is resistant, perhaps via the concomitant upregulation of antioxidant signals such as the superoxide anion scavenger MnSOD (Koliatsos et al., 2004). The second cell, i.e. the relay neuron whose cell body is located in layer IIα, is the target of nitric oxide and the primary site of apoptotic signaling, presumably after initial vacuolization injury to its dendrites that lie in direct proximity to the cell bodies and axon branches of layer I interneurons (Capurso et al., 1997) (Fig. 7). Direct injury to the cell body cannot be ruled out, especially as the latter is located within the 100 μm radius of NO diffusion from the axon branches of layer I interneurons to layer IIα (Capurso et al., 1997). NO can permeate the plasma membrane and destroy post-synaptic structures via the generation of reactive oxygen species (especially peroxynitrite) within the dendrites and cell body (Bonfoco et al., 1995;Koliatsos et al., 2004). Although layer IIα neurons do not express nNOS and, therefore, their ability to generate intracellular NO when exposed to toxic signals is uncertain, the possibility that at least part of the protective BRNI effect observed here is mediated via blockade of low-level NO synthesis in target neurons cannot be entirely ruled out.
Pathological cell-cell interactions involving cortical inhibitory interneurons and pyramidal neurons have also been shown in transgenic mice expressing a pathogenic huntingtin mutant (mhtt-exon1) (Gu et al., 2005). In these mice, the ability of mhtt-exon1 to cause pyramidal cell degeneration depends on its expression in cortical GABAergic interneurons followed by the reduced generation of spontaneous inhibitory postsynaptic currents, a physiological marker of interneuron inhibition, in pyramidal neurons. This work suggests that another possible mechanism of piriform pyramidal cell toxicity by NMDA antagonism, besides NO release, is the deficient inhibition (or an excess of excitation) of piriform pyramids because of reduced activity in GABAergic interneurons.
The vulnerability of projection neurons in paralimbic cortex to pharmacological manipulations of the NMDA receptor, especially with compounds binding at the phencyclidine (PCP) site, was initially discovered by Olney and colleagues who showed that that single doses of PCP, ketamine and MK801 cause reversible vacuolization and structural mitochondrial changes in pyramidal neurons of the posterior cingulate and retrosplenial cortex (Olney et al., 1989). Subsequent work by Corso, Fix and Wozniak and colleagues was interpreted as evidence that PCP and MK801 treatments that were higher-dose or more prolonged compared to those initially implemented by Olney caused degeneration, rather than reversible injury, of vulnerable pyramidal neurons (Fix et al., 1993;Corso et al., 1997;Wozniak et al., 1998). This work was based heavily on the histological demonstration of pyknotic or argyrophilic (“dark”) neurons but also involved, to various degrees, ultrastructural evidence. The type of degeneration of vulnerable neurons is unclear in those studies but it does not appear to have the classical morphological features of apoptosis. In addition, the consistency of degenerative outcomes was variable, e.g. the addition of the muscarinic cholinomimetic pilocarpine was seen to improve the reliability of the injury effect of PCP (Corso et al., 1997). Another report based on silver stains that used the “dark neuron” appearance as histological evidence of degeneration (Horvath et al., 1997) produced mixed results, with piriform pyramidal neurons showing similar vulnerability to the effects of MK801 as retrosplenial/posterior cingulate neurons, and neurons in entorhinal cortex and taenia tecta (the precommissural extension of the hippocampal allocortex) appearing less vulnerable. Horvath and colleagues raised concerns about the use of argyrophilia as a reliable marker of irreversible injury, e.g. granule cells in the hippocampal dentate gyrus became “dark” after MK801 treatment, yet they were not viewed as degenerating nerve cells by the authors. There are important similarities and differences between those and the present study. First, we have not used silver stains in our experiments, because of concerns about the nature and permanence of injury in argyrophilic “dark” neurons. Second, the focus of the present study was the piriform cortex and our methodological tools were optimal for the detection of apoptotic neurons. Although we carefully examined cresyl violet- and active caspase-3-stained sections through the posterior cingulate/retrosplenial cortex, we failed to see classical apoptotic profiles or active caspase-3 (+) neurons in these paralimbic regions. However, we recognize that sections stained with aniline dyes, sufficient as they may be for the demonstration of apoptosis, are not optimal preparations for the demonstration of subtle forms of cytoplasmic vacuolation. In a more recent study of MK801-induced degeneration of limbic and paralimbic cortices, the authors commented on the complexity of “dark” degeneration of piriform neurons after MK801 treatment, mentioning specifically the inconsistency in labeling of layer IIα neurons (Noguchi etal., 2005). We propose that at least one reason for this inconsistency may be the fast apoptotic death of these neurons, as demonstrated in the present study.
Although the vulnerability of pyramidal neurons to the effects of PCP ligands extend over a large portion of limbic and paralimbic cortex, these effects appear to be selective for layers III-IV in the paralimbic retrosplenial cortex and superficial layer II in the limbic piriform cortex, i.e. they show preference for superficially located pyramidal neurons. Compared to pyramidal neurons located in deeper cortical layers, these superficially located pyramids migrate and differentiate late in cortical development (Rakic, 1981) and their vulnerability to NMDA blockade in mature animals can be viewed as a persisting developmental profile of susceptibility to NMDA receptor antagonism (Ikonomidou et al., 1999). In light of the fact that this developmental susceptibility tends to produce a morphologically apoptotic type of degeneration, MK801-induced degeneration of pyramidal neurons in the adult piriform cortex is closer to a developmental degenerative profile. Together, these results underline the ongoing vulnerability of certain classes of late-appearing pyramidal neurons to NMDA blockade in the adult limbic and paralimbic cortex, but also point to the importance of cortical interneurons as mediators of these injurious and in some cases degenerative, effects.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proofbefore it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton SA. Apoptosis and necrosis: two distinct events induced respectively by mild and intense insults with NMDA or nitric oxide/superoxide in cortical cell cultures. Proc Natl Acad Sci USA. 1995;92:7162–7166. doi: 10.1073/pnas.92.16.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Capurso SA, Calhoun ME, Sukhov RR, Mouton PR, Price DL, Koliatsos VE. Deafferentation causes apoptosis in cortical sensory neurons in the adult rat. J Neurosci. 1997;17:7372–7384. doi: 10.1523/JNEUROSCI.17-19-07372.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Corso TD, Sesma MA, Tenkova TI, Der TC, Wozniak DF, Farber NB, Olney JW. Multifocal brain damage induced by phencyclidine is augmented by pilocarpine. Brain Research. 1997;752:1–14. doi: 10.1016/s0006-8993(96)01347-9. [DOI] [PubMed] [Google Scholar]
- 4.Dawson VL, Dawson TM, London ED, Bredt DS, Snyder SH. Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc Natl Acad Sci USA. 1991;88:6368–6371. doi: 10.1073/pnas.88.14.6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fix AS, Horn JW, Wightman KA, Johnson CA, Long GG, Storts RW, Farber N, Wozniak DF, Olney JW. Neuronal Vacuolization and Necrosis Induced by the Noncompetitive N-Methyl-D-Aspartate (Nmda) Antagonist Mk(+)801 (Dizocilpine Maleate) - A Light and Electron-Microscopic Evaluation of the Rat Retrosplenial Cortex. Experimental Neurology. 1993;123:204–215. doi: 10.1006/exnr.1993.1153. [DOI] [PubMed] [Google Scholar]
- 6.Garthwaite J, Garthwaite G, Palmer RMJ, Moncada S. Nmda Receptor Activation Induces Nitric-Oxide Synthesis from Arginine in Rat-Brain Slices. European Journal of Pharmacology-Molecular Pharmacology Section. 1989;172:413–416. doi: 10.1016/0922-4106(89)90023-0. [DOI] [PubMed] [Google Scholar]
- 7.Gu XE, Li CJ, Wei WZ, Lo V, Gong SC, Li SH, Iwasato T, Itohara S, Li XJ, Mody I, Heintz N, Yang XW. Pathological cell-cell interactions elicited by a neuropathogenic form of mutant huntingtin contribute to cortical pathogenesis in HD mice. Neuron. 2005;46:433–444. doi: 10.1016/j.neuron.2005.03.025. [DOI] [PubMed] [Google Scholar]
- 8.Horvath ZC, Czopf J, Buzsaki G. MK-801-induced neuronal damage in rats. Brain Research. 1997;753:181–195. doi: 10.1016/s0006-8993(96)01290-5. [DOI] [PubMed] [Google Scholar]
- 9.Ikonomidou C, Bosch F, Miksa M, Bittigau P, V”ckler J, Dikranian K, Tenkova TI, Stefovska V, Turski L, Olney JW. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science. 1999;283:70–74. doi: 10.1126/science.283.5398.70. [DOI] [PubMed] [Google Scholar]
- 10.Kemp JA, McKernan RM. NMDA receptor pathways as drug targets. NATURE NEUROSCIENCE. 2002;5:1039–1042. doi: 10.1038/nn936. [DOI] [PubMed] [Google Scholar]
- 11.Keynes RG, Duport S, Garthwaite J. Hippocampal neurons in organotypic slice culture are highly resistant to damage by endogenous and exogenous nitric oxide. European Journal of Neuroscience. 2004;19:1163–1173. doi: 10.1111/j.1460-9568.2004.03217.x. [DOI] [PubMed] [Google Scholar]
- 12.Koliatsos VE, Dawson TM, Kecojevic A, Zhou Y, Wang Y-F, Huang K-X. Cortical interneurons become activated by deafferentation and instruct the apoptosis of pyramidal neurons. Proc Natl Acad Sci U S A. 2004;101:14264–14269. doi: 10.1073/pnas.0404364101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koliatsos VE, Price DL, Gouras GK, Cayouette MH, Burton LE, Winslow JW. Highly selective effects of nerve growth factor, brain-derived neurotrophic factor, and neurotrophin-3 on intact and injured basal forebrain magnocellular neurons. J Comp Neurol. 1994;343:247–262. doi: 10.1002/cne.903430206. [DOI] [PubMed] [Google Scholar]
- 14.Liu Z, Gastard M, Verina T, Bora S, Mouton PR, Koliatsos VE. Estrogens modulate experimentally induced apoptosis of granule cells in the adult hippocampus. J Comp Neurol. 2001;441:1–8. doi: 10.1002/cne.1393. [DOI] [PubMed] [Google Scholar]
- 15.Martin LJ, Blackstone CD, Huganir RL, Price DL. Cellular localization of a metabotropic glutamate receptor in rat brain. Neuron. 1992;9:259–270. doi: 10.1016/0896-6273(92)90165-a. [DOI] [PubMed] [Google Scholar]
- 16.Martin LJ, Blackstone CD, Levey AI, Huganir RL, Price DL. AMPA glutamate receptor subunits are differentially distributed in rat brain. Neuroscience. 1993;53:327–358. doi: 10.1016/0306-4522(93)90199-p. [DOI] [PubMed] [Google Scholar]
- 17.Noguchi KK, Nemmers B, Farber NB. Age has a similar influence on the susceptibility to NMDA antagonist-induced neurodegeneration in most brain regions. Developmental Brain Research. 2005;158:82–91. doi: 10.1016/j.devbrainres.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 18.Olney JW, Labruyere J, Price MT. Pathological changes induced in cerebrocortical neurons by phencyclidine and related drugs. Science. 1989;244:1360–1362. doi: 10.1126/science.2660263. [DOI] [PubMed] [Google Scholar]
- 19.Petralia RS, Wang YX, Wenthold RJ. The Nmda Receptor Subunits Nr2A and Nr2B Show Histological and Ultrastructural-Localization Patterns Similar to Those of Nr1. Journal of Neuroscience. 1994a;14:6102–6120. doi: 10.1523/JNEUROSCI.14-10-06102.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petralia RS, Yokotani N, Wenthold RJ. Light and electron microscope distribution of the NMDA receptor subunit NMDAR1 in the rat nervous system using a selective anti- peptide antibody. J Neurosci. 1994b;14:667–696. doi: 10.1523/JNEUROSCI.14-02-00667.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rakic P. Developmental events leading to laminar and areal organization of the neocortex. In: Schmitt FO, Worden FG, Adelman G, Dennis SG, editors. The Organization of the Cerebral Cortex. Cambridge: MIT Press; 1981. pp. 7–28. [Google Scholar]
- 22.Southam E, Garthwaite J. Intercellular Action of Nitric-Oxide in Adult-Rat Cerebellar Slices. Neuroreport. 1991;2:658–660. doi: 10.1097/00001756-199111000-00006. [DOI] [PubMed] [Google Scholar]
- 23.Vincent SR, Johansson O, Hokfelt T, Skirboll L, Elde RP, Terenius L, Kimmel J, Goldstein M. NADPH-diaphorase: a selective histochemical marker for striatal neurons containing both somatostatin- and avian pancreatic polypeptide (APP)- like immunoreactivities. J Comp Neurol. 1983;217:252–263. doi: 10.1002/cne.902170303. [DOI] [PubMed] [Google Scholar]
- 24.West MJ, Slomianka L, Gundersen HJG. Unbiased stereological estimation of the total number of neurons in the subdivisions of the rat hippocampus using the optical fractionator. Anat Rec. 1991;231:482–497. doi: 10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]
- 25.Wozniak DF, Dikranian K, Ishimaru MJ, Nardi A, Corso TD, Tenkova T, Olney JW, Fix AS. Disseminated corticolimbic neuronal degeneration induced in rat brain by MK-801: Potential relevance to Alzheimer’s disease. Neurobiology of Disease. 1998;5:305–322. doi: 10.1006/nbdi.1998.0206. [DOI] [PubMed] [Google Scholar]
- 26.Zhou Y, Chen H, Koliatsos VE. Pharmacological evidence for a glutamatergic activation - nitric oxide synthesis step in the early signaling of transsynaptic apoptosis in limbic cortex. Society for Neuroscience. 2004;Abstracts(No 79416) [Google Scholar]
- 27.Zhou Y, Zhou L, Chen H, Koliatsos VE. An AMPA glutamatergic receptor activation-nitric oxide synthesis step signals transsynaptic apoptosis in limbic cortex. Neuropharmacology. 2006;51:67–76. doi: 10.1016/j.neuropharm.2006.03.002. [DOI] [PubMed] [Google Scholar]