

Abstract
Plasmodium falciparum, the protozoan that causes the most lethal form of human malaria, has been controlled principally by two safe, affordable drugs, chloroquine and sulfadoxine-pyrimethamine (SP). Studies in the laboratory and in the field have demonstrated that resistance to SP depends on non-synonymous point mutations in the dihydrofolate reductase (DHFR), and dihydropteroate synthase (DHPS) coding regions. Parasites that carry dhfr genes with 3 or 4 point mutations (51I/59R/108N triple mutation or 51I/59R/108N/164L quadruple mutation) are resistant to pyrimethamine in vitro and patients infected with these parasites respond poorly to SP treatment.
The wide spread of these pyrimethamine-resistant alleles demonstrates the increased fitness over drug-sensitive alleles in the presence of the drug. However, it is not clear whether these alleles might reduce the fitness of parasites in the absence of drug pressure. As a first step, we compared the kinetic properties of the wild type, and three mutant alleles to determine whether the native DHFR-thymidylate synthase form of the mutant proteins showed compromised activity in vitro. The mutant enzymes had Km values for their substrate, dihydrofolate that were significantly lower than the wild type, kcat values in the same range as the wild type enzyme, and kcat/Km values higher than wild type. In contrast, the Km values for the NADPH cofactor were higher than wild type for the mutant enzymes. These observations suggest that the fitness of these parasites may not be compromised relative to those that carry the wild type allele, even without sustained SP drug pressure.
Keywords: Drug resistance, Antifolate, Relative fitness, Selection
Introduction
HIV, TB, malaria and many bacterial infections can be controlled mainly by drug treatment, and the development of resistance to the drugs is a major clinical problem world wide. To slow the rise in resistant pathogens, it is important to understand the mechanisms by which drug resistance is selected and sustained. Plasmodium falciparum, the protozoan that causes the most lethal form of human malaria, has been controlled principally by two safe, affordable drugs, i.e. chloroquine and sulfadoxine-pyrimethamine (SP). Although overall child mortality due to malaria has declined in many areas of Africa, the development of parasites resistant to chloroquine has been associated with increased mortality in recent years [1,2]. Resistance to each drug is strongly associated with point mutations in specific parasite genes [3-5]. Pyrimethamine is a competitive inhibitor of dihydrofolate reductase (DHFR) and sulfadoxine targets the enzyme dihydropteroate synthase (DHPS). Studies in the laboratory and in the field have demonstrated that resistance to SP depends on non-synonymous point mutations in the DHFR, and DHPS coding regions. Parasites that carry dhfr genes with 3 or 4 point mutations, i.e. 51I/59R/108N triple mutant or 51I/59R/108N/164L quadruple mutant) are resistant to pyrimethamine in vitro and patients infected with these parasites respond poorly or not at all to SP treatment [4,6,7] (See [5] for a comprehensive review).
The completion of the P. falciparum genome made it possible to use microsatellite markers linked to dhfr and dhps to trace the ancestry of SP resistant strains. Using this approach, recent work has demonstrated that highly resistant alleles have not arisen locally by selection to high prevalence in situ. Rather, only a very few SP resistant strains that carry the triple or quadruple mutant allele of dhfr have been selected and these strains have “swept” the P. falciparum populations in low and moderate transmission regions of Southeast Asia, Eastern Africa and South America [8-11]. This may not be the case in higher transmission settings, and there is a need to examine population histories in these areas [12]. These new techniques have changed radically our capacity to understand the selective process, and provide new approaches for surveillance of drug resistant strains.
The wide spread of these pyrimethamine-resistant alleles demonstrates their increased fitness over drug-sensitive alleles in the presence of the drug, but the model raises a key issue. Do these mutations that confer drug resistance reduce the fitness of parasites that carry them if the drugs are withdrawn? If that is the case, then resistant parasite populations might be reduced or even disappear. This reduction in prevalence of a drug resistant parasite has occurred in at least one very well documented case. In Malawi, SP replaced chloroquine as the recommended treatment for falciparum malaria in 1993. This replacement was followed by a rapid replacement of the K76T bearing allele of pfcrt that is the hallmark of chloroquine-resistant strains, with a K76 chloroquine-sensitive allele and a return of clinical chloroquine sensitivity [13,14]. This observation raised the hope that the pyrimethamine-resistant alleles of dhfr might be similarly compromised, and recede from prevalence if SP drug pressure were removed.
The prediction of decreased fitness of parasites that carried the triple or quadruple mutant allele of dhfr was supported by measurement of the DHFR enzyme activity in vitro. Early work by Sirawaraporn and colleagues showed that the catalytic activity (kcat) of the DHFR domain of the triple and quadruple mutant enzymes was severely compromised in vitro compared with the wild type enzyme [15]. However, this deficiency was hard to reconcile with the persistence of the parasites that carried these alleles. In Southeast Asia, SP has not been used extensively for almost 30 years, but parasites that carry the quadruple mutant genotype are still common [16-19], and parasites that carry the triple mutant allele have persisted in Ghana despite very low usage of SP [20]. These observations suggest that the fitness of these parasites may not be compromised relative to those that carry the wild type allele, even without sustained SP drug pressure.
In protozoan parasites, the DHFR enzyme is one domain of a bifunctional protein with thymidylate synthase (TS) [21-23]. Because of the poor expression of P. falciparum proteins in heterologous systems [24], attempts to express the DHFR domain alone were made and the DHFR was purified after denaturation and refolding from inclusion bodies within the E. coli [15]. Since the structure of the native DHFR-TS protein was recently published [23], and it was shown that the two domains could be co-expressed as a DHFR-TS complex [25], we have now reexamined the kinetic parameters of the whole DHFR-TS protein complex that contain the wild-type, triple and quadruple mutant DHFR alleles purified only from the soluble fraction of the E. coli. In addition, we had previously identified a novel 51I/59R/108N/213A allele found at low frequency in field isolates from 4 different locations in Africa, and the DHFR domain of this protein conferred substantial resistance to pyrimethamine when tested in a heterologous yeast system [25]29]. We also compared the kinetic properties of this DHFR-TS protein with other DHFR enzymes that are commonly found in P. falciparum populations.
Materials and Methods
Constructs
A synthetic DHFR construct (codons 1- 231) with the E. coli codon bias [15] was cloned into the carbenicillin resistant pET-17b vector with NdeI and HindIII sites (Novagen, Madison, WI) and an additional N-terminal 6XHisTag. A second construct that contained the joining region (codons 232-324) connected with the TS region (codons 325-607) was cloned into the kanamycin resistant pET-24d (Novagen, Madison, WI) with a C-terminal 6XHisTag. The three mutant constructs (51I/59R/108N, 51I/59R/108N/164L and 51I/59R/108N/213A) were created using QuikChange II with the wild type synthetic DHFR construct as a template (Stratagene, Cedar Creek, TX).
Growth in E. coli
The constructs harboring the dhfr and jr-ts sequences were co-transformed into BL21(DE3)pLysS E. coli cells (Novagen, Madison, WI) and grown overnight at 37°C in 50 mL Luria-Bertani broth (LB) supplemented with 60 μ/mL carbenicillin, 30 μg/mL kanamycin and 50 μg/mL chloramphenicol. Four liters of LB supplemented with the same concentrations of antibiotics were inoculated with 1:100 dilution of the saturated culture and grown at 37°C to an OD600 of 0.6 – 0.8.
Protein expression was induced by addition of 1 mM isopropyl-β-D-thio-galactopyranoside (IPTG) and the culture was allowed to grow at 22°C for 16 hours. The induced cells were harvested, washed in 20 mM Tris buffer, pH 8.0 and the pelleted cells were stored at −80° C until purification.
Protein purification
Protein purification was based on Shallom et al [26]. Cells were lysed with (per half liter) 20 mL BugBuster Protein Extraction Reagent (Novagen, Madison, WI), 500 units benzonase nuclease, > 99% Purity (Novagen, Madison, WI), aprotinin (1 μg/mL), leupeptin (1 μg/mL), pepstatin A (1 μg/mL) and 50 mM phenylmethanesulfonyl fluoride (PMSF). Cell debris was pelleted and the soluble fraction was adjusted to 20% glycerol and 20 mM imidazole. The column (Econo-10 columns) containing Ni-NTA resin (Qiagen, Valencia, CA) was packed (50 mM NaH2PO4 / 300 mM NaCl) and shielded (50 mM NaH2PO4 / 300 mM NaCl / 20% glycerol / 10 mM imidazole). The adjusted lysate was passed through the column in a cold room at a rate of 1 mL/min. The column was washed (50 mM NaH2PO4 / 300 mM NaCl / 20% glycerol / 50 mM imidazole) and bound protein was eluted with 10 mL elution buffer (50 mM NaH2PO4 /300 mM NaCl / 20% glycerol /250 mM imidazole). For storage, 10 mM dithiothreitol (DTT) was added and purified protein was stored at 4° C. Protein concentrations of the fractions were determined by the Bradford assay [27]. A sample of each fraction was analyzed before addition of DTT on a non-reducing 4-15% polyacrylamide gel, and after reduction, on a 12% polyacrylamide gel using standard protocols. The average yield of DHFR-TS protein was 800 micrograms from 4L culture.
Enzyme Assay
The activity of DHFR was determined spectrophotometrically according to the method of Sirawaraporn [28] by measuring the oxidation of NADPH to NADP+ at 340 nm using a microplate reader (Thermoelectron, Labsystems Multiskan MCC/240). The activity assay contained 100 μM NADPH, 50 mM TES, pH 7.0, 1 mM EDTA, pH 8.0, 75 mM 2-mercaptoethanol, 1 mg/mL bovine serum albumin and ∼0.005 unit of enzyme. The reaction was initiated with 100 μM dihydrofolate (DHF). One unit of DHFR enzyme activity is defined as the amount of enzyme required to produce 1 μmole product/min at 25°C.
Steady-state kinetic parameters (Km and Vmax) were determined by varying the concentrations of DHF or NADPH between 3.5 μM and 225 μM with the other (NADPH or DHF, respectively) remaining constant at 100 μM and the enzyme was added to initiate the reaction. Absorbance values were converted to concentrations using Beer-Lambert's law. The rates of reactions were plotted in Kaleidograph (Synergy Software, Reading, PA) and kinetic parameters were calculated using the Michaelis-Menten curve fit.
Results
The protein is purified as a DHFR-TS complex
Because of the difficulties of expressing large quantities of native DHFR-TS protein in E. coli [24], previous studies on the kinetic properties of the P. falciparum DHFR enzyme have focused mainly on using partially purified enzyme preparations [29] or DHFR domain alone purified and refolded from inclusion bodies from E. coli [15]. To circumvent this problem, we capitalized on an observation from the laboratory of Pradipsingh Rathod: when the DHFR and TS domains are coexpressed in E. coli, the two domains form a tight complex of 2 TS and 2 DHFR domains, like the native protein [26]. We transformed into BL21(DE3)pLysS E. coli separate plasmids that encoded the DHFR domain and the joining region/TS domains, and purified the complex from the soluble fraction using 6XHis tags on the proteins. Figure 1A shows that the protein purified from E. coli had the expected molecular size for this complex, 132 kDa. Upon reduction, two components of the appropriate sizes, 27 kDa for the DHFR domain and 40 kDa for the bridge/TS domain were observed (Figure 1B).
Figure 1. Molecular size of the DHFR-TS complex.
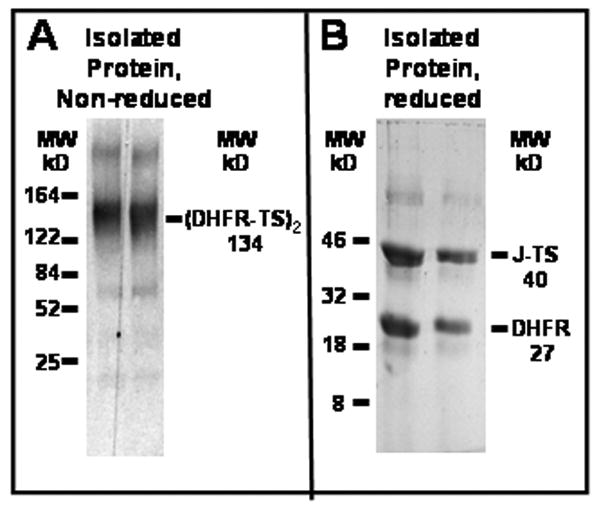
The DHFR-TS complex was isolated as described in the methods, and analyzed by SDS-polyacrylamide gel electrophoresis. A. Non reducing conditions, B. Reducing conditions. Two micrograms of the first and second fractions eluted from the nickel column were added to loading buffer and separated on polyacrylamide gels as described in the methods.
The activities of wild type and mutant enzymes are indistinguishable
Our goal was to compare the kinetic properties of the wild type, and three mutant alleles to determine whether the native form of the mutant proteins also showed compromised activity in vitro. Each DHFR domain and the bridge-TS domain were co-expressed in E. coli and the complex purified from the soluble fraction. We determined the Km and Vmax for both the substrate, dihydrofolate (DHF) and the cofactor, NADPH and the catalytic activity, kcat and the kcat/Km for DHF for each complex. We tested complexes that carried the DHFR domains of the wild type, the triple mutant (51I/59R/108N), the quadruple mutant (51I/59R/108N/164L) including a novel quadruple mutant, (51I/59R/108N/213A). The kinetic data of the wild-type and mutant enzymes are summarized in Table 1.
Table 1.
Kinetic parameters of the wild type and three pyrimethamine-resistant DHFR enzymes. Values are the average of at least 7 experiments ± the standard deviation.
| Genotype | kcat (sec−1) | KmDHF (μM) | Km NADPH (μM) | kcat/Km |
|---|---|---|---|---|
| WILD TYPE | 1.24 ± 0.11 | 44.4 ± 12.1 | 23.2 ± 9.5 | 0.028 |
| 51I/59R/108N | 1.23 ± 0.35 | 20.9 ± 5.5 | 37.9 ± 11.9 | 0.059 |
| 51I/59R/108N/164L | 1.42 ±0.57 | 20.7 ± 6.8 | 37.3 ± 13.2 | 0.069 |
| 51I/59R/108N/213A | 0.68 ± 0.17 | 10.2 ± 5.1 | 30.9 ± 12.2 | 0.066 |
The kinetics data from the present study showed that the Km values for DHF of the mutant enzymes were 2-4 fold lower than that of the wild type (p < 0.001, 2 tailed t-test). The kcat value for the wild-type was comparable to that of the mutant enzymes. As a consequence of the 2-fold higher of calculated enzyme efficiency, the calculated kcat/Km values for the mutant enzymes were 2-fold higher that that of the wild-type enzyme. In contrast, the Km values for NADPH cofactor of both mutant enzymes were slightly higher than that of the wild type enzyme (p= 0.0261 for triple and p = 0.0535 for quadruple).
The DHFR-TS that carried the novel 51I/59R/108N/213A quadruple DHFR domain was analyzed. The Km value for DHF was found to be about 2-fold lower than that for the triple and quadruple mutant enzymes and about 4-fold lower than that for the wild-type enzyme (p < 0.01 for all comparisons). However, the kcat values for all the three proteins were compromised. As a result the kcat/Km values for the mutant enzymes were similar and were significantly better than that of the wild type (p = 0.017).
Discussion
The demonstration that the triple and quadruple mutant DHFR-TS complex enzymes are more active in vitro than the wild type is important for two reasons. First, based on the assay of the isolated DHFR domains, it has been widely assumed that both mutant enzymes were far less efficient than the wild type. This assumption and a some field studies [30,31] supported the hope that in the absence of drug selection, parasites that carry these alleles would have compromised fitness relative to the wild type. Our data show that both enzymes are more efficient in vitro than the wild type. Thus, it seems more likely that these alleles may have been rapidly selected and widely disseminated because they are both resistant to antifolates, and encode enzymes with adequate activity. Second, their adequate enzyme activity may explain the persistence of the highly mutant alleles in P. falciparum populations in Southeast Asia, even under conditions of low pyrimethamine usage [16,17,32,33].
The Km values of the mutant enzymes for both substrates determined in the present study are within the same range compared to those previously reported for the DHFR domain alone and the partially purified DHFR-TS enzyme [29]. The discrepancy between the Km values of the wild-type DHFR domain reported earlier [15] and the DHFR-TS complex in the present study could be attributed to the different structural topology of the two proteins particularly in different experimental conditions. However, the kcat values differ from those that we calculated. The relative efficiency of the enzyme, kcat/Km, was almost 13-fold and 6-fold lower for the triple and quadruple mutants, respectively when the isolated DHFR domain was assayed [15]. In contrast, our data suggest that the mutant enzymes are a bit more efficient than the wild type. There are two principal differences between the two studies. First in this work, we have assayed the DHFR activity in the context of the DHFR-TS complex. We have noticed that the DHFR-TS complex is stable at 4°C for at least 3 weeks, supporting the idea that the enzyme is in its native conformation. In contrast, the DHFR domain alone is relatively unstable in vitro, and repeated attempts to purify and concentrate the domain for crystallography studies produced disordered aggregates, suggesting that it may not be as stable as its native form (JMW, data not shown). Second, in the prior studies, the DHFR domain was isolated from inclusion bodies, denatured and renatured prior to assay, whereas we have assayed only the soluble fraction isolated from the bacterial lysates. Both of these differences make it difficult to compare the two results, but they are likely to affect the concentration of active enzyme, and would both affect the calculation of the kcat. In any case, it is possible that the activity of the DHFR-TS enzyme may reflect more accurately the behavior of the endogenous enzyme.
The triple and quadruple mutant alleles are routinely found at high levels where ever SP has been intensively used [18]. However, various groups have used molecular methods to identify rare novel alleles of dhfr in field isolates from several locations [12,16,25,34-37]. The effect of these alleles on pyrimethamine-resistance in parasites is not known, although several alleles have been shown to confer pyrimethamine-resistance in a heterologous system. The failure of these rare alleles to come to prominence in the P. falciparum population is likely to depend mostly on stochastic processes; they simply fail to run the gauntlet of transmission [38]. However, the 51I/59R/108N/213V allele was observed in 4 widely separated African sites. The overall efficiency of the enzyme kcat/Km, is comparable to the common resistant variants because the far lower Km for DHF compensates for the lower kcat. However, these differences may impair the performance of the enzyme in vivo, and provide a possible explanation for the allele remaining at low levels in the P. falciparum population.
The relationship between the in vitro activity of the purified protein and the overall fitness of the parasites that depend upon that enzyme is unknown, and probably unknowable. However, the very widespread distribution of parasites that carry the triple mutant allele, certainly suggests that this genotype may not severely compromise the fitness of the parasites that carry it. It has been suggested that the success of the parasites that carry the triple mutant allele could reflect contributions of other loci that compensate for a less than optimal DHFR activity [8-11,14]. However, if the DHFR activity of the drug resistant enzymes is equivalent to wild type, then there is no need to invoke compensatory loci.
When chloroquine was successfully withdrawn in Malawi in 1993, the prevalence of the K76T allele of Pfcrt declined precipitously and there was a concomitant recovery of the clinical effectiveness of chloroquine treatment [13]. There have been suggestions of similar declines in chloroquine resistance in other areas (reviewed in [14]). These observations raised the hope that similar decreases in resistance might accompany withdrawal of other drugs. However, the cross resistance between the widely used antibiotic, trimethoprim and pyrimethamine already suggests that a true reduction in antifolate drug pressure might be difficult to achieve [39]. Moreover, if the parasites that carry pyrimethamine-resistant enzymes do not show lower fitness than their wild type counterparts, simply reducing drug pressure by withdrawing DHFR inhibitors would not be a successful strategy for “resurrecting” drugs of this class as effective antimalarials.
Acknowledgments
This work was supported by NIH grant AI 55604 awarded to CHS.
Abbreviations
- DHFR
dihydrofolate reductase
- DHF
dihydrofolate
- NADP
Nicotinamide adenine dinucleotide
- SP
sulfadoxine-pyrimethamine
- TS
thymidylate synthase
- kD
Kilodalton
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Trape JF. The public health impact of chloroquine resistance in Africa. Am J Trop Med Hyg. 2001;64:12–17. doi: 10.4269/ajtmh.2001.64.12. [DOI] [PubMed] [Google Scholar]
- 2.Snow RW, Trape JF, Marsh K. The past, present and future of childhood malaria mortality in Africa. Trends Parasitol. 2001;17:593–597. doi: 10.1016/s1471-4922(01)02031-1. [DOI] [PubMed] [Google Scholar]
- 3.Plowe CV. Monitoring antimalarial drug resistance: making the most of the tools at hand. J Exp Biol. 2003;206:3745–3752. doi: 10.1242/jeb.00658. [DOI] [PubMed] [Google Scholar]
- 4.Hyde JE. Mechanisms of resistance of Plasmodium falciparum to antimalarial drugs. Microbes Infect. 2002;4:165–174. doi: 10.1016/s1286-4579(01)01524-6. [DOI] [PubMed] [Google Scholar]
- 5.Gregson A, Plowe CV. Mechanisms of Resistance of Malaria Parasites to Antifolates. Pharmacol Rev. 2005;57:117–145. doi: 10.1124/pr.57.1.4. [DOI] [PubMed] [Google Scholar]
- 6.Sibley CH, Hyde JE, Sims PFG, Plowe CV, Kublin JG, Mberu EK, Cowman AF, Winstanley PA, Watkins WM, Nzila AM. Pyrimethamine/sulfadoxine resistance in Plasmodium falciparum: What next? Trends in Parasitology. 2001;17:582–588. doi: 10.1016/s1471-4922(01)02085-2. [DOI] [PubMed] [Google Scholar]
- 7.Talisuna AO, Nalunkuma-Kazibwe A, Langi P, Mutabingwa TK, Watkins WW, Marck EV, Egwang TG, D'Alessandro U. Two mutations in dihydrofolate reductase combined with one in the dihydropteroate synthase gene predict sulphadoxine-pyrimethamine parasitological failure in Ugandan children with uncomplicated falciparum malaria. Infect Genet Evol. 2004;4:321–327. doi: 10.1016/j.meegid.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 8.Cortese JF, Caraballo A, Contreras CE, Plowe CV. Origin and dissemination of Plasmodium falciparum drug-resistance mutations in South America. J Infect Dis. 2002;186:999–1006. doi: 10.1086/342946. [DOI] [PubMed] [Google Scholar]
- 9.Roper C, Pearce R, Nair S, Sharp B, Nosten F, Anderson T. Intercontinental Spread of Pyrimethamine-Resistant Malaria. Science. 2004;305:1124. doi: 10.1126/science.1098876. [DOI] [PubMed] [Google Scholar]
- 10.Roper C, Pearce R, Bredenkamp B, Gumede J, Drakeley C, Mosha F, Chandramohan D, Sharp B. Antifolate antimalarial resistance in southeast Africa: a population-based analysis. Lancet. 2003;361:1174–1181. doi: 10.1016/S0140-6736(03)12951-0. [DOI] [PubMed] [Google Scholar]
- 11.Nair S, Williams JT, Brockman A, Paiphun L, Mayxay M, Newton PN, Guthmann JP, Smithuis FM, Hien TT, White NJ, Nosten F, Anderson TJ. A selective sweep driven by pyrimethamine treatment in Southeast Asian malaria parasites. Mol Biol Evol. 2003;20:1526–1536. doi: 10.1093/molbev/msg162. [DOI] [PubMed] [Google Scholar]
- 12.McCollum AM, Poe AC, Hamel M, Huber C, Zhou Z, Shi YP, Ouma P, Vulule J, Bloland P, Slutsker L, Barnwell JW, Udhayakumar V, Escalante AA. Antifolate Resistance in Plasmodium falciparum: Multiple Origins and Identification of Novel dhfr Alleles. J Infect Dis. 2006;194:189–197. doi: 10.1086/504687. [DOI] [PubMed] [Google Scholar]
- 13.Kublin JG, Cortese JF, Njunju EM, RA M, Wirima JJ, Kazembe PN, Djimde AA, Kouriba B, Taylor TE, Plowe CV. Reemergence of Chloroquine-Sensitive Plasmodium falciparum Malaria after Cessation of Chloroquine Use in Malawi. J Infect Dis. 2003;187:1870–1875. doi: 10.1086/375419. [DOI] [PubMed] [Google Scholar]
- 14.Laufer M, Plowe C. Withdrawing antimalarial drugs: impact on parasite resistance and implications for malaria treatment policies. Drug Resist Updat. 2004;7:279–288. doi: 10.1016/j.drup.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 15.Sirawaraporn W, Sathitkul T, Sirawaraporn R, Yuthavong Y, Santi DV. Antifolate-resistant mutants of Plasmodium falciparum dihydrofolate reductase. Proc Natl Acad Sci U S A. 1997;94:1124–1129. doi: 10.1073/pnas.94.4.1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang P, Lee CS, Bayoumi R, Djimde A, Doumbo O, Swedberg G, Dao LD, Mshinda H, Tanner M, Watkins WM, Sims PFG, Hyde JE. Resistance to antifolates in Plasmodium falciparum monitored by sequence analysis of dihydropteroate synthetase and dihydrofolate reductase alleles in a large number of field samples of diverse origins. Mol Biochem Parasitol. 1997;89:161–177. doi: 10.1016/s0166-6851(97)00114-x. [DOI] [PubMed] [Google Scholar]
- 17.Zindrou S, Dung NP, Sy ND, Skold O, Swedberg G. Plasmodium falciparum: Mutation pattern in the dihydrofolate reductase thymidylate synthase genes of Vietnamese isolates, a novel mutation, and coexistence of two clones in a Thai patient. Exp Parasitol. 1996;84:56–64. doi: 10.1006/expr.1996.0089. [DOI] [PubMed] [Google Scholar]
- 18.Wongsrichanalai C, Pickard AL, Wernsdorfer WH, Meshnick SR. Epidemiology of drug-resistant malaria. Lancet Infect Dis. 2002;2:209–218. doi: 10.1016/s1473-3099(02)00239-6. [DOI] [PubMed] [Google Scholar]
- 19.Basco LK, de Pecoulas PE, LeBras J, Wilson CM. Plasmodium falciparum: Molecular characterization of multidrug-resistant Cambodian isolates. Exp Parasitol. 1996;82:97–103. doi: 10.1006/expr.1996.0013. [DOI] [PubMed] [Google Scholar]
- 20.Marks F, Evans J, Meyer CG, Browne EN, Flessner C, von Kalckreuth V, Eggelte TA, Horstmann RD, May J. High Prevalence of Markers for Sulfadoxine and Pyrimethamine Resistance in Plasmodium falciparum in the Absence of Drug Pressure in the Ashanti Region of Ghana. Antimicrob Agents Chemother. 2005;49:1101–1105. doi: 10.1128/AAC.49.3.1101-1105.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bzik DJ, Li WB, Horii T, Inselburg J. Molecular cloning and sequence analysis of the Plasmodium falciparum dihydrofolate reductase-thymidylate synthase gene. Proc Natl Acad Sci U S A. 1987;84:8360–8364. doi: 10.1073/pnas.84.23.8360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen G, Zolg J. Purification of the bifunctional thymidylate synthase-dihydrofolate reductase complex from the human malaria parasite Plasmodium falciparum. Mol Pharmacol. 1987;32:723–730. [PubMed] [Google Scholar]
- 23.Yuvaniyama J, Chitnumsub P, Kamchonwongpaisan S, Vanichtanankul J, Sirawaraporn W, Taylor P, Walkinshaw MD, Yuthavong Y. Insights into antifolate resistance from malarial DHFR-TS structures. Nat Struct Biol. 2003;10:357–365. doi: 10.1038/nsb921. [DOI] [PubMed] [Google Scholar]
- 24.Sirawaraporn W, Sirawaraporn R, Cowman AF, Yuthavong Y, Santi DV. Heterologous expression of active thymidylate synthase-dihydrofolate reductase from Plasmodium falciparum. Biochemistry (Mosc) 1990;29:10779–10785. doi: 10.1021/bi00500a009. [DOI] [PubMed] [Google Scholar]
- 25.Bates SJ, Winstanley PA, Watkins WM, Alloueche A, Bwika J, Happi TC, Kremsner PG, Kublin JG, Premji Z, Sibley CH. Rare, highly pyrimethamine-resistant alleles of the Plasmodium falciparum dihydrofolate reductase gene from 5 African sites. J Infect Dis. 2004;190:1783–1792. doi: 10.1086/425078. [DOI] [PubMed] [Google Scholar]
- 26.Shallom S, Zhang K, Jiang L, Rathod PK. Essential Protein-Protein Interactions between Plasmodium falciparum Thymidylate Synthase and Dihydrofolate Reductase Domains. J Biol Chem. 1999;274:37781–37786. doi: 10.1074/jbc.274.53.37781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein dye-binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 28.Sirawaraporn W, Prapunwattana P, Sirawaraporn R, Yuthavong Y, Santi DV. The dihydrofolate reductase domain of Plasmodium falciparum thymidylate synthase-dihydrofolate reductase. Gene synthesis, expression, and anti-folate-resistant mutants. J Biol Chem. 1993;268:21637–21644. [PubMed] [Google Scholar]
- 29.Chen G, Mueller C, Wendlinger M, Zolg J. Kinetic and molecular properties of the dihydrofolate reductase from pyrimethamine-sensitive and pyrimethamine-resistant clones of the human malaria parasite Plasmodium falciparum. Mol Pharmacol. 1987;31:430–437. [PubMed] [Google Scholar]
- 30.Alifrangis M, Lemnge MM, Ronn AM, Segeda MD, Magesa SM, Khalil IF, Bygbjerg IC. Increasing prevalence of wildtypes in the dihydrofolate reductase gene of Plasmodium falciparum in an area with high levels of sulfadoxine/pyrimethamine resistance after the introduction of treated bed nets. Am J Trop Med Hyg. 2003;69:238–243. [PubMed] [Google Scholar]
- 31.Osman ME, Mockenhaupt FP, Bienzle U, Elbashir MI, Giha HA. Field-based evidence for linkage of mutations associated with chloroquine (pfcrt/pfmdr1) and sulfadoxine-pyrimethamine (pfdhfr/pfdhps) resistance and for the fitness cost of multiple mutations in P. falciparum. Infect Genet Evol. 2007;7:52–59. doi: 10.1016/j.meegid.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 32.Ngo T, Duraisingh M, Reed M, Hipgrave D, Biggs B, Cowman AF. Analysis of pfcrt, pfmdr1, dhfr, and dhps mutations and drug sensitivities in Plasmodium falciparum isolates from patients in Vietnam before and after treatment with artemisinin. Am J Trop Med Hyg. 2003;68:350–356. [PubMed] [Google Scholar]
- 33.Masimirembwa CM, Phuong-dung N, Phuc BQ, Duc-Dao L, Sy ND, Skold O, Swedberg G. Molecular epidemiology of Plasmodium falciparum antifolate resistance in Vietnam: genotyping for resistance variants of dihydropteroate synthase and dihydrofolate reductase. Int J Antimicrob Agents. 1999;12:203–211. doi: 10.1016/s0924-8579(99)00061-8. [DOI] [PubMed] [Google Scholar]
- 34.Cortese JF, Plowe CV. Antifolate resistance due to new and known Plasmodium falciparum dihydrofolate reductase mutations expressed in yeast. Mol Biochem Parasitol. 1998;94:205–214. doi: 10.1016/s0166-6851(98)00075-9. [DOI] [PubMed] [Google Scholar]
- 35.Mookherjee S, Howard V, Nzila-Mouanda A, Watkins W, Sibley CH. Identification and analysis of dihydrofolate reductase alleles from Plasmodium falciparum present at low frequency in polyclonal patient samples. Am J Trop Med Hyg. 1999;61:131–140. doi: 10.4269/ajtmh.1999.61.131. [DOI] [PubMed] [Google Scholar]
- 36.Hastings MD, Bates SJ, Blackstone EA, Monks SM, Mutabingwa TK, Sibley CH. Highly pyrimethamine-resistant alleles of dihydrofolate reductase in isolates of Plasmodium falciparum from Tanzania. Trans R Soc Trop Med Hyg. 2002;96:674–676. doi: 10.1016/s0035-9203(02)90349-4. [DOI] [PubMed] [Google Scholar]
- 37.Wilson PE, Kazadi W, Alker AP, Meshnick SR. Rare Congolese Plasmodium falciparum DHFR alleles. Mol Biochem Parasitol. 2005;144:227–229. doi: 10.1016/j.molbiopara.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 38.Hastings I. Complex dynamics and stability of resistance to antimalarial drugs. Parasitology. 2006:1–10. doi: 10.1017/S0031182005009790. [DOI] [PubMed] [Google Scholar]
- 39.Iyer JK, Milhous WK, Cortese JF, Kublin JG, Plowe CV. Plasmodium falciparum cross-resistance between trimethoprim and pyrimethamine. Lancet. 2001;358:1066–1067. doi: 10.1016/S0140-6736(01)06201-8. [DOI] [PubMed] [Google Scholar]