

Abstract
Mammalian cells have been presumed to repair potentially lethal chromosomal double-strand breaks (DSBs) in large part by processes that do not require homology to the break site. This contrasts with Saccharomyces cerevisiae where the major DSB repair pathway is homologous recombination. Recently, it has been determined that DSBs in genomic DNA in mammalian cells can stimulate homologous recombination as much as 3 or 4 orders of magnitude, suggesting that homology-directed repair may play an important role in the repair of chromosomal breaks. To determine whether mammalian cells use recombinational repair at a significant level, we have analyzed the spectrum of repair events at a defined chromosomal break by using direct physical analysis of repair products. When an endonuclease-generated DSB is introduced into one of two direct repeats, homologous repair is found to account for 30–50% of observed repair events. Both noncrossover and deletional homologous repair products are detected, at approximately a 1:3 ratio. These results demonstrate the importance of homologous recombination in the repair of DSBs in mammalian cells. In the remaining observed repair events, DSBs are repaired by nonhomologous processes. The nonhomologous repair events generally result in small deletions or insertions at the break site, although a small fraction of events result in larger chromosomal rearrangements. Interestingly, in two insertions, GT repeats were integrated at one of the broken chromosome ends, suggesting that DSB repair can contribute to the spread of microsatellite sequences in mammalian genomes.
Keywords: homologous recombination, DNA end-joining, single-strand annealing, I-SceI, microsatellite repeats
Repair of chromosome breaks is necessary for the maintenance of genome integrity in all organisms. In Saccharomyces cerevisiae double-strand breaks (DSBs) are repaired primarily by homologous recombination (1). Homologous repair frequently occurs by gene conversion, a conservative process in which little or no sequence information is lost during the repair process (2, 3). A nonconservative type of recombinational repair also exists, termed single-strand annealing (4). In single-strand annealing, a DSB in or near one of two directly repeated sequences leads to a recombination intermediate in which complementary strands from each repeated sequence are annealed. The product of such a repair event contains only one copy of the repeated sequences, with a deletion of sequences originally present between the two repeats. Nonhomologous repair of DSBs occurs infrequently in yeast and is detected only when there is no homology from which to repair a broken chromosome, or when the homologous recombination machinery in a cell is disabled (4).
In contrast to yeast, mammalian cells have been presumed to repair chromosomal breaks primarily by nonhomologous processes (5). The evidence for this is indirect, coming from transfection studies in which it is found that nonhomologous integration of introduced DNA predominates over homologous integration. Recent studies, however, in which DSBs have been experimentally introduced into mammalian genomes have demonstrated that chromosomal DSBs in mammalian cells, as in yeast, are highly recombinogenic, stimulating recombination as much as 3–4 orders of magnitude (6). This stimulation is seen whether the homology exists in an allelic position (7), as a tandemly repeated chromosomal sequence (8–10), or as an exogenously introduced DNA (i.e., gene targeting; refs. 8 and 11–14).
To resolve these apparent contradictions, we have attempted to analyze, using an unbiased approach, all of the major types of repair detectable at a defined DSB in a mammalian genome. The DSB was introduced into one of two chromosomal repeats in hamster cells by the rare-cutting I-SceI endonuclease. Physical analysis of repair products demonstrates a major contribution of homologous recombination to DSB repair in mammalian cells, with up to 50% of the observed repair occurring by recombination. Homologous repair results in both noncrossover and deletional events. Interestingly, one type of nonhomologous repair includes insertion of microsatellite repeats at a broken chromosomal end.
MATERIALS AND METHODS
Cell Transfections.
Transfections were done with uncut plasmids using 1.6 × 107 (or, for clonal analysis, 3 × 108) CHO-K1 DRA10 cells in PBS. Cells were electroporated at 250 V and 960 μF with 30 μg (or 100 μg, where indicated) of plasmid DNA, either pCMV3xnls-I-SceI or pCMV-lacZ. The I-SceI expression vector, pCMV3xnls-I-SceI, is a modified version of pCMV-I-SceI (15), containing a triplicated nuclear localization signal fused to I-Sce I (G. Donoho, M.J., and P. Berg, unpublished results). In cases in which drug selection was imposed, cells were selected in 1 mg/ml of G418 with or without hygromycin (0.5 mg/ml) beginning 24 h after electroporation. In experiments carried out without selection, clones derived from single cells were isolated by plating dilutions of cells into 96-well plates and examining individual wells immediately after dilution for the presence of a single cell, and after a few days for the presence of a single colony.
DNA Manipulations.
Southern analysis was performed by using 8 μg of genomic DNA according to standard procedures (16). Probes were the 5′ neo gene fragment (XhoI–Nar I) or the entire neo gene fragment (XhoI–BamHI). PCR experiments were carried out by using primer 1, neo5′Pst (5′CTGTCCGGTGCCCTGAATGAA), which hybridizes 410 bp upstream of the I-SceI cleavage site, and primer 2, neo3′Bam (5′CGGGATCCGAACAAACGACCCAA), which hybridizes 302 bp downstream of the cleavage site.
RESULTS
Homology-Directed Repair of DSBs.
The CHO-K1 DRA10 hamster cell line has one copy of the DRneo repair substrate stably integrated into its genome (8). This substrate contains two directly repeated copies of the neo gene separated by a hygR gene (Fig. 1A). One copy, called S2neo, is mutated at an NcoI restriction site by deletion of 4 bp of neo sequences and insertion of the 18-bp I-SceI endonuclease cleavage site (13). Downstream of S2neo, and in direct orientation with it, is a 0.7-kb 3′ neo gene fragment. The 3′ neo gene fragment contains a wild-type sequence at the NcoI site, which can be used to correct the mutation in the S2neo gene. It also contains a silent base-pair mutation 22 bp downstream of the NcoI site, which creates an SspI restriction site polymorphism (11). DSB-promoted homologous recombination between an I-SceI-cleaved S2neo gene and a 3′ neo gene on the same chromatid or on a sister chromatid will result in a neo+ gene. Recombination can occur by gene conversion or deletional “popout” recombination (Fig. 1B). In this system, we define gene conversions as noncrossover events in which the 3′ neo gene acts as a donor of information. Popout recombination events are defined as events that lead to a deletion of one of the neo repeats and the intervening sequence, without implying a specific mechanism for the event (see Discussion).
Figure 1.

DSB repair substrate DRneo. (A) The DRneo substrate contains the 18-bp cleavage site for the rare-cutting endonuclease I-SceI (black bar) inserted in the S2neo gene. The homology between the S2neo gene and the 685-bp 3′ neo gene is indicated by the stippling. (B) Two outcomes of DSB-induced homologous recombination that produce a neo+ gene.
Gene conversion and popout recombination can be distinguished by retention or loss of hygromycin resistance, respectively, after DSB repair (Fig. 1B). To examine DSB-promoted recombination events, CHO-K1 DRA10 cells were electroporated with an I-SceI expression vector, pCMV3xnlsI-SceI, or a control vector, pCMV-lacZ. Cells were selected in G418 only, to detect all neo+ clones, or G418 and hygromycin, to detect neo+ clones derived from noncrossover gene conversion events. As seen previously, DSBs induced by I-SceI expression were found to stimulate homologous recombination between the neo repeats more than 200-fold (ref. 8; Table 1). The frequency of neo+ clones was approximately 4-fold higher than the frequency of neo+hyg+ clones, indicating that popout recombinants predominate over simple gene convertants (Table 1). Because the G418-only selection includes both neo+hyg+ and neo+hyg− colonies, popout recombinants can be deduced to be approximately 3-fold more abundant than noncrossover gene conversion events. Two other CHO-K1 clones with an integrated DRneo repair substrate gave rise to a similar excess of popout recombinants after DSB repair, as have murine cell lines that have been tested (M.H. and M.J., unpublished results).
Table 1.
DSB-promoted recombination by popout recombination and gene conversion
| Transfected DNA | Frequency neo+ colonies | Frequency neo+hyg+ colonies |
|---|---|---|
| pCMV3xnlsI-SceI | 4.0 × 10−3 | 0.9 × 10−3 |
| pCMV-lacZ | ≤2 × 10−5 | ≤2 × 10−5 |
These results were confirmed by Southern analysis of genomic DNA from pools of cells selected in G418. Recombination by either mechanism would result in the presence of an NcoI site at the former position of the I-SceI site, giving rise to a 0.9-kb XhoI–NcoI band (Fig. 1B). Genomic DNA from a pool of cells selected with G418 only gives rise to the expected 0.9-kb band, demonstrating that neo+ clones are derived from homologous recombination. Gene conversion without crossing over would be expected to maintain both neo gene repeats on a 4.0-kb XhoI–HindIII fragment, whereas popout recombination would delete one neo repeat and the hygR gene, resulting in a 1.1-kb XhoI–HindIII band (Fig. 1B). Genomic DNA from the pool of neo+ cells gives rise to both the 4.0- and 1.1-kb XhoI–HindIII fragments (Fig. 2), with the 1.1-kb fragment being more intense than the 4.0-kb fragment.
Figure 2.
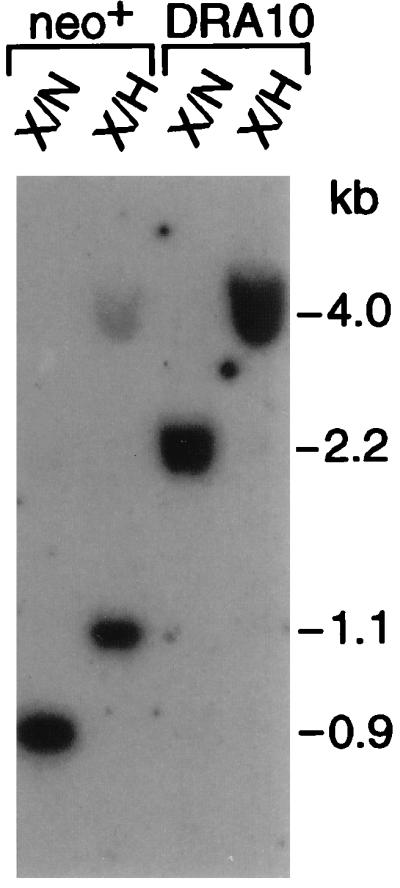
Southern blot analysis of pooled DSB-induced recombinants. Genomic DNA from the parental CHO-K1 DRA10 cell line and a pool of neo+ recombinants derived from expression of I-SceI was cleaved with either XhoI–NcoI (X/N) or XhoI–HindIII (X/H). The 5′ neo gene probe (probe A, see Fig. 1) has the same amount of homology to both types of recombination products.
Physical Analysis of DSB Repair Products After I-SceI Expression.
Because the S2neo gene is nonrevertable, selecting for a neo+ gene after I-SceI cleavage eliminates cells that have undergone nonhomologous repair of the chromosome break. To detect all classes of repair events in which the I-SceI site is lost, we developed nonselective strategies. We developed a PCR assay in which we could physically monitor repair products at time points after I-SceI expression, even without a requirement for cellular viability. Because nonhomologous repair products found in mammalian cells frequently involve small sequence changes at the cleavage site (e.g., ref. 11), we expected that we would be able to PCR-amplify many of the nonhomologous repair products at the neo locus, as well as the homologous products. PCR was performed on genomic DNA from pools of cells electroporated with the I-SceI expression vector and then grown in nonselective media for 0, 4, and 24 h until DNA recovery. To reduce the amplification of genomic DNA that retained the I-SceI site, genomic DNA was precleaved in some cases with I-SceI before PCR.
PCR was performed by using primers that were 410 bp upstream and 302 bp downstream of the I-SceI cleavage site (Fig. 3A). The 5′ primer is upstream of the homology region, whereas the 3′ primer is within the homology region to be able to amplify both types of homologous repair products. I-SceI cleavage of the amplified product indicated that a major portion of the cells had retained the cleavage site, either because the genome was never cleaved or because it was cleaved but then repaired to retain the site (Fig. 3B). Precleavage of the genomic DNA with I-SceI significantly reduced this product although it did not eliminate it. NcoI digestion of the amplified product from genomic DNA isolated 24 h after electroporation gave rise to readily detectable fragments of the size expected from homologous repair. These fragments were not detectable at 0 or 4 h after electroporation, demonstrating a requirement for a DSB to induce recombination. Double digestion with NcoI and I-SceI uncovered a band 24 h postelectroporation that was resistant to both enzymes. This band was not seen at 0 h but is apparent at a reduced amount 4 h after electroporation.
Figure 3.

Physical analysis of DSB repair events. (A) Scheme for PCR analysis. Genomic DNA was prepared from pools of cells that were transfected with the I-SceI expression vector and incubated in nonselective media for various times. The neo gene was subsequently amplified with primers 1 and 2, and the amplified product was cleaved with NcoI and I-SceI to detect homologous (NcoI+/I-SceI−) and nonhomologous (NcoI−/I-SceI−) repair products. Nonhomologous repair could result in deletions (Δ) or insertions (++). (B) PCR analysis of genomic DNA that was not cut in vitro by I-SceI (Upper) or was cut in vitro with I-SceI (Lower) before PCR amplification. After PCR amplification, DNA was electrophoresed either uncleaved or cleaved with the indicated endonucleases and then probed with the entire neo gene probe (see Fig. 1, probe B). Genomic DNA preparations were 0, 4, or 24 h after transfection, as indicated.
Quantitation of the NcoI+/I-SceI− (homologous) and NcoI−/I-SceI− (nonhomologous) products was performed by using PhosphorImager analysis to determine the relative amount of the two types of repair products. Comparing results from three independent experiments, we find that the NcoI+/I-SceI− repair products are between 30% and 50% of the total observed repair products, suggesting a significant contribution of homologous recombination to DSB repair.
Random Clone Analysis of Unselected DSB Repair Events.
Because PCR analysis would not detect gross changes to the neo locus, clonal analysis also was performed in the absence of selection to physically analyze all major types of repair products. Cells were electroporated with a large amount (100 μg) of either the I-SceI expression vector or the control lacZ vector and then cloned in 96-well plates in media that contained neither hygromycin nor G418. Wells were examined after electroporation for the presence of a single cell and after 3 or 4 days for the presence of a single colony. Single clones were expanded for genomic DNA preparation.
A total of 48 clones were examined from electroporation of the I-SceI vector and 21 clones from electroporation of the lacZ vector. Approximately one-third of the clones from electroporation with the I-SceI expression vector were found to have undergone loss of the I-SceI site (Fig. 4A), whereas none of the clones derived from the control lacZ electroporation had lost the site (Fig. 4B). In the control lacZ electroporation, all 21 clones contained the parental 3.1- and 0.9-kb XhoI–I-SceI–HindIII fragments. These two bands were unaltered in 30 of the clones electroporated with the I-SceI vector (e.g., Fig. 4A, clones 1–4, 6, 8, 12, 13, and 34). The other 18 clones had altered bands, indicative of cleavage of the I-SceI site and repair to result in its loss. Five clones had a single band of 4.0 kb, indicating loss of the I-SceI site without any gross change to the locus (e.g., clones 5, 10, and 11). Three other clones had a single band of 1.1 kb, indicative of popout recombination (e.g., clones 9 and 14). In contrast to these eight clones, nine others had a mixture of bands. These mixed clones were of two types. Five contained both the parental bands and a repair product of either 4.0 or 1.1 kb and four contained two different repair products. For example, clone 19 had both the 4.0-kb repair band and the parental 3.1- and 0.9-kb bands (Fig. 5). When these mixed clones were subcloned, the subclones had either one of the two genotypes (Fig. 5). Because wells were carefully screened after electroporation for a single cell, we interpret these mixed clones as being derived from an electroporated cell that had undergone I-SceI cleavage after DNA replication. In addition to these 17 clones with predictable types of repair, one clone was found to have a substantially altered neo locus. In this case, one large fragment was seen that hybridized to the neo probe (Fig. 4A; clone 33), suggesting that there was a deletion of greater than 0.9 kb or that there was some other gross nonhomologous rearrangement at the locus.
Figure 4.

Southern blot analysis of randomly isolated clones. Genomic DNA from cells that were electroporated with pCMV3xnlsI-SceI (A) or pCMVlacZ (B) and cloned in nonselective media was cleaved with XhoI–I-SceI–HindIII to detect popout (PO), gene conversion (GC), and nonhomologous end-joining (EJ) products. The variation in intensities of the hybridization signal in some lanes is caused by unevenness in the amount of genomic DNA loaded. The entire neo gene was used as probe. In clone 33, an undetermined type of repair event occurred, indicated by a ?.
Figure 5.

Southern blot analysis of subclones from a clone containing a mixed genotype. Genomic DNA was cleaved with XhoI–I-SceI–HindIII and probed with the entire neo gene.
The 22 repair products from the 18 clones were examined in more detail. The 4.0-kb repair product could be derived either from nonhomologous end-joining or from gene conversion. Southern blot analysis was performed by using NcoI digestion of genomic DNA to distinguish these two types of events. Three clones were found to have converted the I-SceI site to an NcoI site (data not shown). These clones were also resistant to G418 and hygromycin, indicating that they had undergone gene conversion. Eleven clones had not converted the cleavage site to NcoI (data not shown), implying that the DNA ends were nonhomologously rejoined. Supporting this, the clones were sensitive to G418, although they maintained resistance to hygromycin. One of these 11 clones was found to have undergone two distinct end-joining events. Similarly, clones (or subclones from mixed clones) with the 1.1-kb band were analyzed by Southern blotting and drug selection. These clones were resistant to G418 and sensitive to hygromycin, as expected, and the remaining neo gene repeat contained the NcoI site.
Of the 48 analyzed clones, none were found to have completely lost the neo hybridization signal, as would have been obtained with a deletion of 4 kb or more. If this type of deletion had occurred after DNA replication, however, it would have been masked by the hybridization signal of the other chromosome. To check for this possibility, a few clones with the parental DRneo locus were subcloned. All of these subclones were found to have retained the entire locus (data not shown). Considering that only one of 22 repair events resulted in a gross nonhomologous rearrangement, these types of DSB repair are apparently rare, comprising 5% or fewer of the repair events at this locus.
Sequence Analysis of Nonhomologous End-Joining Events.
To examine the nature of the nonhomologous rejoining events, PCR was performed on genomic DNA from cell clones to amplify the neo locus and the amplified fragments were sequenced. Seven clones were found to contain small deletions at the cleavage site (Fig. 6A). The smallest deletion was 1 bp from the last base of one of the I-SceI overhangs. The largest deletion was 21 bp and included nucleotides on both sides of the cleavage site.
Figure 6.

Sequence analysis of nonhomologous repair products. (A) Sequences of nonhomologous rejoining products derived from the S2neo gene. The underlined nucleotides in deletion products 5 and 7.2 indicate sequence overlap between the two ends at the junction. Bases from the ATAA I-SceI overhang are duplicated in insertions 11, 10, and 16X (indicated in lowercase letters at the break site). Note that some clones have two different repair products (i.e., clone 7, deletion 7.2 and insertion 7.1; clone 16, insertion 16X and a PO event not shown; and clone 35, deletion 35.1 and a PO event not shown). (B) Diagram of insertion products. Insertions are indicated by the stippled bars except for the insertions of the GT repeats (lined bars) and the simian virus 40 (SV40) origin region (arrows in 16X). The SV40 sequences are derived from three nearby regions that were joined together at a 6-bp sequence overlap and a 13-bp palindromic repeat, as shown.
In addition to clones with deletions, five clones were found to contain insertions that ranged in size from 45 to 205 bp. Sequences were determined for each of these inserts (Fig. 6; data not shown). Two inserts contained GT dinucleotide repeats, one with 19 repeats and the other with 23 repeats. The GT repeats in both inserts were directly incorporated into the neo locus without intervening sequences. At the junction of the break site and the GT repeat in both clones, 1 bp could be derived from either the sequence at the break site or the first bp of the GT repeat (Fig. 6B), which may indicate that one base at the chromosomal break site paired with one base of the GT repeat to initiate the repair event. In both clones, the inserts contained additional sequences at the other junction.
Two other inserts were 45 and 48 bp long. To determine the complexity of genomic DNA from which these sequences were derived, the inserts were used to probe CHO-K1 hamster DNA. In both cases, the inserts were from single copy sequences (data not shown). The inserts had ORFs (data not shown), although the significance of this is not clear because the inserts were short. The fifth insert originated from the transfected I-SceI expression vector. Vector sequences in the insert were not contiguous, but came from three nearby regions in the simian virus 40 origin of replication contained within the plasmid (Fig. 6B). (Note that this is not a functional origin of replication in hamster cells.)
DISCUSSION
By using direct physical analysis of repair products, we have examined DSB repair in a defined chromosomal context in a mammalian cell line and determined that homologous recombination is a major pathway for DSB repair. Homologous recombination, as previously demonstrated and as shown here, is stimulated more than 100-fold by a DSB at the genomic locus. Physical examination of repair products by PCR across the break site or by Southern blot analysis of randomly generated clones demonstrates that homology-directed repair occurs in one-third to one-half of the events. Because the two homologous chromosomal repeats used in our assay differ by the presence of the 18-bp I-SceI cleavage site and a nearby silent mutation, it is possible that levels of recombination could be even higher when a break occurs in one of two identical sequences.
The random clone analysis is the most unbiased study of DSB repair in mammalian cells performed to date. Of 22 observed repair events, nine were homologous and 13 were nonhomologous (Fig. 7). For the homologous events, popout recombination was observed to predominate over noncrossover gene conversion, and this finding was confirmed by selective strategies. The conservative DSB repair model proposed for yeast recombination predicts an equal frequency of crossover and noncrossover events (2), although other models predict a bias toward noncrossover events (3). The analysis of conservative recombination events between introduced plasmids and genomic DNA in mammalian cells appears to support a preference for noncrossover events (refs. 17 and 18; C. Richardson, J. Winderbaum, and M.J., unpublished results). The excess of popout recombinants from direct repeat recombination suggests that many of these recombinants are not generated by gene conversion events that involve crossing over but instead by the nonconservative single-strand annealing pathway (19). The random clone analysis tends to further support single-strand annealing for generating popout recombinants, because the reciprocal product is not observed in any of the clones. Whereas a nonconservative pathway would result in loss of the sequence between the repeats, conservative recombination between sister chromatids would result in the deletion product and the reciprocal triplication product. None of the six clones that underwent popout recombination contains the reciprocal product (Fig. 4A; data not shown). Similarly, conservative recombination between repeats on the same chromatid would lead to an excised circle that could reintegrate at another chromosomal location. None of the popout recombinants showed evidence of excised circle integration, although we cannot rule out that excised circles would have been degraded. Interestingly, the noncrossover gene conversion products in all three cases were part of mixed clones, suggesting that the clones underwent conversion after the locus was replicated. Further analysis will be valuable in determining whether conservative recombination events are regulated during the cell cycle.
Figure 7.

Summary of DSB repair events derived from random clone analysis. Number of each type of repair event is indicated. PO, popout recombination; GC, noncrossover gene conversion; Δ, deletional rejoining; ++, insertional rejoining; Gr, gross chromosomal rearrangement.
The very high induction of homologous recombination that is observed in mammalian cells when a chromosome is broken suggests that homology-directed repair is not restricted to direct repeats but rather is a more general repair mechanism, perhaps reflective of an important contribution of sister chromatid recombination to repair. Once DNA is replicated, sister chromatids provide a general homologous repair template for all chromosomal sequences. Because sister chromatids are identical to each other, there is no barrier to recombination as would exist between diverged repetitive elements (e.g., refs. 14 and 20 and references therein). Homologous chromosomes, like sister chromatids, can provide a general repair template for all chromosomal sequences, except an XY pair. However, even though allelic recombination is induced by DSBs, the level remains low compared with other types of DSB-induced recombination (7), suggesting that homologs are only rarely used.
By Southern blot analysis, we can detect an I-SceI-cleaved chromosomal band as early as 4 h after electroporation (unpublished results). Because the cleaved band is never more than a few percent of total at various times after transfection, all chromosomal sites may not be cleaved during the course of transient I-SceI expression or they may be restored to the original sequence. Restoration of the I-SceI site could occur through recombination with an intact sister chromatid or, simply, precise ligation of the broken ends. Repair events that restore the original sequence may play an important role in DSB repair, although they are necessarily excluded from the analysis presented here in which loss of the I-SceI site is assayed. It is possible that some clones had undergone multiple rounds of cleavage and precise repair, either through ligation or recombination, before loss of the I-SceI site. To address this, it will be necessary to synchronize cleavage of the chromosome in a majority of cells.
In addition to homologous repair, we observe that nonhomologous repair is also a major repair pathway in mammalian cells. The events are primarily small deletions and insertions. Deletion junctions are broadly similar to those that have been previously reported in mouse cells at repaired chromosomal I-SceI sites (11, 21), although fewer rejoining events at microhomologies were found in the hamster cells than in the mouse cells. Interestingly, two inserts contained microsatellite GT repeats directly joined to one end of the break site. A GT repeat at a break site has been detected in one other study, in this case (GT)15 joined to restriction enzyme-cleaved genomic DNA (22). Although GT repeats are highly repetitive, they comprise only 0.1% of mammalian genomic DNA (estimating 100 K repeats of 30 bp each; ref. 23). Thus, they appear to be overrepresented in insert junctions described to date in DSB repair. This raises the possibility that GT repeats are preferred genomic sites to be copied during DSB repair and that DSB repair contributes to their spread in genomes. Consistent with this, the positions of GT repeats in mammalian genomes are not extensively conserved, with only a fraction of (GT)n sites having arisen before the divergence of primates and rodents (24). The potential for these sequences to form unusual structures (25) may promote their use during repair.
The spectrum of DSB repair events we observed in our random clone analysis is broader than those seen in other DSB repair studies. In one study, which relied on more gross chromosomal rearrangements to score nonhomologous repair, DSBs were suggested to stimulate nonhomologous rearrangements to a 10-fold greater extent than homologous recombination (9). However, this is apparently because of a higher baseline rate of spontaneous homologous recombination to spontaneous nonhomologous rearrangements (9), rather than a 10-fold greater preference for nonhomologous repair. It may be expected that the proportion of various types of repair events will vary somewhat with the chromosomal context of a DSB. For example, our higher ratio of popout to conversion events than previously seen (9) may be because of the closer proximity of our neo gene repeats.
The substantial use of two major repair pathways in mammalian cells, homologous and nonhomologous repair, contrasts with what is seen in S. cerevisiae, where homologous repair greatly predominates. The genome of budding yeast is extremely compact, having few and very short introns, short intergenic regions, and few repetitive elements. Being unicellular, the yeast cell is also its own germ line. Thus, in yeast nonhomologous repair that leads to deletions or insertions is likely to be functionally mutagenic. Because mammalian genomes have abundant repetitive elements and large introns and intergenic regions and the germ line is set aside early in development, nonhomologous repair is much less likely to have serious deleterious consequences. When sister chromatids are available, homologous repair may be favored to suppress mutagenesis. However, when sister chromatids are not available for repair, nonhomologous repair may be favored to suppress potentially deleterious outcomes of other types of homologous recombination. For example, somatic recombination between alleles is a common mechanism for loss of heterozygosity in tumor cells (26) and recombination between Alu repeats is known to generate a number of genetic diseases, including cancers (e.g., ref. 27). Considering the recent cloning of genes involved in DSB repair (5, 28) and the development of tools to study repair (6), it should be possible to further dissect the contribution of these two pathways of repair.
Acknowledgments
We thank Scott Keeney and members of the Jasin laboratory for comments on the manuscript. This work was supported by grants from the American Cancer Society (NP-82674) and the National Institutes of Health (GM54688) to M.J.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviation: DSB, double-strand break.
References
- 1.Petes T D, Malone R E, Symington L S. Recombination in Yeast. Plainview, NY: Cold Spring Harbor Lab. Press; 1991. [Google Scholar]
- 2.Szostak J W, Orr-Weaver T L, Rothstein R J, Stahl F W. Cell. 1983;33:25–35. doi: 10.1016/0092-8674(83)90331-8. [DOI] [PubMed] [Google Scholar]
- 3.Stahl F. Cell. 1996;87:965–968. doi: 10.1016/s0092-8674(00)81791-2. [DOI] [PubMed] [Google Scholar]
- 4.Haber J E. BioEssays. 1995;17:609–620. doi: 10.1002/bies.950170707. [DOI] [PubMed] [Google Scholar]
- 5.Jackson S P, Jeggo P A. Trends Biochem Sci. 1995;20:412–415. doi: 10.1016/s0968-0004(00)89090-8. [DOI] [PubMed] [Google Scholar]
- 6.Jasin M. Trends Genet. 1996;12:224–228. doi: 10.1016/0168-9525(96)10019-6. [DOI] [PubMed] [Google Scholar]
- 7.Moynahan M E, Jasin M. Proc Natl Acad Sci USA. 1997;94:8988–8993. doi: 10.1073/pnas.94.17.8988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liang F, Romanienko P J, Weaver D T, Jeggo P A, Jasin M. Proc Natl Acad Sci USA. 1996;93:8929–8933. doi: 10.1073/pnas.93.17.8929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sargent R G, Brenneman M A, Wilson J H. Mol Cell Biol. 1997;17:267–277. doi: 10.1128/mcb.17.1.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taghian DG, Nickoloff J A. Mol Cell Biol. 1997;17:6386–6393. doi: 10.1128/mcb.17.11.6386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rouet P, Smih F, Jasin M. Mol Cell Biol. 1994;14:8096–8106. doi: 10.1128/mcb.14.12.8096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choulika A, Perrin A, Dujon B, Nicolas J-F. Mol Cell Biol. 1995;15:1963–1973. doi: 10.1128/mcb.15.4.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smih F, Rouet P, Romanienko P J, Jasin M. Nucleic Acids Res. 1995;23:5012–5019. doi: 10.1093/nar/23.24.5012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elliott B, Richardson C, Winderbaum J, Nickoloff J A, Jasin M. Mol Cell Biol. 1998;18:93–101. doi: 10.1128/mcb.18.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rouet P, Smih F, Jasin M. Proc Natl Acad Sci USA. 1994;91:6064–6068. doi: 10.1073/pnas.91.13.6064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maniatis T, Fritsch E F, Sambrook J. Molecular Cloning: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1982. [Google Scholar]
- 17.Jasin M, deVilliers J, Weber F, Schaffner W. Cell. 1985;43:695–703. doi: 10.1016/0092-8674(85)90242-9. [DOI] [PubMed] [Google Scholar]
- 18.Jasin M, Berg P. Genes Dev. 1988;2:1353–1363. doi: 10.1101/gad.2.11.1353. [DOI] [PubMed] [Google Scholar]
- 19.Lin F-L, Sperle K, Sternberg N. Mol Cell Biol. 1990;10:103–112. doi: 10.1128/mcb.10.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chambers S R, Hunter N, Louis E J, Borts R H. Mol Cell Biol. 1996;16:6110–6120. doi: 10.1128/mcb.16.11.6110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lukacsovich T, Yang D, Waldman A S. Nucleic Acids Res. 1994;22:5649–5657. doi: 10.1093/nar/22.25.5649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Phillips J W, Morgan W F. Mol Cell Biol. 1994;14:5794–5803. doi: 10.1128/mcb.14.9.5794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hamada H, Petrino M G, Takunaga T. Proc Natl Acad Sci USA. 1982;79:6465–6469. doi: 10.1073/pnas.79.21.6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stallings R L, Ford A F, Nelson D, Torney D C, Hildebrand C E, Moyzis R K. Genomics. 1991;10:807–815. doi: 10.1016/0888-7543(91)90467-s. [DOI] [PubMed] [Google Scholar]
- 25.Nordheim A, Rich A. Proc Natl Acad Sci USA. 1983;80:1821–1825. doi: 10.1073/pnas.80.7.1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lasko D, Cavenee W, Nordenskjöld M. Annu Rev Gen. 1991;25:281–314. doi: 10.1146/annurev.ge.25.120191.001433. [DOI] [PubMed] [Google Scholar]
- 27.Schichman S A, Caligiuri M A, Strout M P, Carter S L, Gu Y, Canaani E, Bloomfield C D, Croce C M. Cancer Res. 1994;54:4277–4280. [PubMed] [Google Scholar]
- 28.Shinohara A, Ogawa T. Trends Biochem. 1995;20:387–391. doi: 10.1016/s0968-0004(00)89085-4. [DOI] [PubMed] [Google Scholar]