

Abstract
We have recently identified a novel basal transcription complex, TAC, that is present and active in embryonal carcinoma (EC) cells but not in other adult cells such as COS7. In the search for factors involved in TAC formation, we found that expression of the adenoviral 12S E1A oncoprotein abolishes TAC formation in EC cells. This effect of E1A depends on its N-terminal domain that is essential for cell differentiation and that targets the transcriptional coactivators p300 and PCAF. Expression of p300 lacking its major E1A interaction domain, CH3, restores TAC formation in the presence of E1A, in a bromodomain- and HAT domain-dependent manner. Consistently, the unprocessed TFIIAαβ precursor that is selectively assembled into TAC is acetylated preferentially compared with the processed subunits present in ‘free’ TFIIA. Intriguingly, expression of p300 in COS7 cells that do not contain detectable levels of TAC instigates formation of TAC from endogenous components. Our data suggest that p300 plays a role in formation of the TBP–TFIIA-containing basal transcription complex, TAC.
Keywords: E1A/EC cells/p300/TAC
Introduction
Transcription initiation of protein-encoding genes by RNA polymerase II (Pol II) critically depends on the TATA-binding protein (TBP). In yeast, at least two forms of transcriptionally active TBP appear to be present: a TAF-containing and a TAF-free form (Kuras et al., 2000; Li et al., 2000). The TAF-containing form is TFIID, a multiprotein complex that consists of TBP and a number (10–13) of TBP-associated factors (TAFIIs) (reviewed in Lee and Young, 1998). The TAF-free form appears to be ‘free’ TBP or an as yet unidentified TBP-containing complex. In line with the existence of a TBP-sans-TAFs in yeast, a TBP–TFIIA-containing complex (TAC) has been identified recently in embryonal carcinoma (EC) cells (Mitsiou and Stunnenberg, 2000). Whether TAF-containing and TAF-free TBP forms are selectively recruited by distinct (co)activators remains to be elucidated.
The mammalian proteins p300 and CBP, collectively referred to as p300/CBP, are transcriptional coactivators that are highly related in structure and function (Chrivia et al., 1993; Eckner et al., 1994). Both proteins play important roles in cell growth, transformation and development (reviewed in Goodman and Smolik, 2000). p300 and CBP contain multiple functional domains that are highly conserved and serve as binding sites for a plethora of factors (reviewed in Shikama et al., 1997). The so-called CH3 region is the site for interaction with many transcription factors, including the adenovirus E1A oncoprotein, the coactivator PCAF and the SV40 large T antigen (Eckner et al., 1994, 1996; Arany et al., 1995; Yang et al., 1996). Interaction of p300/CBP with general transcription factors such as TFIIB and RNA Pol II-containing complexes suggest that p300/CBP may play a direct role in transcription initiation by serving as a molecular bridge between upstream transcriptional activators and the basal machinery (Kwok et al., 1994; Nakajima et al., 1997a,b; Felzien et al., 1999). p300/CBP possesses intrinsic histone acetyltransferase (HAT) activity (Bannister and Kouzarides, 1996; Ogryzko et al., 1996) and it has been demonstrated that p300 is recruited to chromatin templates in vitro by direct interaction with activators to stimulate transcription by histone acetylation (Kundu et al., 2000). p300/CBP has been shown further to acetylate transcriptional activators, general transcription factors and chromatin-associated proteins (reviewed in Sterner and Berger, 2000).
EC cells are pluripotent stem cells derived from teratocarcinomas and resemble early embryonic cells. EC cells are widely used as a model system to study the regulation of gene expression during very early developmental stages of mammals. F9 and P19 EC cells can be induced to differentiate by expression of the adenoviral 12S E1A oncoprotein that depends on the N-terminal part as well as the conserved region 1 (CR1) of E1A protein (Slack et al., 1995). Interestingly, the same regions of E1A are involved in binding to the transcriptional coactivators p300/CBP and PCAF (Wang et al., 1993; Arany et al., 1995; Lundblad et al., 1995; Reid et al., 1998; Chakravarti et al., 1999), suggesting a direct, functional link between these coactivators and EC cell differentiation. Although it seems clear that E1A induces differentiation of EC cells by regulating p300/CBP activity or function (Kawasaki et al., 1998; Ugai et al., 1999), downstream effectors of p300/CBP that redirect gene expression in EC cells have not been identified.
We recently have identified a TBP–TFIIA-containing complex (TAC) that lacks classical TAFs and that is present in EC but not in other adult cells such as COS7 (Mitsiou and Stunnenberg, 2000). TAC is associated with chromatin in vivo and plays a functional role in transcriptional activation in EC cells. In the present study, we show that expression of the adenoviral 12S E1A oncoprotein in P19 EC cells affects TAC by destabilizing it or by preventing its formation/accumulation. This effect of E1A requires its N-terminal domain that targets the transcriptional coactivators p300/CBP and PCAF. Interestingly, co-expression of p300 nullifies the effect of E1A on TAC accumulation in a bromodomain- and HAT domain-dependent manner. Consistently, we found that the TFIIAαβ precursor assembled into TAC is acetylated in vivo in P19 EC cells. Expression of p300 that is unable to interact with E1A (p300del30) also restores TAC formation, showing that p300 is downstream of E1A. Our data suggest that E1A interacts with p300 and inhibits its function, resulting in abolition of TAC in EC cells. Furthermore, we show that expression of p300 in COS7 cells results in formation of TAC from endogenous components. We suggest that p300 regulates the formation and consequently the function of TAC in mammalian cells.
Results
Adenoviral 12S E1A oncoprotein abolishes TAC formation in P19 EC cells
We previously have identified a TBP–TFIIA-containing complex, TAC, that is present at relatively high levels in EC cells but not in other adult cells such as COS7, HeLa or L cells. TAC contains the TFIIAγ subunit and the unprocessed form of TFIIAαβ along with TBP, and it is devoid of classical TAFs. TAC is assembled efficiently upon transfection of its individual components, i.e. hTBP and hTFIIAαβ + γ (hTFIIA for short) in P19 EC but not in COS7 cells (Mitsiou and Stunnenberg, 2000). In this study, we exploited this phenomenon to identify auxiliary factors required for the formation of TAC.
To assess whether the presence of TAC correlates with the pluripotent state of P19 EC cells, differentiation of P19 EC cells was induced by expression of the adenoviral 12S E1A oncoprotein. As described previously, co-expression of hTBP and hTFIIA resulted in stabilization of hTBP due to formation of a complex with TFIIA, the so-called ‘exogenous’ TAC (Figure 1A; Mitsiou and Stunnenberg, 2000). Co-expression of E1A abolished stabilization of hTBP (Figure 1A, upper panel, compare lanes 1–6 with lanes 7–12), indicating that TAC does not accumulate in the presence of E1A. Immunoprecipitations using a monoclonal antibody against TBP confirmed that hTFIIAαβ and hTFIIAγ are indeed stably associated with hTBP in the absence but not in the presence of E1A (Figure 1C, compare lanes 1 and 2; data not shown). In COS7, HeLa or L cells, TAC is not formed upon co-expression of hTBP and hTFIIA, and consequently expression of E1A did not affect accumulation of hTBP levels (Figure 1A, lower panel and data not shown).

Fig. 1. Expression of 12S E1A protein abolishes TAC accumulation and activity in an N-terminal-dependent manner. (A) Stabilization of hTBP by increasing amounts of hTFIIAαβ + γ is abolished upon co-expression of 12S E1A in P19 EC cells. Extracts from P19 EC or COS7 cells transfected with expression plasmids encoding hTBP, Myc-tagged hTFIIAαβ, HA-tagged hTFIIAγ and 12S E1A were analyzed by SDS–PAGE and immunoblotting using the anti-TBP antibody SL39. Amounts of extract loaded for each sample were adjusted according to the expression levels of the CAT internal control. endTBP = endogenous TBP. (B) Schematic diagram of 12S E1A protein. The shaded boxes represent conserved regions CR1 and CR2. The bars represent interaction domains of E1A with p300/CBP, PCAF and retinoblastoma family proteins (pRb), as indicated. (C) The N-terminal part of 12S E1A is required for TAC destabilization. Extracts from P19 EC cells transfected with expression plasmids encoding hTBP, hTFIIAαβ + γ and 12S E1A (wild-type or deletions), as indicated, were subjected to immunoprecipitation using SL39. The immunoprecipitates were eluted with synthetic peptide and were analyzed by immunoblotting using antibodies against TBP (SL39) and hTFIIAαβ (Myc). (D) The N-terminal part of 12S E1A is required for repression of TAC-dependent transactivation. P19 EC cells were transfected with the RARE-M1-tk-luc reporter, the pSV2-CAT internal control plasmid and expression plasmids as in (C). Luciferase values obtained from cell extracts were normalized relative to expression of the CAT internal control.
To assess the domain(s) of E1A required for its effect on TAC accumulation, we used a series of deletion mutants of 12S E1A (Figure 1B). Immunoprecipitation experiments showed that accumulation of exogenous TAC was unaffected upon co-expression of E1A carrying an N-terminal deletion (del4–25) (Figure 1C, compare lanes 1 and 4). Co-expression of E1A with deletions in CR1 and in the region preceding CR2 inhibited TAC accumulation, similar to wild-type E1A (compare lane 1 with lanes 2, 3 and 5–10). To corroborate and extend these observations, we tested the effect of E1A on TAC activity. Transient transfection assays were performed using a luciferase reporter containing the tk promoter with a mutated TATA box (TGTAAA M1-TATA) and an upstream retinoic acid (RA)-responsive element (RARE), in combination with a hTBP with altered DNA binding specificity (hTBP-spm3; Strubin and Struhl, 1992; Mitsiou and Stunnenberg, 2000). As shown previously, ligand-induced and RA receptor-mediated activation of transcription was stimulated by co-expression of hTBP-spm3 and hTFIIA (Figure 1D; Mitsiou and Stunnenberg, 2000). Co-expression of wild type E1A nullified TAC-dependent transcription, whereas E1A del4–25 did not affect transcription; a shorter N-terminal deletion (del1–14) resulted in partial inhibition. All other mutants reduced the transcriptional activity of TAC to levels similar to that observed with wild-type E1A, paralleling their effect on accumulation of TAC. Taken together, the above data argue that expression of E1A prevents accumulation of TAC and that this effect requires the extreme N-terminal part of the E1A protein.
TAC is formed in differentiated EC cells
Expression of E1A in EC cells is known to trigger their differentiation, and hence the effect of E1A on TAC formation/accumulation might be indirect, i.e. the consequence of differentiation of the cells. Therefore, we tested whether TAC can still be detected in P19 EC cells induced to differentiate by RA or dimethylsulfoxide (DMSO). Visceral endoderm-like (END-2) and mesoderm-like (MES-1) cells, isolated from RA- and DMSO-treated P19 EC cells (Mummery et al., 1985, 1986), were used in transient transfection experiments. Figure 2 shows that co-expression of hTBP and hTFIIA resulted in stabilization of hTBP in END-2 and MES-1 cells (compare lanes 1 and 7 with lanes 3 and 10, respectively). Immunoprecipitations using a monoclonal antibody against hTFIIAαβ further confirmed that TBP is stably associated with hTFIIA in both cell lines (lanes 6 and 12). These results suggest that differentiation of P19 EC cells per se does not abolish TAC formation and that the presence of TAC possibly correlates with the embryonic rather than the pluripotent state of P19 EC cells.

Fig. 2. TAC is formed in differentiated EC cells. Extracts from MES-1 and END-2 cells were transfected with expression plasmids as indicated and subjected to immunoprecipitation using the Myc antibody. Peptide elution with synthetic peptide was followed by immunoblotting using SL39. endTBP = endogenous TBP.
p300 is required for TAC formation in P19 EC cells
Our observation that the N-terminal region of E1A is critical for abolition of TAC combined with numerous observations reporting the involvement of the N-terminus of E1A in interactions with the transcriptional coactivators p300/CBP and PCAF (Wang et al., 1993; Arany et al., 1995; Chakravarti et al., 1999) urged us to assess the role of p300, CBP and/or PCAF in TAC formation in P19 EC cells.
Co-expression of p300 with E1A restored TAC accumulation (Figure 3B, compare lanes 2 and 3), whereas neither CBP, that is closely related to p300, nor PCAF nullified the effect of E1A on TAC (lane 5 and data not shown). These findings could imply that p300 is involved in TAC formation and that E1A inhibits its function. Alternatively, E1A affects TAC formation directly, and co-expression of p300 sequesters or modifies E1A to prevent its effect on TAC. To discriminate between these possibilities, a p300 protein was tested that lacks part of the CH3 region (p300del30; Figure 3A) and that is unable to interact efficiently with E1A (Eckner et al., 1994; also Figure 4C and data not shown). If p300 sequesters E1A and consequently nullifies the effect of E1A on TAC formation, then expression of p300del30 should not re-facilitate TAC formation. However, co-expression of p300del30 yielded TAC to levels that were even higher than obtained with wild-type p300 (Figure 3B, compare lanes 3 and 4). It should be noted that both p300 wt and p300del30 were expressed at similar levels (data not shown). These observations suggest that E1A is acting directly or indirectly on p300 and that p300 is involved in TAC formation. In line with this conclusion, expression of p300 in the absence of E1A may result in elevated levels of TAC. Indeed, co-expression of hTBP and hTFIIA together with p300 wt or p300del30 resulted in a moderate but reproducible increase of TAC levels (Figure 3C, compare lane 1 with lanes 2 and 3). Co-expression of CBP only slightly if at all increased TAC levels (compare lanes 1 and 4), indicating that CBP does not affect TAC formation in P19 EC cells. Taken together, these data show that p300 is involved in TAC formation in P19 EC cells.

Fig. 3. p300 is involved in TAC formation in P19 EC cells. (A) Schematic diagram of wild-type (wt) and mutant variants of human p300. Interacting proteins are listed above the figure. Functional domains are: N-terminal region (NR), CREB-binding domain, bromodomain, cysteine–histidine-rich regions (CH1, CH2 and CH3), histone acetyltransferase domain (HAT) and glutamine-rich (Q-rich) region. The N-terminal truncations of p300 are: ΔN244, ΔN514 and ΔN870 lacking amino acids 1–243, 1–513 and 1–869, respectively; del30, ΔBromo, ΔSRC and ΔQ lack amino acids 1737–1809, 1071–1241, 2042–2157 and 2042–2375, respectively. The six amino acid substitutions in the MutAT2 protein are shown. (B) p300 restores TAC formation in the presence of 12S E1A in P19 EC cells. Extracts from P19 EC cells transfected with expression plasmids encoding hTBP and hTFIIAαβ + γ, in the presence or the absence of 12S E1A, HA-tagged p300 (wt or del30) and CBP, were subjected to immunoprecipitation using SL39. Peptide elution and western blot analysis were performed as in Figure 1C. (C) p300 increases TAC formation in P19 EC cells. Extracts from P19 EC cells transfected with expression plasmids encoding hTBP and hTFIIAαβ + γ, in the presence or absence of HA-tagged p300 (wt or del30) and CBP, were subjected to immunoprecipitation and western blot analysis as in (B).

Fig. 4. Domains of p300 required for TAC formation in P19 EC cells. (A) Effect of the N-terminal truncations of p300 on TAC formation in the presence of 12S E1A in P19 EC cells. Extracts from P19 EC cells transfected with expression plasmids encoding hTBP and hTFIIAαβ + γ, in the presence or absence of 12S E1A and Myc-tagged p300 (wt and del30, full-length or N-terminal truncations), were subjected to immunoprecipitation using the HA antibody. Peptide elution was followed by immunoblotting using SL39. (B) Expression of p300 in P19 EC cells. Extracts from P19 EC cells transfected with expression plasmids encoding Myc-tagged p300 (as in A) were subjected to immunoblotting using an antibody against p300 (Myc). (C) E1A interacts with the CH3 domain of p300 in an N-terminal-dependent manner. Extracts from P19 EC cells transfected with plasmids expressing12S E1A (wild-type or del4–25) and Myc-tagged p300 ΔN870 (wt or del30) were subjected to immunoprecipitation using the anti-E1A antibody M73. The immunoprecipitates were analyzed by immunoblotting as in (B). The input represents one-tenth of the samples analyzed for immunoprecipitation. (D) Effect of C-terminal mutants of p300 on TAC formation in the presence of 12S E1A in P19 EC cells. Extracts from P19 EC cells transfected with expression plasmids encoding hTBP and hTFIIAαβ + γ, in the presence or absence of 12S E1A and Myc-tagged p300 (wt, ΔSRC or ΔQ), were subjected to immunoprecipitation and western blotting as in (A). (E) The HAT domain and bromodomain of p300 are required for TAC formation in the presence of 12S E1A in P19 EC cells. Extracts from P19 EC cells transfected with expression plasmids encoding hTBP and hTFIIAαβ + γ, in the presence or absence of 12S E1A and Myc-tagged p300 (wt or mutants), were subjected to immunoprecipitation using the Myc antibody and western blot analysis as in (A). endTBP = endogenous TBP.
p300 domains required for TAC formation
To delineate the domain(s) of p300 involved in TAC formation, a series of N-terminal deletion mutants were used (Figure 3A). Extracts from transfected cells were assessed for TAC accumulation in the presence or absence of 12S E1A and p300 derivatives. Accumulation of TAC was restored upon co-expression of p300 lacking the N-terminal region (NR) and the CH1 domain (Figure 4A, compare lane 3 with lanes 4 and 5). In contrast, expression of p300 lacking amino acids 1–869 did not restore TAC accumulation (lane 6), indicating that the region between amino acids 514 and 869 is important for TAC formation. Note that similar levels of p300 proteins (full-length and N-terminal truncations) were expressed in P19 EC cells by transfecting appropriate amounts of each expression plasmid (Figure 4B).
To provide further evidence that the p300 derivatives affect TAC formation, the N-terminal truncations were also created in the context of p300del30 protein (Figure 3A). Co-expression of p300del30 lacking the NR and the CH1 domains restored TAC as the parental p300del30 protein (Figure 4A, compare lane 7 with lanes 8 and 9). As observed with truncations of p300 wt, TAC accumulation was abolished upon co-expression of p300del30 lacking amino acids 1–869 (lane 10), reinforcing the notion that amino acids 514–869 of p300 are required for TAC formation. Immunoprecipitations using an antibody against E1A showed that wild-type p300 as well as the N-terminal truncations interacted with wild-type E1A but not with E1A carrying the N-terminal deletion (Figure 4C, lane 8; data not shown). However, p300del30 (full-length and truncations) did not interact with E1A (compare lanes 4 and 9; data not shown), in agreement with reports showing that the CH3 region of p300 is the primary binding site for E1A (Eckner et al., 1994).
Next we tested the effect of mutations in the C-terminal region of p300 on TAC formation. Expression of a p300 lacking the site for the binding of the SRC/p160 family of coactivator proteins (ΔSRC, Figure 3A) re-facilitated TAC formation in the presence of E1A (Figure 4D, compare lanes 2 and 3 with lane 4). In contrast, a p300 mutant lacking the majority of the C-terminal Q-rich domain (ΔQ, Figure 3A) did not restore TAC formation (lane 5).
Furthermore, expression of a p300 mutant containing six amino acid substitutions in the HAT domain that does not exhibit HAT activity (p300 MutAT2, Figure 3A; Kraus et al., 1999) did not restore TAC accumulation in the presence of E1A (Figure 4E, compare lanes 2 and 3 with lane 5). In addition, expression of p300 lacking the bromodomain (ΔBromo, Figure 3A) did not re-facilitate TAC formation in the presence of E1A (lane 4). Given that p300 ΔBromo is not able to acetylate nucleosomal histones (Kraus et al., 1999), it is feasible that deletion of the bromodomain affects the HAT activity of p300. At present, we cannot exclude that the bromodomain mediates protein–protein interactions that are critical for TAC formation. The above data provide evidence that p300 plays a critical role in TAC formation involving the region between amino acids 514 and 869, the HAT domain and bromodomain as well as the Q-rich domain.
The TFIIAαβ precursor assembled into TAC is acetylated
The critical role of p300 and its HAT activity on TAC formation opened up the possibility that one or more components of TAC are acetylated.
We first tested whether components of native TAC are acetylated in P19 EC cells. Endogenous TAC was purified by Ni2+-NTA affinity chromatography followed by immunopurification (Mitsiou and Stunnenberg, 2000). Western blot analysis using an antibody directed against acetyl-lysine residues detected acetylation of endogenous mouse TFIIAαβ precursor (mTFIIAαβ) in Ni2+-NTA eluates as well as in immunopurified native TAC (Figure 5A, lanes 3–5). Acetylation of mTFIIAα was also detected in the Ni2+-NTA eluates but not in immunopurified TAC (compare lane 3 with lanes 4 and 5). Taking into account that the unprocessed mTFIIAαβ was much less abundant than the processed mTFIIAα subunit (lane 2), the mTFIIAαβ appeared to be hyperacetylated in P19 EC cells. Acetylated mTFIIAβ, mTFIIAγ or mTBP were not detected either in the Ni2+-NTA eluates or in immunopurified TAC (Figure 5A and data not shown).

Fig. 5. Acetylation of TAC components. (A) Extracts from P19 EC were subjected to affinity chromatography using an Ni2+-NTA–agarose column. Ni2+-NTA eluates were subjected to immunoprecipitation under low (lane 4) or high stringency conditions (lane 5) using SL39 as in Figure 1C. Western blot analysis was performed using polyclonal antibodies against TFIIAαβ (β- and αN-specific, lanes 1 and 2) or acetyl-lysine residues (lanes 3–5). The Ni2+-NTA input (lanes 1–3) represents one-fifth of the eluates analyzed for immunoprecipitation. mTFIIA = endogenous mouse TFIIA. (B and C) Extracts from P19 EC cells transfected with expression plasmids encoding hTBP and hTFIIAαβ + γ, as indicated, and pulsed with [3H]sodium acetate were subjected to immunoprecipitation using the Myc antibody. Peptide elution was followed by SDS–PAGE and fluorography (B) or immunoblotting using antibodies against TBP (SL39), hTFIIAαβ and hTFIIAα (Myc) and hTFIIAγ (HA) (C). The asterisks indicate non-related acetylated proteins that were immunoprecipitated.
Having established that the TFIIAαβ precursor is acetylated preferentially in native TAC and given the role of p300 and its HAT activity in TAC formation, we tested whether components of TAC assembled de novo from transfected proteins are acetylated. To gain independent support for acetylation of TAC components, we used in vivo metabolic labeling with [3H]sodium acetate followed by immunopurification of de novo assembled TAC. Apart from labeling acetylated lysine residues, metabolic labeling may also result in incorporation of label into modified N-terminal methionines. Consequently, we cannot directly deduce from the fluorograph whether the observed labeling of hTFIIAαβ, hTFIIAα, hTFIIAγ and hTBP resulted from acetylation of lysines or modification of N-terminal methionines (Figure 5B, lanes 2 and 3). However, given that the hTFIIAαβ precursor was present at much lower levels compared with the processed hTFIIAα subunit (Figure 5C), it can be concluded that the hTFIIAαβ was acetylated preferentially on lysine residues, in agreement with the results obtained for endogenous mTFIIAαβ (Figure 5A). Moreover, co-expression of hTBP resulted in increased acetylation of the hTFIIAαβ precursor (compare lanes 2 and 3) in line with its assembly into TAC (Mitsiou and Stunnenberg, 2000). Labeling of hTFIIAβ that it is generated by proteolytic cleavage and hence does not contain an N-terminal methionine was not detected (Figure 5B, expected position indicated by arrowhead). This finding is consistent with the fact that the antibody against the acetylated lysine residues did not detect endogenous mTFIIAβ (Figure 5A). Given that endogenous mTBP and mTFIIAγ were not detected by the anti-acetylated lysine antibody, we tentatively conclude that the in vivo labeling of hTFIIAγ and hTBP is most probably due to modification of their N-terminal methionine. Immunoprecipitation of hTFIIA from COS7 cells did not reveal significant acetylation of any of the hTFIIA subunits (data not shown), consistent with the fact that TAC cannot be detected in these cells (Mitsiou and Stunnenberg, 2000). Taken together, the above data show that the TFIIAαβ precursor assembled into TAC is acetylated preferentially.
Expression of p300 in COS7 cells yields de novo assembly of TAC
We have shown previously that TAC is present and can be readily formed from transfected components in P19 EC but not in other adult cells such as COS7, HeLa or L cells (Mitsiou and Stunnenberg, 2000). Given the observation that p300 is involved in TAC formation in P19 EC cells, we tested whether expression of p300 yields detectable levels of TAC in COS7 cells. Co-expression of p300 with hTBP and hTFIIA in COS7 cells resulted in co-immunoprecipitation of hTBP and hTFIIAαβ (Figure 6A, compare lanes 1 and 2), indicative of the formation of TAC. To corroborate and extend these observations, the effect of the three N-terminal truncations of p300 on TAC formation in COS7 cells was tested. Figure 6B shows that deletion of the NR and CH1 domains did not affect p300-dependent formation of TAC (compare lane 2 with lanes 3 and 4). However, TAC was not formed upon co-expression of p300 lacking amino acids 1–869 (lane 5), in agreement with the observations made in P19 EC cells (Figure 4A). In light of the effect of E1A on TAC accumulation in P19 EC cells, we considered the possibility that the SV40 large T antigen could have exerted the same effect in COS7 cells, resulting in undetectable TAC levels, and that p300 sequesters large T antigen. However, co-expression of p300del30 that is unable to interact with large T antigen (Eckner et al., 1996) facilitates TAC formation in COS7 cells, similar to p300 wt (data not shown). In addition, expression of large T antigen in P19 EC cells did not affect formation of TAC (data not shown). These results indicate that p300 is involved in TAC formation in COS7 cells.

Fig. 6. De novo assembly of TAC upon expression of p300 in COS7 cells. (A) Expression of p300 results in TAC formation in COS7 cells. Extracts from COS7 cells transfected with expression plasmids encoding hTBP and hTFIIAαβ + γ, in the presence or absence of HA-tagged p300, were subjected to immunoprecipitation using SL39 or the Myc antibody. Peptide elution with the respective synthetic peptides was followed by immunoblotting as described in Figure 1C. (B) Effect of the N-terminal truncations of p300 on TAC formation in COS7 cells. Extracts from COS7 cells transfected with expression plasmids encoding hTBP and hTFIIAαβ + γ, in the presence or the absence of Myc-tagged p300 (full-length or N-terminal truncations), were subjected to immunoprecipitation using the Myc antibody. Peptide elution and western blot analysis were performed as described in Figore 3A. (C) TAC formed in COS7 cells in the presence of p300 mediates transcriptional activation in vivo. COS7 cells were transfected with the Gal4-TATA-luc reporter, the pSV2-CAT internal control plasmid and expression plasmids encoding HA-tagged p300, hTBP, Myc-tagged hTFIIAαβ and HA-tagged hTFIIAγ. Luciferase values were expressed as in Figure 1D.
Next, we tested whether TAC formed in COS7 cells in the presence of p300 can support transcription of a reporter gene in vivo. To circumvent that p300 affects transcription due to its recruitment as a bridging factor to the promoter by upstream transcription activators, the ability of de novo assembled TAC to support basal transcription in COS7 cells was assessed using a minimal promoter. Transient transfection assays were performed using a luciferase reporter containing five Gal4-binding sites upstream of the wild-type TATA box (Gal4-TATA-luc). In the absence of a Gal4 activator, transcription will depend solely on the TATA box. Co-expression of hTBP and hTFIIA in COS7 cells did not yield appreciable levels of transcription from this minimal promoter (Figure 6C). Importantly, co-expression of p300 with hTBP and hTFIIA resulted in 10-fold activation of transcription.
The above data provide evidence that TAC is formed and is transcriptionally active in COS7 cells from exogenously provided TBP and TFIIA upon co-expression of p300.
Elevation of p300 levels in COS7 cells results in formation of native TAC
Since p300 facilitates formation of functional TAC from exogenous, transfected components, expression of p300 could also lead to TAC formation from endogenous TBP and TFIIA in COS7 cells. To test this hypothesis, COS7 cells were transfected with expression plasmids encoding p300 and green fluorescent protein (GFP) and sorted by fluorescence-activated cell sorting (FACS) analysis based on GFP expression (Figure 7A). Extract was prepared from GFP-positive cells and used for immunoprecipitation with a monoclonal antibody against endogenous TBP (Figure 7B). Significantly, the endogenous TFIIAαβ precursor was co-immunoprecipitated with TBP upon expression of p300 (lane 2), showing that expression of p300 results in formation of native TAC from endogenous components. As expected, the processed TFIIAα subunit (expected position indicated by arrowhead) was not precipitated because it is not assembled into TAC. In non-transfected COS7 cells, i.e. in the absence of p300, TFIIAαβ was not immunoprecipitated by TBP (compare lanes 1 and 2). Finally, we tested whether native TAC formed upon expression of p300 in COS7 cells could facilitate transcription from the GAL4-TATA-luc reporter. In the absence of p300, the minimal promoter was inactive but, upon expression of p300, transcription was boosted up to 5-fold (Figure 7C). These findings provide strong evidence that p300 is involved in formation of native TAC in COS7 cells.
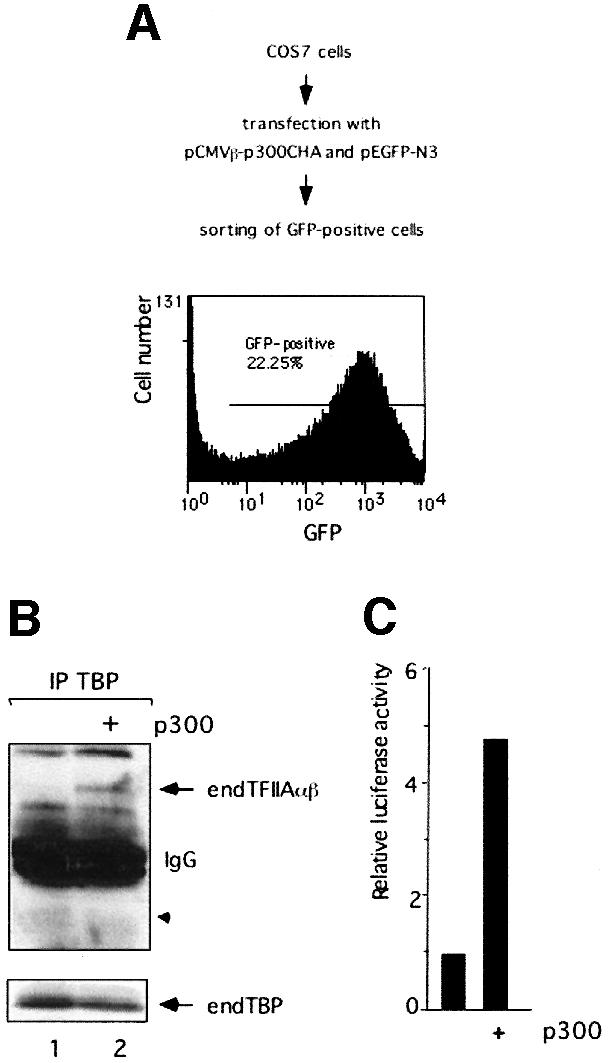
Fig. 7. Formation of native TAC upon expression of p300 in COS7 cells. (A) FACS analysis of COS7 cells co-expressing p300 and GFP, and sorted by FACS based on GFP expression. By selecting optimal parameters, a fraction of positive cells that was >95% pure was obtained. (B) Expression of p300 results in formation of native TAC in COS7 cells. Cell extracts were subjected to immunoprecipitation using the anti-TBP antibody SL27. Lane 1, non-transfected COS7 cells; lane 2, COS7 cells transfected and sorted as described in (A). Western blot analysis was performed using antibodies against TFIIAαβ (α-specific) and TBP (SL39). endTFIIAαβ and endTBP represent endogenous proteins. (C) Native TAC formed in COS7 cells upon expression of p300 is transcriptionally active. COS7 cells were transfected with the Gal4-TATA-luc reporter, the pSV2-CAT internal control plasmid and the expression plasmid encoding HA-tagged p300. Luciferase values were expressed as in Figure 1D.
Discussion
p300 governs TAC formation in P19 EC cells
Our data have provided several lines of evidence to suggest that p300 is involved in formation of TAC in P19 EC cells. First, expression of E1A abolishes TAC formation and this effect requires the N-terminal domain of E1A (Figure 1). Secondly, expression of p300 in P19 EC cells results in elevated TAC levels, and co-expression of p300 along with E1A restores TAC formation (Figure 3). Thirdly, co-expression of p300 lacking its interaction domain for E1A (p300del30) also re-facilitates TAC formation (Figures 3 and 4). If E1A would act on TAC formation independently of p300, and p300 would ‘merely’ sequester and neutralize the effect of E1A, then p300del30 should have been unable to re-facilitate TAC formation. We confirmed that deletion of amino acids 1737–1809 in the CH3 region of p300 (del30) indeed prevents interaction between p300 and E1A (Figure 4C and data not shown) in line with other studies (Eckner et al., 1994). It has, however, also been reported that E1A can interact with p300 through the N-terminal region (amino acids 1–450) and possibly through a region in the C-terminus (approximately amino acids 2000–2200) (Kurokawa et al., 1998; Lipinski et al., 1999). The fact that expression of p300 wt or p300del30 lacking amino acids 1–513 is able to restore TAC formation efficiently in the presence of E1A (Figure 4A) excludes the possibility that the N-terminal part of p300 sequesters and nullifies the effect of E1A. In addition, the C-terminal part of p300 (amino acids 2000–2200) does not seem to contribute to an interaction between E1A and p300 in our experiments (Figure 4C and data not shown), consistent with previous reports (Eckner et al., 1994). Finally, expression of p300 in COS7 cells facilitates formation and transcriptional activity of TAC. Collectively, our experiments suggest that E1A affects TAC formation by binding directly to p300, thereby inhibiting p300 function including its HAT activity. Reduced HAT activity of p300 may result in deacetylation of the TFIIAαβ precursor and destabilization of TAC (see below).
p300 facilitates TAC formation in COS7 cells
Our experiments reveal that p300 is either the missing factor required for TAC formation in adult cells or can substitute for such a factor (Figures 6 and 7). We have shown previously that native TAC was undetectable in COS7 cells and that de novo assembly of TAC from transfected components does not occur (Mitsiou and Stunnenberg, 2000). We now show that co-expression of p300 not only results in de novo assembly of TAC from co-transfected components but, most importantly, facilitates formation of native TAC composed of endogenous genome-encoded TBP, TFIIAαβ and -γ in COS7 cells. We can exclude the possibility that the SV40 large T antigen inhibits TAC formation in COS7 cells and that p300 nullifies this effect. First, expression of p300 lacking its large T antigen interaction domain (p300del30) facilitates TAC formation in COS7 cells and, secondly, expression of large T antigen in P19 EC cells did not affect TAC formation. It is thus conceivable that TAC is not formed in COS7 and other adult cells (such as HeLa or L) because they do not express sufficient levels of p300 or an activity that p300 can substitute for. However, TAC may be present in other adult cell lines expressing high levels of p300.
Role of p300 in TAC formation and function
Although we have provided evidence that p300 is required for TAC formation, the molecular mechanisms of TAC formation/function and the role of p300 in this process are not yet clear.
The precise role of p300 can only be speculated upon at present. It is likely that p300 is directly involved in TAC formation by interacting with TAC components. Observations made by us and others are indicative of such an interaction. It has been reported that p300 interacts with TBP and that the N-terminal part of p300 (amino acids 1–743) is involved in this interaction (Abraham et al., 1993; Yuan et al., 1996). If so, this may explain our finding that p300 lacking amino acids 1–869 is not able to facilitate TAC formation.
Given that the HAT activity of p300 is required for TAC formation and that the TFIIAαβ precursor is acetylated in P19 EC cells, it is conceivable that acetylation of TFIIAαβ precursor by p300 is a prerequisite for TAC formation. Acetylation of TFIIA in EC cells may alter its interactions with TBP. Since TAC contains the TFIIAαβ precursor but not the processed forms TFIIAα and TFIIAβ (Mitsiou and Stunnenberg, 2000), it is also likely that acetylation regulates the processing of the TFIIAαβ precursor and thereby its assembly into TAC. The precise role of acetylation in TAC formation and transcriptional activity will have to be assessed with detailed mutational and biochemical analyses. Since the Q-rich domain of p300 is also required for TAC formation (Figure 4D), it is tempting to speculate that p300 interacts with TAC components via its Q-rich domain, whereas the HAT domain acetylates TFIIAαβ. Such a role for p300 in DNA-binding/activity of IRF-3 has already been proposed (Suhara et al., 2002). At present, we do not exclude the possibility that other factors may act downstream of p300 to mediate its effect on TAC formation.
An intriguing question is whether p300 is ‘solely’ required for formation or also for function of TAC. Given the plethora of transcription factors that interact with p300 and the proposed bridging factor function of p300, it seems likely that p300 recruited by upstream factors physically and directly participates in transcription by recruiting TAC to nucleate formation of a transcription-competent complex. In line with such a scenario, a direct physical interaction between p300 and TBP has been reported previously (Abraham et al., 1993; Yuan et al., 1996). Since p300 has been shown to interact with the PCAF/GCN5 coactivator (Yang et al., 1996), it is also conceivable that p300 bridges an interaction between TAC and the recently identified TBP-free, TAFII-containing complexes (SAGA in yeast and TFTC, PCAF, GCN5 or STAGA in human cells; reviewed in Bell and Tora, 1999) via their PCAF component. This hypothesis is also supported by genetic data from yeast indicating that members of the SPT family of proteins, which are components of the SAGA complexes, physically and functionally interact with TBP and TFIIA (Eisenmann et al., 1992; Barlev et al., 1995; Madison and Winston, 1997; Roberts and Winston, 1997; Sterner et al., 1999).
In conclusion, our data argue that p300 governs TAC formation and suggest that regulation of TAC formation and function by p300 may contribute to transcription of a subset of specific genes. The presence of TAC in pluripotent embryonal cells such as embryonic stem (ES; our unpublished data) and EC cells (NT2 and P19) as well as in their differentiated embryonal derivatives MES-1 and END-2, but not in some adult cells (such as COS7, HeLa or L), and the involvement of p300 in TAC formation are all suggestive of a role for TAC during early embryogenesis. At present, we cannot exclude the possibility that TAC is present and active at low levels in other adult cells, in particular those containing high levels/activity of p300. Further analysis using ES cells and specific inactivation of p300 and/or CBP may unravel the precise role of p300 in TAC formation/function. Identification of target genes should provide insight into TAC-dependent regulation of specific gene expression patterns.
Materials and methods
Plasmids and antibodies
Expression plasmids encoding hTBPs (wt and spm3), Myc-tagged hTFIIAαβ and hemagglutinin (HA)-tagged hTFIIAγ, reporter plasmid RARE-M1-tk-luc and internal control plasmid pSV2-CAT have been described (Mitsiou and Stunnenberg, 2000). Plasmid Gal4-TATA-luc is a luciferase reporter containing five Gal4-binding sites upstream of the wild-type TATA box. The following expression plasmids for the 12S E1A wild-type and mutant proteins were used: dl520 (wt), dl1504 (del1–14), dl1101/520 (del4–25), dl1102/520 (del26–35), dl1103/520 (del30–49), dl1104/520 (del48–60), dl1105/520 (del70–81), dl1106/520 (del90–105) and dl1107/520 (del111–123) (Jelsma et al., 1989).
Expression plasmids encoding human p300 were as follows. pCMVβ-p300CHA and pCMVβ-p300del30CHA expressing HA-tagged p300 and p300del30, respectively, were provided by R.Eckner (Eckner et al., 1994). pSG8-p300-Myc and pSG8-p300del30-Myc expressing Myc-tagged p300 and p300del30, respectively, were constructed by subcloning the NotI–NheI fragment from plasmids pCMVβ-p300CHA and CMVβ-p300del30CHA into the NotI–NheI site of pSG8-Myc (pSG8 empty expression vector containing the Myc epitope in the NheI–HindIII site). The pSG8-p300-Myc and pSG8-p300del30-Myc plasmids were also used to generate the three N-terminal truncations of p300 and p300del30 (ΔN244, ΔN514 and ΔN870). pSG8-p300ΔBromo-Myc, pSG8-p300ΔSRC-Myc and pSG8-p300MutAT2-Myc expressing Myc-tagged p300 lacking amino acids 1071–1241 and 2042–2157, and p300 containing six amino acid substitutions (H1415A, E1423A, Y1424A, L1428S, Y1430A and H1434A), respectively, were constructed by subcloning the NotI–NheI fragment from plasmids pBluescript-p300ΔBromo, pBluescript-p300ΔSRC and pBluescript-p300MutAT2, provided by W.L.Kraus (Kraus et al., 1999), into the NotI–NheI site of pSG8-Myc. pSG8-p300ΔQ-Myc expressing Myc-tagged p300 lacking amino acids and 2042–2375 was constructed by deleting the sequence encoding amino acids 2157–2375 from plasmid pSG8-p300ΔSRC-Myc.
Expression plasmid pRC/RSV-mCBP·HA encoding HA-tagged mouse CBP was provided by T.Kouzarides. Expression plasmid pEGFP-N3 encoding enhanced green fluorescent protein (EGFP) was purchased from Clontech.
The monoclonal antibodies SL39, SL27, Myc and HA and the polyclonal antibodies against hTFIIAαβ (α-specific, αN-specific and β-specific) have been described previously (Ruppert et al., 1996; Mitsiou and Stunnenberg, 2000). The following antibodies were also used: M73 monoclonal antibody against E1A (ascites, provided by E.Harlow) and a polyclonal antibody against acetyl-lysine-containing proteins (anti-acetyl-Lys; Upstate).
Cell culture, transient transfections, in vivo labeling and FACS analysis
P19 EC and COS7 cells were maintained as described (Berkenstam et al., 1992). P19 EC derivatives END-2 and MES-1, provided by C.L.Mummery, have been described (Mummery et al., 1985, 1986). For transfections, each 6 cm dish received: 5 µg of luciferase reporter plasmid, 0.5 µg of the internal control plasmid pSV2-CAT, 1 µg of plasmids expressing hTBPs, 2 µg of plasmids expressing hTFIIAαβ and hTFIIAγ, 1 µg of plasmids expressing 12S E1A (wild-type or mutants), 0.2 µg of plasmid expressing GFP, 1 µg of plasmids expressing p300 ΔN870 and p300del30 ΔN870 and 5 µg of plasmids expressing all the other p300 proteins (wild-type and derivatives) and CBP, as indicated. In Figure 1A, increasing amounts (0.25, 0.50, 1.00 and 2.00 µg) of plasmids expressing hTFIIAαβ and hTFIIAγ were used. At 16–18 h after transfection, cells were washed twice with phosphate-buffered saline and incubated in fresh medium in the presence (in the case of the RARE-M1-tk-luc reporter) or in the absence of 1 × 10–6 M 9-cis-RA for 24 h before harvesting. For in vivo labeling, cells were incubated in fresh medium containing 1 mCi/ml of [3H]sodium acetate (16.1 Ci/mmol, ICN) for 1 h before harvesting.
For FACS analysis, COS7 cells were co-transfected with expression plasmids encoding p300 and GFP. The forward-scatter (FS), side-scatter (SS) and cell fluorescence were analyzed using a Coulter Epics Altra flowcytometer equipped with an Enterprise II laser running at 100 mW (Argon). Viable, GFP-positive cells were detected using a band-pass filter 525/30 nm for green fluorescence and a dichroic mirror of 550 nm, and sorted at high speed (15 000 cells/s).
Protein extracts, luciferase and CAT assays, Ni2+-NTA–agarose affinity chromatography, immunoprecipitation, SDS–PAGE, immunoblotting and fluorography
Preparation of cell extract, luciferase and chloramphenicol acetyltransferase (CAT) assays, purification of TFIIA by Ni2+-NTA–agarose affinity chromatography, immunoprecipitation, peptide elution, SDS–PAGE and immunoblotting were carried out as previously described (Mitsiou and Stunnenberg, 2000).
For fluorography, proteins in the gel labeled with [3H]sodium acetate were fixed with 30% methanol and 10% glacial acetic acid for 1 h, then the gel was enhanced by incubating with EN3HANCE (NEN) for 1.5 h, immersed in 2% glycerol and 5% glacial acetic for 1 h and dried onto 3MM paper. Dried gels were subjected to autoradiography at –70°C for 10–15 days.
Acknowledgments
Acknowledgements
We thank C.Logie, P.Verrijzer and members of the Stunnenberg laboratory for continued discussions and critical reading of the manuscript, R.Eckner, W.L.Kraus, E.Harlow, T.Kouzarides, P.M.Lieberman, C.L.Mummery and N.Hernandez for kindly providing plasmids, antibodies and cells, and A.Pennings for FACS analysis. D.J.M. was supported by a Marie Curie Fellowship of the European Community programme ‘Training and Mobility of Researchers’ under contract number ERB-FMBI-CT97-2728. This project was supported by the EU-TMR Network grant ERB-FMRX-CT96-0064.
References
- Abraham S.E., Lobo,S., Yaciuk,P., Wang,H.G.H. and Moran,E. (1993) p300 and p300-associated proteins, are components of TATA-binding protein (TBP) complexes. Oncogene, 8, 1639–1647. [PubMed] [Google Scholar]
- Arany Z., Newsome,D., Oldread,E., Livingston,D.M. and Eckner,R. (1995) A family of transcriptional adaptor proteins targeted by the E1A oncoprotein. Nature, 374, 81–84. [DOI] [PubMed] [Google Scholar]
- Bannister A.J. and Kouzarides,T. (1996) The CBP co-activator is a histone acetyltransferase. Nature, 384, 641–643. [DOI] [PubMed] [Google Scholar]
- Barlev N.A., Candau,R., Wang,L., Darpino,P., Silverman,N. and Berger,S.L. (1995) Characterization of physical interactions of the putative transcriptional adaptor, ADA2, with acidic activation domains and TATA-binding protein. J. Biol. Chem., 270, 19337–19344. [DOI] [PubMed] [Google Scholar]
- Bell B. and Tora,L. (1999) Regulation of gene expression by multiple forms of TFIID and other novel TAFII-containing complexes. Exp. Cell Res., 246, 11–19. [DOI] [PubMed] [Google Scholar]
- Berkenstam A., Ruiz,M.M., Barettino,D., Horikoshi,M. and Stunnenberg,H.G. (1992) Cooperativity in transactivation between retinoic acid receptor and TFIID requires an activity analogous to E1A. Cell, 69, 401–412. [DOI] [PubMed] [Google Scholar]
- Chakravarti D., Ogryzko,V., Kao,H.Y., Nash,A., Chen,H., Nakatani,Y. and Evans,R.M. (1999) A viral mechanism for inhibition of p300 and PCAF acetyltransferase activity. Cell, 96, 393–403. [DOI] [PubMed] [Google Scholar]
- Chrivia J.C., Kwok,R.P.S., Lamb,N., Hagiwara,M., Montminy,M.R. and Goodman,R.H. (1993) Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature, 365, 855–859. [DOI] [PubMed] [Google Scholar]
- Eckner R., Ewen,M.E., Newsome,D., Gerdes,M., DeCaprio,J.A., Lawrence,J.B. and Livingston,D.M. (1994) Molecular cloning and functional analysis of the adenovirus E1A-associated 300-kD protein (p300) reveals a protein with properties of a transcriptional adaptor. Genes Dev., 8, 869–884. [DOI] [PubMed] [Google Scholar]
- Eckner R., Ludlow,J.W., Lill,N.L., Oldread,E., Arany,Z., Modjtahedi,N., DeCaprio,J.A., Livingston,D.M. and Morgan,J.A. (1996) Association of p300 and CBP with simian virus 40 large T antigen. Mol. Cell. Biol., 16, 3454–3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenmann D.M., Arndt,K.M., Ricupero,S.L., Rooney,J.W. and Winston,F. (1992) SPT3 interacts with TFIID to allow normal transcription in Saccharomyces cerevisiae. Genes Dev., 6, 1319–1331. [DOI] [PubMed] [Google Scholar]
- Felzien L.K., Farrell,S., Betts,J.C., Mosavin,R. and Nabel,G.J. (1999) Specificity of cyclin E–Cdk2, TFIIB and E1A interactions with a common domain of the p300 coactivator. Mol. Cell. Biol., 19, 4241–4246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman R.H. and Smolik,S. (2000) CBP/p300 in cell growth, transformation and development. Genes Dev., 14, 1553–1577. [PubMed] [Google Scholar]
- Jelsma T.N., Howe,J.A., Mymryk,J.S., Evelegh,C.M., Cunniff,N.F.A. and Bayley,S.T. (1989) Sequences in E1A proteins of human adenovirus 5 required for cell transformation, repression of a transcriptional enhancer and induction of proliferating cell nuclear antigen. Virology, 171, 120–130. [DOI] [PubMed] [Google Scholar]
- Kawasaki H., Eckner,R., Yao,T.P., Taira,K., Chiu,R., Livingston,D.M. and Yokoyama,K.K. (1998) Distinct roles of the co-activators p300 and CBP in retinoic-acid-induced F9-cell differentiation. Nature, 393, 284–289. [DOI] [PubMed] [Google Scholar]
- Kraus W.L., Manning,E.T. and Kadonaga,J.T. (1999) Biochemical analysis of distinct activation functions in p300 that enhance transcription initiation with chromatin templates. Mol. Cell. Biol., 19, 8123–8135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundu T.K., Palhan,V.B., Wang,Z., An,W., Cole,P.A. and Roeder,R.G. (2000) Activator-dependent transcription from chromatin in vitro involving targeted histone acetylation by p300. Mol. Cell, 6, 551–561. [DOI] [PubMed] [Google Scholar]
- Kuras L., Kosa,P., Mencia,M. and Struhl,K. (2000) TAF-containing and TAF-independent forms of transcriptionally active TBP in vivo. Science, 288, 1244–1248. [DOI] [PubMed] [Google Scholar]
- Kurokawa R., Kalafus,D., Ogliastro,M.H., Kioussi,C., Xu,L., Torchia,J., Rosenfeld,M.G. and Glass,C.K. (1998) Differential use of CREB binding protein–coactivator complexes. Science, 279, 700–703. [DOI] [PubMed] [Google Scholar]
- Kwok R.P.S., Lundblad,J.R., Chrivia,J.C., Richards,J.P., Bächinger,H.P., Brennan,R.G., Roberts,S.G.E., Green,M.R. and Goodman,R.H. (1994) Nuclear protein CBP is a coactivator for the transcription factor CREB. Nature, 370, 223–226. [DOI] [PubMed] [Google Scholar]
- Lee T.I. and Young,R.A. (1998) Regulation of gene expression by TBP-associated proteins. Genes Dev., 12, 1398–1408. [DOI] [PubMed] [Google Scholar]
- Li X.-Y., Bhaumik,S.R. and Green,M.R. (2000) Distinct classes of yeast promoters revealed by differential TAF recruitment. Science, 288, 1242–1244. [DOI] [PubMed] [Google Scholar]
- Lipinski K.S., Fax,P., Wilker,B., Hennemann,H., Brockmann,D. and Esche,H. (1999) Differences in the interactions of oncogenic adenovirus 12 early region 1A and nononcogenic adenovirus 2 early region 1A with the cellular coactivators p300 and CBP. Virology, 255, 94–105. [DOI] [PubMed] [Google Scholar]
- Lundblad J.R., Kwok,R.P.S., Laurance,M.E., Harter,M.L. and Goodman,R.H. (1995) Adenoviral E1A-associated protein p300 as a functional homologue of the transcriptional co-activator CBP. Nature, 374, 85–88. [DOI] [PubMed] [Google Scholar]
- Madison J.M. and Winston,F. (1997) Evidence that Spt3 functionally interacts with Mot1, TFIIA and TATA-binding protein to confer promoter-specific transcriptional control in Saccharomyces cerevisiae. Mol. Cell. Biol., 17, 287–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsiou D.J. and Stunnenberg,H.G. (2000) TAC, a TBP-sans-TAFs complex containing the unprocessed TFIIAαβ precursor and the TFIIAγ subunit. Mol. Cell, 6, 527–537. [DOI] [PubMed] [Google Scholar]
- Mummery C.L., Feijen,A., van der Saag,P.T., van den Brink,C.E. and de Laat,S.W. (1985) Clonal variants of differentiated P19 embryonal carcinoma cells exhibit epidermal growth factor receptor kinase activity. Dev. Biol., 109, 402–410. [DOI] [PubMed] [Google Scholar]
- Mummery C.L, Feijen,A., Moolenaar,W.H., van den Brink,C.E. and de Laat,S.W. (1986) Establishment of a differentiated mesodermal line from P19 EC cells expressing functional PDGF and EGF receptors. Exp. Cell Res., 165, 229–242. [DOI] [PubMed] [Google Scholar]
- Nakajima T., Uchida,C. anderson,S.F., Lee,C.G., Hurwitz,J., Parvin,J.D. and Montminy,M. (1997a) RNA helicase A mediates association of CBP with RNA polymerase II. Cell, 90, 1107–1112. [DOI] [PubMed] [Google Scholar]
- Nakajima T., Uchida,C. anderson,S.F., Parvin,J.D. and Montminy,M. (1997b) Analysis of a cAMP-responsive activator reveals a two-component mechanism for transcriptional induction via signal-dependent factors. Genes Dev., 11, 738–747. [DOI] [PubMed] [Google Scholar]
- Ogryzko V.V., Schiltz,R.L., Russanova,V., Howard,B.H. and Nakatani,Y. (1996) The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell, 87, 953–959. [DOI] [PubMed] [Google Scholar]
- Reid J.L., Bannister,A.J., Zegerman,P., Martinez-Balbas,M.A. and Kouzarides,T. (1998) E1A directly binds and regulates the P/CAF acetyltransferase. EMBO J., 17, 4469–4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts S.M. and Winston,F. (1997) Essential functional interactions of SAGA, a Saccharomyces cerevisiae complex of Spt, Ada and Gcn5 proteins, with the Snf/Swi and Srb/mediator complexes. Genetics, 147, 451–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruppert S.M., McCulloch,V., Meyer,M., Bautista,C., Falkowski,M., Stunnenberg,H.G. and Hernandez,N. (1996) Monoclonal antibodies directed against the amino-terminal domain of human TBP cross-react with TBP from other species. Hybridoma, 15, 55–68. [DOI] [PubMed] [Google Scholar]
- Shikama N., Lyon,J. and La Thangue,N.B. (1997) The p300/CBP family: integrating signals with transcription factors and chromatin. Trends Cell Biol., 7, 230–236. [DOI] [PubMed] [Google Scholar]
- Slack R.S., Craig,J., Costa,S. and McBurney,M.W. (1995) Adenovirus 5 E1A induced differentiation of P19 embryonal carcinoma cells requires binding to p300. Oncogene, 10, 19–25. [PubMed] [Google Scholar]
- Sterner D.E. and Berger,S.L. (2000) Acetylation of histones and transcription-related factors. Microbiol. Mol. Biol. Rev., 64, 435–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterner D.E., Grant,P.A., Roberts,S.M., Duggan,L.J., Belotserkovskaya, R., Pacella,L.A., Winston,F., Workman,J.L. and Berger,S.L. (1999) Functional organization of the yeast SAGA complex: distinct components involved in structural integrity, nucleosome acetylation and TATA-binding protein interaction. Mol. Cell. Biol., 19, 86–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strubin M. and Struhl,K. (1992) Yeast and human TFIID with altered DNA-binding specificity for TATA elements. Cell, 68, 721–730. [DOI] [PubMed] [Google Scholar]
- Suhara W., Yoneyama,M., Kitabayashi,I. and Fujita,T. (2002) Direct involvement of CREB-binding protein/p300 in sequence-specific DNA binding of virus-activated interferon regulatory factor-3 holocomplex. J. Biol. Chem., 277, 22304–22313. [DOI] [PubMed] [Google Scholar]
- Ugai H., Uchida,K., Kawasaki,H. and Yokoyama,K.K. (1999) The coactivators p300 and CBP have different functions during the differentiation of F9 cells. J. Mol. Med., 77, 481–494. [DOI] [PubMed] [Google Scholar]
- Wang H.G.H., Rikitake,Y., Carter,M.C., Yaciuk,P., Abraham,S.E., Zerler,B. and Moran,E. (1993) Identification of specific adenovirus E1A N-terminal residues critical to the binding of cellular proteins and to the control of cell growth. J. Virol., 67, 476–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X.-J., Ogryzko,V.V., Nishikawa,J., Howard,B.H. and Nakatani,Y. (1996) A p300/CBP-associated factor that competes with the adenoviral oncoprotein E1A. Nature, 382, 319–324. [DOI] [PubMed] [Google Scholar]
- Yuan W., Condorelli,G., Caruso,M., Felsani,A. and Giordano,A. (1996) Human p300 protein is a coactivator for the transcription factor MyoD. J. Biol. Chem., 271, 9009–9013. [DOI] [PubMed] [Google Scholar]