

Abstract
Mitochondrial outer-membrane permeabilization by pro-apoptotic Bcl-2 family members plays a crucial role in apoptosis induction. However, whether this directly causes the release of the different mitochondrial apoptogenic factors simultaneously is currently unknown. Here we report that in cells or with isolated mitochondria, pro-apoptotic Bcl-2 proteins cause the release of cytochrome c, Smac/Diablo and HtrA2/Omi but not endonuclease G (EndoG) and apoptosis-inducing factor (AIF). In cells treated with Bax/Bak-dependent pro-apoptotic drugs, neither the caspase inhibitor zVAD-fmk nor loss of Apaf-1 affected the efflux of cytochrome c, Smac/Diablo and HtrA2/Omi, but both prevented the release of EndoG and AIF. Our findings identify the mitochondrial response to pro-apoptotic stimuli as a selective process leading to a hierarchical ordering of the effectors involved in cell death induction. Moreover, as in Caenorhabditis elegans, EndoG and AIF act downstream of caspase activation. Thus EndoG and AIF seem to define a ‘caspase-dependent’ mitochondria-initiated apoptotic DNA degradation pathway that is conserved between mammals and nematodes.
Keywords: AIF/cytochrome c/EndoG/HtrA2/Omi/Smac/Diablo
Introduction
Programmed cell death (PCD) and its main phenotype, apoptosis, is a cell suicide program essential for development and for adult tissue homeostasis of all metazoan animals (Jacobson et al., 1997). Defaults (inhibition or exacerbation) in PCD are involved in several pathologies such as neurodegenerative diseases, cancers and AIDS (Thompson, 1995; Kroemer and Reed, 2000). The stereotypical death throes of a cell undergoing apoptosis include DNA fragmentation, nuclear condensation, cell shrinkage, blebbing and phosphatidylserine externalization, and these features are orchestrated by the activity of a family of cysteine proteases called caspases (Thornberry and Lazebnik, 1998).
A mitochondria-dependent step, involving outer- membrane permeabilization, is associated with most pro-apoptotic stimuli. This process is controlled by the pro- and anti-apoptotic members of the Bcl-2 family and leads to the cytosolic release of mitochondrial intermembrane space proteins that can trigger either caspase activation or caspase-independent death pathways (Gross et al., 1999; Martinou and Green, 2001; Zamzami and Kroemer, 2001). Mitochondrial proteins that cause caspase-dependent cell death include cytochrome c (P.Li et al., 1997) which triggers caspase-9 activation by binding and activating the apoptosis protease activating factor-1 (Apaf-1), and Smac/Diablo (Du et al., 2000; Verhagen et al., 2000) and HtrA2/Omi (Suzuki et al., 2001; Hegde et al., 2002; Verhagen et al., 2002) which potentiate caspase activation by binding inhibitor of apoptosis proteins (IAPs) and blocking their caspase inhibitory activity. Mitochondria have also been reported to contain the caspase-independent death effectors apoptosis-inducing factor (AIF) and endonuclease G (EndoG). AIF induces chromatin condensation and large-scale DNA fragmentation (50 kbp) when released into the cytosol (Susin et al., 1999). During apoptosis, EndoG, like AIF, translocates to the nucleus where it causes oligonucleosomal DNA fragmentation (L.Y.Li et al., 2001). Subsequent studies have demonstrated that EndoG catalyzes both high molecular weight DNA cleavage and oligonucleosomal DNA breakdown in a sequential fashion. Moreover, EndoG cooperates with exonuclease and DNase I to facilitate DNA processing (Widlak et al., 2001).
The mechanisms by which the pro-apoptotic Bcl-2 family members induce the release of mitochondrial proteins remain controversial (Gross et al., 1999; Martinou and Green, 2001; Zamzami and Kroemer, 2001). One proposed model involves rupture of the mitochondrial outer membrane as a consequence of mitochondrial swelling after the opening of the permeability transition pore (PTP) (Marzo et al., 1998; Vander Heiden and Thompson, 1999; Zamzami and Kroemer, 2001) and, according to another model, pro-apoptotic Bcl-2 proteins like Bax/Bak induce a selective process of outer-membrane permeabilization through the formation of channels or pores allowing the selective release of proteins soluble in the inner-membrane space such as cytochrome c (Eskes et al., 1998; Desagher and Martinou, 2000; Martinou and Green, 2001). Recently it has been shown that outer-membrane permeabilization by the pro-apoptotic Bcl-2 family members does not require the mitochondrial matrix, the inner membrane or other mitochondrial proteins (Kuwana et al., 2002).
Important questions that remain unresolved are whether pro-apoptotic Bcl-2 members induce the release of all the known mitochondrial apoptogenic factors (Wang, 2001; Van Loo et al., 2002) and the temporal sequence of their release. Indeed, numerous studies have reported that cytochrome c release is a direct consequence of the Bax/Bak-mediated mitochondrial permeabilization (Eskes et al., 1998; Jurgensmeier et al., 1998; Desagher et al., 1999; Finucane et al., 1999; Antonsson et al., 2000) but little is currently known about the release of Smac/Diablo, HtrA2/Omi, AIF or EndoG. Nevertheless, it has recently been reported that Bax/Bak-mediated mitochondrial permeabilization does not directly induce AIF release and that AIF release occurs downstream of cytochrome c release (Arnoult et al., 2002).
Here we report that cell death stimuli induce a hierarchical release of the mitochondrial factors involved in cell death induction. Indeed, we show that the mitochondrial outer-membrane permeabilization induced by Bax-, tBid- or Bax/Bak-dependent pro-apoptotic drugs results in the release of cytochrome c, Smac/Diablo and HtrA2/Omi, but that subsequent caspase activation is required to induce the translocation of EndoG in addition to AIF into the cytosol. Our results provide a paradigm for mitochondria-dependent death pathways in which EndoG or AIF cannot substitute for caspase executioners since their intracytosolic release requires caspase activation.
Results
Bax and tBid induce the release of cytochrome c, Smac/Diablo and HtrA2/Omi, but not EndoG and AIF, from isolated mitochondria
To investigate whether pro-apoptotic Bcl-2 members like Bax may induce the mitochondrial release of all known apoptogenic factors simultaneously (Wang, 2001; Van Loo et al., 2002), we incubated freshly isolated HeLa cell mitochondria with recombinant Bax (Eskes et al., 1998; Antonsson et al., 2000; Arnoult et al., 2002). First, isolated mitochondria were incubated for 30 min with increasing amounts (0, 25, 50, 100 and 200 nM) of recombinant oligomeric Bax prior to western blot analysis of cytochrome c, Smac/Diablo, EndoG, HtrA2/Omi and AIF in both the mitochondrial supernatants and pellets. VDAC, an intrinsic component of the outer membrane, was used as a control for loading. For the detection of Smac/Diablo and EndoG, we used two commercially available polyclonal antibodies, both working in immunofluorescence and western blotting (see Supplementary figure S1 available at The EMBO Journal Online). We also produced a rabbit polyclonal antibody raised against HtrA2/Omi (Supplementary figure S1). Cytochrome c and AIF were both detected with monoclonal antibodies (Arnoult et al., 2002). Mitochondrial release of cytochrome c, Smac/Diablo and HtrA2/Omi was maximal at a Bax concentration of 100 nM, with almost no cytochrome c, Smac/Diablo or HtrA2/Omi remaining in the mitochondria pellet (Figure 1A). A kinetic analysis (5, 10 and 30 min) with 200 nM of oligomeric Bax indicated that cytochrome c, Smac/Diablo and HtrA2/Omi translocation from the mitochondria was a rapid event, complete within 30 min (Figure 1B). In contrast, Bax did not induce any detectable EndoG and AIF release (nor any detectable EndoG or AIF decrease in the mitochondrial pellet) after 30 min incubation with oligomeric Bax at concentrations up to 200 nM (Figure 1A and B). Monomeric Bax (Suzuki et al., 2000) did not induce any cytochrome c, Smac/Diablo, HtrA2/Omi, EndoG or AIF release (data not shown), consistent with previous findings indicating that Bax requires oligomerization in order to induce cytochrome c release from isolated mitochondria (Antonsson et al., 2000) and that Bax is present in an oligomeric form in the mitochondrial outer membranes of apoptotic cells (Mikhailov et al., 2001).
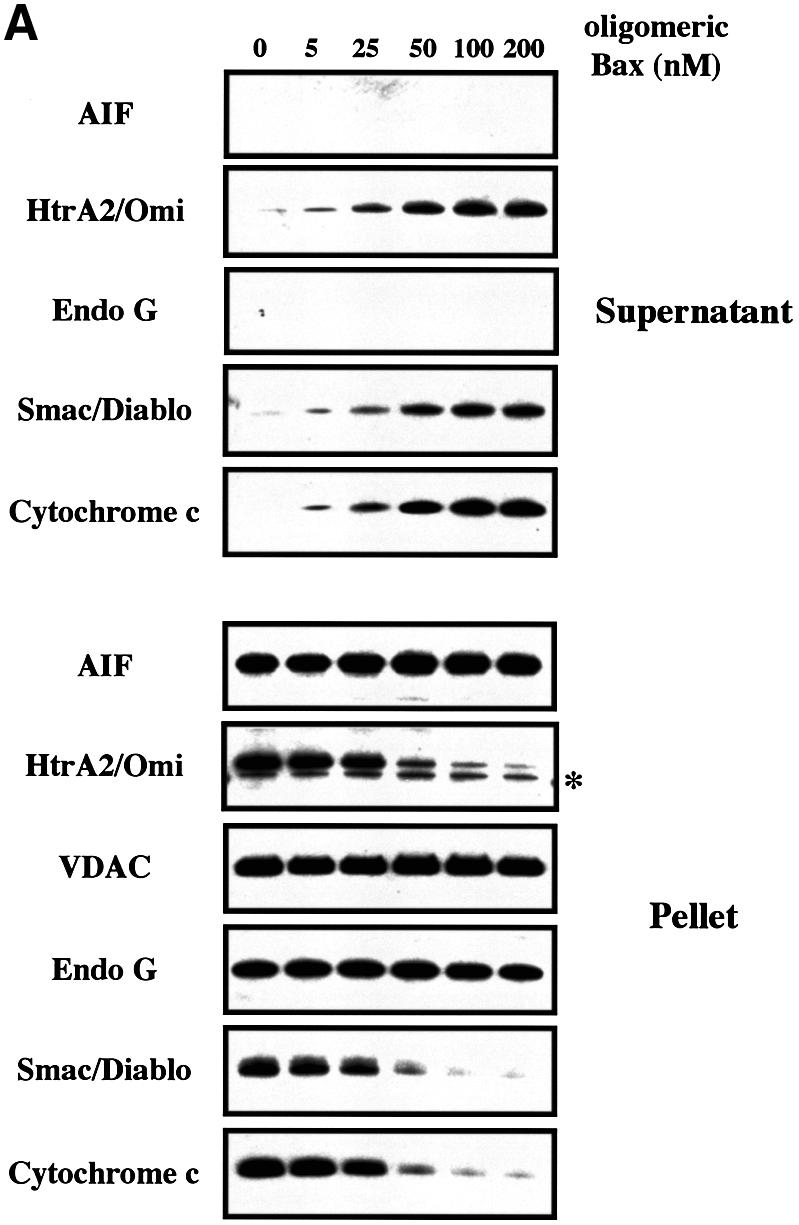


Fig. 1. Recombinant pro-apoptotic Bcl-2 members induce the release of cytochrome c, Smac/Diablo and HtrA2/Omi but not that of EndoG and AIF from isolated mitochondria. (A) Mitochondria isolated from HeLa cells were incubated for 30 min at 30°C with different concentrations (nM) of recombinant oligomeric Bax. Mitochondrial pellets and supernatant fractions were separated by SDS–PAGE (Tricine Gel), and their respective cytochrome c, Smac/Diablo, HtrA2/Omi, EndoG and AIF contents were analyzed by western blotting. (B) Mitochondria isolated from HeLa cells were incubated with 200 nM oligomeric recombinant Bax or with control buffer (none) at 30°C and the mitochondrial pellet and supernatant were analyzed at different time points (min), as in (A). (C) Mitochondria isolated from HeLa cells were incubated for 15 min with different concentrations (nM) of recombinant tBid and the mitochondrial pellet and supernatant were analyzed as in (A). In (A), (B) and (C), equal loading of the mitochondrial pellet was controlled using VDAC. The asterisks in (A), (B) and (C) indicate an additional band due to the previous VDAC detection.
We also incubated isolated mitochondria for 15 min with increasing amounts (0, 0.001, 0.01, 0.1, 1, 10 and 100 nM) of the caspase-8 cleaved active form of Bid (tBid) (Gross et al., 1999). tBid induces the oligomerization of Bax or Bak in the mitochondrial outer membrane to allow cytochrome c release (Desagher et al., 1999; Wei et al., 2001). Mitochondrial efflux of cytochrome c, Smac/Diablo and HtrA2/Omi was maximal at a tBid concentration of 1 nM, with almost no cytochrome c, Smac/Diablo or HtrA2/Omi remaining in the mitochondria pellet (Figure 1C). However, we still did not detect any EndoG or AIF in the mitochondria supernatant or any decrease in the pellet (Figure 1C).
Thus our results suggest that the pro-apoptotic Bcl-2 members permeabilize the mitochondrial outer membrane allowing the co-release of cytochrome c, Smac/Diablo and HtrA2/Omi but not that of EndoG and AIF. Li and coworkers (L.Y.Li et al., 2001) reported that recombinant tBid and Bim can induce in vitro EndoG release from isolated mouse liver mitochondria. Using our experimental conditions, the response of mitochondria from primary mouse hepatocytes to either recombinant tBid or recombinant oligomeric Bax was also assessed, but no release of EndoG and AIF was detected whereas a mitochondrial efflux of cytochrome c, Smac/Diablo and HtrA2/Omi was observed (Supplementary figure S2). Interestingly, mouse liver mitochondria seem to be more resistant to recombinant tBid than HeLa cell mitochondria. Indeed, while incubation of HeLa cell mitochondria with 1 nM tBid for 15 min induced a nearly complete release of cytochrome c, Smac/Diablo and HtrA2/Omi (Figure 1C), incubation of mouse liver mitochondria with 100 nM tBid for 30 min did not trigger such a mitochondrial efflux of these factors (Supplementary figure S2).
Next, we asked if the loss of mitochondrial mem brane potential (ΔΨm) is required for the release of the different mitochondrial apoptogenic factors. Increasing concentrations up to 50 nM of recombinant oligomeric Bax or 0.1 nM of recombinant tBid induced a significant (>50%) release of cytochrome c, Smac/Diablo and HtrA2/Omi without any change in ΔΨm (Figure 2). Nevertheless, with 100 nM of Bax, 1 nM of tBid or higher concentrations of recombinant proteins, a ΔΨm loss occurred and this was associated with a complete or nearly complete release (>90%) of cytochrome c, Smac/Diablo and HtrA2/Omi (Figure 2). These data suggest that a ΔΨm loss is required for the release of the remaining pool of cytochrome c, Smac/Diablo and HtrA2/Omi after the initial release following the Bax/ Bak-mediated mitochondrial outer-membrane perme abilization. This is in agreement with the ‘two-step release’ of cytochrome c recently proposed (Ott et al., 2002; Scorrano et al., 2002). Finally, while 100 nM of Bax, 1 nM of tBid or higher concentrations triggered a ΔΨm loss (Figure 2), AIF and EndoG were not released at these concentrations (Figure 1), suggesting that a ΔΨm loss is not required for the release of these mitochondrial apoptogenic factors.


Fig. 2. ΔΨm analysis in isolated mitochondria incubated with recombinant pro-apoptotic Bcl-2 members. Mitochondria isolated from HeLa cells were incubated with different concentrations (nM) of (A) recombinant oligomeric Bax or (B) recombinant tBid as in Figure 1. Then, ΔΨm was measured by flow cytometry using Rh123 (50 nM) as a probe. Incubation of isolated mitochondria with CCCP (10 µM) was used as a control. As in Figure 1, a fraction of mitochondrial pellets and supernatant fractions was also analyzed by western blotting. Blue arrows indicate a significant release of cytochrome c, Smac/Diablo and HtrA2/Omi (>50% release) and red arrows indicate complete or nearly complete release (>90% release).
Our observation that recombinant oligomeric Bax or tBid trigger a ΔΨm loss on isolated mitochondria is not in agreement with results published by D.R.Green and colleagues. Indeed, they reported that in their in vitro models, neither recombinant protein disrupted ΔΨm while cytochrome c is released (Finucane et al., 1999; Ricci et al., 2003). However, they indicated that this observed effect requires that the mitochondria be maintained at high density (Ricci et al., 2003), since dilution of the treated organelles causes a ΔΨm loss as the cytochrome c concentration falls below a critical level (Waterhouse et al., 2001).
Caspase inhibitor zVAD-fmk prevents the mitochondrial release of EndoG and AIF, but not that of cytochrome c, Smac/Diablo and HtrA2/Omi, during Bax overexpression
To confirm our in vitro results, HeLa cells were transiently transfected with an expression vector encoding GFP-Bax in the presence of zVAD-fmk to prevent caspase activation. Then, the cells were immunostained with specific antibodies for cytochrome c, Smac/Diablo, EndoG or AIF (Figure 3A). We observed that caspase inhibition by zVAD-fmk did not prevent cytochrome c and Smac/Diablo release in cells expressing GFP-Bax whereas EndoG and AIF were still present in the mitochondria in most of the transfected cells (Figure 3A and B). Moreover, GFP-Bax transfected cells did not show any signs of nuclear apoptosis when caspases were inhibited (Figure 3A), confirming our observation that EndoG and AIF, two mitochondrial nuclear effectors of apoptosis (Susin et al., 1999; L.Y.Li et al., 2001), have not been released. Expression of GFP-Bax in the absence of caspase inhibitor led to diffuse cytochrome c, Smac/Diablo, EndoG or AIF in the cytosol (Figure 3A and B). However, we observed that the number of GFP-Bax transfected cells showing diffuse AIF or Endo G is weaker than GFP-Bax transfected cells with diffuse cytochrome c or Smac/Diablo (Figure 3B).



Fig. 3. Caspase inhibition by zVAD-fmk prevents the mitochondrial release of EndoG and AIF but not that of cytochrome c and Smac/Diablo during Bax overexpression. (A) GFP-Bax expression and immunostaining of cytochrome c, Smac/Diablo, EndoG and AIF together with nuclear Hoechst staining in HeLa cells 18 h after transient transfection with a vector encoding GFP-Bax in the absence or in the presence of the caspase inhibitor z-VAD-fmk (100 µM). (B) Quantitative analysis of the numbers of GFP-Bax transfected cells with intracytosolic release of cytochrome c, Smac/Diablo, EndoG or AIF in the absence or presence of z-VAD-fmk. Each histogram indicates mean ± SD of three fields of at least 100 cells within a representative experiment.
Owing to a high non-specific background, our anti-HtrA2 antibody could only detect overexpressed HtrA2/Omi by immunofluorescence. In HtrA2/Omi overexpressing cells, we also observed that zVAD-fmk had no effect on its cytosolic relocalization following Bax expression (data not shown).
Our data demonstrate that mitochondrial insertion of Bax is sufficient to induce the release of cytochrome c, Smac/Diablo and HtrA2/Omi but not EndoG and AIF.
Cell stress-associated EndoG and AIF release requires active caspases unlike cytochrome c, Smac/Diablo and HtrA2/Omi
We next examined the ability of zVAD-fmk to modulate the release of mitochondrial apoptogenic factors in response to pro-apoptotic drugs such as staurosporine and actinomycin D. Both drugs require the presence of either Bax or Bak to induce mitochondrial release of cytochrome c and initiate cell death (Wei et al., 2001). As anticipated, HeLa cells pretreated with zVAD-fmk were protected from apoptosis induced by both drugs as assessed by the number of apoptotic nuclei (Figure 4A), and zVAD-fmk also inhibited caspase-9 and caspase-3 activation and the cleavage of the caspase substrate PARP (Figure 4B). As previously reported (Kluck et al., 1997; Adrain et al., 2001), the accumulation of cytochrome c within the cytosol of cells undergoing apoptosis in response to these cytotoxic drugs was insensitive to caspase inhibition by zVAD-fmk (Figure 4C). In comparison with the cytosolic fractions of apoptotic cells, the cytosolic fraction of cells induced to undergo apoptosis in the presence of zVAD-fmk showed a small decrease in the amount of Smac/Diablo while HtrA2/Omi was barely detectable (Figure 4C). Nevertheless, these differences were not due to an inhibition of Smac/Diablo and HtrA2/Omi release because analysis of the mitochondrial pellet confirmed that both factors were released in the presence or absence of caspase inhibitor. Our results suggest that caspase inhibition by zVAD-fmk induces the degradation of HtrA2/Omi and Smac/Diablo (to a lesser degree) once they are released in the cytosol. Recently, it was reported that the proteasome degrades Smac/Diablo during apoptosis (MacFarlane et al., 2002), and thus further studies are required to check if the proteasome also degrades HtrA2/Omi and if caspase inhibition accelerates this process.



Fig. 4. Cell-stress-associated EndoG and AIF release is prevented by zVAD-fmk, unlike cytochrome c, Smac/Diablo and HtrA2/Omi. (A) Percentages of cells with apoptotic nuclei (percentage of apoptotic nuclei) in HeLa cells treated for 7 and 9 h with staurosporine (STS, 2 µM) or actinomycin D (ActD, 20 µM) in the absence or presence of zVAD-fmk (100 µM). (B) Cells were treated as in (A). Total cell extracts were analyzed by western blotting for caspase-9 and caspase-3 processing and PARP cleavage. (C) HeLa cells were treated as in (A). Cytosolic fraction and heavy membrane fraction were analyzed by western blotting for the presence of cytochrome c, Smac/Diablo, HtrA2/Omi, EndoG and AIF. As control for loading, actin was used in the cytosolic fraction and Cox IV in the heavy membrane fraction. (D) HeLa cells were treated as in (A) and then ΔΨm was assessed by flow cytometry using DiOC6 (50 nM). Top: % indicates percentage of ΔΨm loss. Bottom: histogram showing the percentage of ΔΨm loss in three independent experiments. The asterisks indicate additional non-specific bands.
In contrast with cytochrome c, Smac/Diablo or HtrA2/Omi, the presence of EndoG or AIF in the cytosolic fraction or their mitochondrial efflux was repressed in zVAD-fmk-treated cells, strongly suggesting that caspase activation was required to mediate EndoG and AIF release in response to the cytotoxic drugs (Figure 4C). Furthermore, after apoptosis induction without caspase inhibition, while the mitochondrial efflux of cytochrome c, Smac/Diablo and HtrA2/Omi was nearly complete, the EndoG and AIF release was significant but not complete (Figure 4C). This observation suggests again that the mechanism for EndoG and AIF release is different from that of the pro-apoptotic Bcl-2 family mediated mitochondrial outer-membrane permeabilization required for cytochrome c, Smac/Diablo and HtrA2/Omi release (Figure 1).
ΔΨm was also studied under these conditions (Figure 4D). As previously reported (Goldstein et al., 2000; Waterhouse et al., 2001), we observed that zVAD-fmk prevented ΔΨm loss in actinomycin-D-treated cells. It is likely that a ΔΨm loss also occurred in actinomycin-D-treated cells in the presence of caspase inhibitor, but when the ΔΨm was assessed (Figure 4D) it had already recovered as previously described (Waterhouse et al., 2001). Fluorescence experiments confirmed that cytochrome c and Smac/Diablo were diffuse in the cytosol of zVAD-fmk/actinomycin-D-treated cells while the ΔΨm was high (Supplementary figure S3). In contrast, in the same cells with high ΔΨm, AIF and EndoG were still present in the mitochondria (Supplementary figure S3).
Surprisingly, we observed that zVAD-fmk had no effect on the ΔΨm loss in staurosporine-treated cells (Figure 4D and Supplementary figure S3). This observation allowed us to confirm that the release of AIF and EndoG is independent of ΔΨm loss. Indeed, staurosporine-treated cells in the presence of zVAD-fmk showed a ΔΨm loss (Figure 4D and Supplementary figure S3) while AIF and EndoG were clearly retained within the mitochondria (Supplementary figure S3). Our observation that AIF and EndoG were not released from mitochondria with low ΔΨm correlated with our in vitro results (Figures 1 and 2).
In order to confirm the cell fractionation results (Figure 4C), double immunostainings were also performed on HeLa cells pretreated with zVAD-fmk and induced to undergo apoptosis with staurosporine or actinomycin D. We observed that while cytochrome c had a diffuse staining pattern in most of the cells, EndoG or AIF was clearly retained within the mitochondria (Figure 5A and B). In contrast, we did not observe any inhibition of Smac/Diablo or HtrA2/Omi release (Figure 5C and data not shown).



Fig. 5. Cell-stress-associated cytochrome c and Smac/Diablo release is insensitive to zVAD-fmk, unlike EndoG and AIF. HeLa cells were treated for 8 h with actinomycin D (20 µM) or staurosporine (2 µM) in the presence of zVAD-fmk (100 µM). The cells were immunostained with (A) anti-cytochrome c and anti-EndoG antibodies or (B) anti-cytochrome c and anti-AIF or (C) anti-cytochrome c and anti-Smac/Diablo together with Hoechst nuclear staining. Then a quantitative analysis of the numbers of actinomycin D- or staurosporine-treated cells in the presence of z-VAD-fmk (100 µM) with intracytosolic release of (A) cytochrome c and/or EndoG or (B) AIF or (C) Smac/Diablo was performed. Each histogram indicates mean ± SD of three fields of at least 100 cells within a representative experiment.
Our observation that the mitochondrial release of Smac/Diablo was unaffected by zVAD-fmk (Figures 4C and 5C) was inconsistent with a recent report which showed that Smac/Diablo release was prevented by this caspase inhibitor (Adrain et al., 2001). However, our result was confirmed using another Smac/Diablo-specific antibody (mAb; Zymed Laboratories) by western blots after cell fractionation and by immunofluorescence (data not shown).
Therefore our data demonstrate that while cytochrome c, Smac/Diablo and HtrA2/Omi release is insensitive to caspase inhibition by zVAD-fmk, active caspases seem to be required for EndoG and AIF release.
EndoG and AIF are not required for staurosporine-mediated caspase-independent chromatin condensation
We observed in independent experiments that while caspase inhibition by zVAD-fmk prevented EndoG and AIF release, it did not prevent perinuclear chromatin condensation (type I nuclear apoptosis; Susin et al., 2000) in staurosporine-treated cells (Figure 5). To confirm that neither EndoG nor AIF was involved in this caspase-independent nuclear phenotype, we performed immunostaining of cytochrome c, AIF and EndoG together with nuclear labeling in zVAD-fmk/staurosporine-treated cells. Most of the cells demonstrated type I nuclear apoptosis and diffuse cytochrome c, but EndoG and AIF were clearly still present in the mitochondria (Figure 6A), suggesting that neither factor was involved in this caspase-independent chromatin condensation though the mitochondrial outer membrane was permeabilized. Moreover, this nuclear phenotype does not seem to be dependent on mitochondrial permeabilization because chromatin condensation without cytochrome c release was observed in some cells (data not shown) and this nuclear phenotype is also observed in cells overexpressing Bcl-2 (D.Arnoult and R.J.Youle unpublished results).



Fig. 6. Bax/Bak-mediated mitochondrial permeabilization does not induce the release of nuclear effectors of apoptosis. (A) HeLa cells were either left untreated (Control) or treated for 8 h with staurosporine (2 µM) or actinomycin D (20 µM) in the presence of zVAD-fmk (100 µM). Then cells were immunostained with a sheep anti-cytochrome c, a mouse anti-AIF and a rabbit anti-EndoG together with Hoechst nuclear staining. (B) Flow cytometry analysis of DNA degradation in isolated nuclei from CEM cells after incubation for 6 h with supernatants from isolated HeLa mitochondria that had been incubated for 30 min with either 200 nM oligomeric recombinant Bax (Mitochondria + Bax) or control buffer (Control). As a positive control for mitochondrial effectors of DNA degradation, whole mitochondria extracts (Mitochondrial Rupture) were used (% indicates percentage of DNA degradation). (C) EndoG and AIF are not soluble in the mitochondrial inner-membrane space. HeLa mitochondria, mitoplasts and mitoplasts treated with sodium carbonate (Na2CO3) were resolved by SDS–PAGE, and their respective contents in cytochrome c, EndoG, AIF and Cox IV were analyzed by western blotting. To show that EndoG is not simply precipitated by carbonate, mitoplasts were also treated with sodium carbonate and proteinase K.
Bax/Bak does not trigger the mitochondrial release of nuclear effectors of apoptosis
We also performed triple immunostaining on zVAD-fmk/actinomycin-D-treated cells. Although most of the cells have a diffuse cytochrome c in the cytosol, they did not show any EndoG and AIF release or chromatin condensation (Figure 6A). This observation suggested that Bax/Bak did not induce mitochondrial release of any caspase-independent nuclear effectors of apoptosis. To check this hypothesis, we incubated isolated nuclei from CEM cells for 6 h with supernatants from isolated mitochondria that had been incubated with either recombinant oligomeric Bax (200 nM) or medium alone. The supernatant of Bax-treated mitochondria did not contain any detectable EndoG or any significant AIF (Figure 6B). As a positive control for mitochondrial effectors of DNA degradation, we used whole mitochondrial extracts (lysis by sonication) which contained AIF and EndoG (Figure 6B). Flow cytometry analysis of the isolated nuclei indicated that while whole mitochondrial extracts induced nuclear DNA degradation, no significant DNA degradation was induced by the supernatants of Bax-treated mitochondria when compared with supernatants of untreated mitochondria (Figure 6B). Thus Bax did not induce a detectable release of any nuclear effectors of apoptosis from the mitochondria, consistent with our findings in Bax-overexpressing cells (Figure 3A) that treatment with a caspase inhibitor prevented nuclear apoptosis. Also, we did not observe significant nuclease activity in the supernatant of tBid-treated mitochondria (data not shown).
Mitochondrial rupture by sonication led to complete cytochrome c release but only a small portion of AIF and EndoG (Figure 6B), suggesting that, in contrast with cytochrome c, these factors are not soluble in the mitochondrial inner-membrane space. Analysis of mitoplasts showed that they contained the same amount of AIF and EndoG as whole mitochondria (Figure 6C) while a significant part of cytochrome c was lost. When we treated the mitoplasts with carbonate, the remaining cytochrome c was lost and most of the AIF whereas EndoG was unaffected. Together, these results suggest that AIF is peripherally associated with the mitochondrial inner membrane as previously reported (Arnoult et al., 2002) while EndoG may be localized in the mitochondrial matrix or tightly bound to the inner membrane and may thus explain why pro-apoptotic Bcl-2 family mediated mitochondrial permeabilization does not lead to the release of these factors (Figures 1 and 6B).
EndoG and AIF release requires caspase activation downstream of Bax/Bak-mediated mitochondrial permeabilization
Based on the use of the broad caspase inhibitor zVAD-fmk, our findings suggest that EndoG and AIF release occurs in a caspase-dependent manner. To confirm this, we used Apaf-1 –/– mouse embryonic fibroblasts (MEFs) (Cecconi et al., 1998). These MEFs have been described as having a defect in caspase activation following a stress signal owing to a lack of apoptosome formation; thus these cells were shown to be more resistant to apoptosis (Cecconi et al., 1998; Yoshida et al., 1998). We also noticed that Apaf-1 –/– MEFs were more resistant to staurosporine- or actinomycin-D-induced apoptosis than the Apaf-1 +/– MEFs (Supplementary figure S4).
We performed cytochrome c/AIF and cytochrome c/EndoG immunostaining on Apaf-1 –/– or +/– MEFs that were induced to undergo apoptosis with staurosporine or actinomycin D. We observed that in both types of MEFs, the Bax/Bak-mediated mitochondrial permeabilization occurred normally as assessed by the diffuse cytochrome c, whereas the release of EndoG and AIF was inhibited in Apaf-1 –/– but not in Apaf-1 +/– MEFs (Figure 7A and B). Therefore this finding confirms that EndoG and AIF release requires caspase activation downstream of Bax/Bak-mediated mitochondrial outer-membrane permeabilization. Finally, unlike EndoG and AIF, the release of Smac/Diablo and HtrA2/Omi was unaffected in Apaf-1 –/– MEFs (data not shown).




Fig. 7. EndoG and AIF release requires caspase activation downstream of cytochrome c release. Apaf +/– or –/– MEFs were treated for 24 h with staurosporine (STS, 10 µM) or actinomycin D (ActD, 40 µM). Next, the cells were immunostained with either (A) anti-cytochrome c and anti-EndoG antibodies or (B) anti-cytochrome c and anti-AIF together with Hoechst nuclear staining. Then a quantitative analysis of the numbers of actinomycin D or staurosporine-treated MEFs with intracytosolic release of cytochrome c and/or (A) EndoG or (B) AIF was performed. Each histogram indicates mean ± SD of three fields of at least 100 cells within a representative experiment.
Together, our results suggest that the Bax/Bak-mediated mitochondrial outer-membrane permeabilization is sufficient to induce the release of cytochrome c, Smac/Diablo and HtrA2/Omi while downstream caspase activation is required for the release of EndoG and AIF.
Discussion
Mitochondria contain several apoptogenic factors (cytochrome c, Smac/Diablo, HtrA2/Omi, AIF, EndoG) (Wang, 2001; Van Loo et al., 2002), and it has been proposed that Bcl-2 family members control apoptosis by regulating the release of these factors (Gross et al., 1999; Hengartner, 2000; Martinou and Green, 2001; Zamzami and Kroemer, 2001). Here we report that pro-apoptotic Bcl-2 members induce a hierarchical release of the apoptogenic factors. Indeed, our results suggest that pro-apoptotic Bcl-2 members trigger a selective mitochondrial efflux of cytochrome c, Smac/Diablo and HtrA2/Omi which, when in the cytosol, allow caspase activation (Wang, 2001; Van Loo et al., 2002). Then, active caspase-mediated attack of a (unidentified) mitochondrial component may result in the opening of an EndoG- and/or AIF-conducting pore or process and, once released in the cytosol, both factors can participate in the nuclear apoptosis (Susin et al., 1999; L.Y.Li et al., 2001).
Further experiments are required to determinate if Smac/Diablo and HtrA2/Omi are colocalized with cytochrome c within the mitochondria. Indeed, while a part of cytochrome c is soluble in the inner-membrane space, another part is bound to its respiratory partners and cardiolipin (Doran and Halestrap, 2000) and cytochrome c is also localized in the mitochondrial cristae (Scorrano et al., 2002). Thus it was shown that the release of cytochrome c occurs in two steps where remodeling of mitochondrial cristae and/or lipid peroxidation is required to allow the complete release of cytochrome after the initial mitochondrial outer-membrane permeabilization (Ott et al., 2002; Scorrano et al., 2002). In agreement with this model, we observed in vitro with isolated mitochondria treated with Bax or tBid that a ΔΨm loss may be required for the complete release of cytochrome c, Smac/Diablo and HtrA2/Omi. Indeed, while a significant part of cytochrome c, Smac/Diablo and HtrA2/Omi is released independently of any ΔΨm change, a complete or nearly complete release of those apoptogenic factors is observed only after a drop of ΔΨm. Interestingly, it was very recently reported that ΔΨm regulates matrix configuration (Gottlieb et al., 2003), and thus the ΔΨm loss that we observed may be required for the remodeling of the mitochondrial structure (Scorrano et al., 2002), allowing the release of the remaining pool of cytochrome c, Smac/Diablo and HtrA2/Omi. Furthermore, the ΔΨm loss that we observed may also lead to lipid peroxidation which is also required for the release of the second pool of cytochrome c (and probably Smac/Diablo and HtrA2/Omi) after the initial mitochondrial outer-membrane permeabilization (Ott et al., 2002). Finally, while in our model, a ΔΨm loss may be required for the release of the last pool of cytochrome c, Smac/Diablo and HtrA2/Omi, this ΔΨm loss does not seem to be involved in the release of the other mitochondrial apoptogenic factors, i.e. AIF and EndoG. Thus our data support a model in which the different mitochondrial apoptogenic factors are released in three steps. In the first step, Bax/Bak-mediated mitochondrial outer-membrane permeabilization leads to the release of a significant part of the cytochrome c, Smac/Diablo and HtrA2/Omi. Secondly, a ΔΨm loss occurs. This ΔΨm loss may be required for the release of the last pool of cytochrome c, Smac/Diablo and HtrA2/Omi. Never theless, the two other apoptogenic factors AIF and EndoG are unaffected by the ΔΨm loss and are still localized in the mitochondria. During a third step, once released into the cytosol, cytochrome c, Smac/Diablo and HtrA2/Omi trigger caspase activation. Then the action of zVAD-fmk inhibitable caspases is required to alter the physical association of AIF and EndoG with the inner membrane to enable their relocation to the cytosol.
CAD, the main effector involved in oligonucleosomal DNA degradation, requires the caspase-specific cleavage of its inhibitor ICAD to be active (Nagata, 2000), and Acinus, which is involved in chromatin condensation, is activated after proteolytic cleavage by caspase-3 (Sahara et al., 1999). These data and our observations that EndoG and AIF release requires caspase activation downstream of the Bax/Bak-mediated mitochondrial outer-membrane permeabilization allow an explanation of why caspase inhibition by zVAD-fmk prevents nuclear apoptosis in Bax-overexpressing cells or in actinomycin-D-treated cells. However, in staurosporine-treated cells, a perinuclear chromatin condensation (type I nuclear apoptosis; Susin et al., 2000) still occurs in the presence of zVAD-fmk while EndoG and AIF are clearly retained in the mitochondria, suggesting an involvement of other caspase-independent nuclear effector(s) of apoptosis in this case. These putative effectors are probably not released from the mitochondria as a consequence of the Bax/Bak-mediated outer-membrane permeabilization because we did not detect any nuclease activity in the supernatant of mitochondria incubated with recombinant oligomeric Bax and a caspase-independent type I nuclear apoptosis could be seen in cells where the cytochrome c was not yet released in the cytosol (data not shown) or in cells overexpressing Bcl-2 (D.Arnoult and R.J.Youle, unpublished results). Furthermore, in contrast with Bax-overexpressing cells or cells treated with actinomycin D or etoposide (data not shown), zVAD-fmk does not prevent cell shrinkage upon staurosporine treatment, suggesting that the cell shrinkage may be responsible for the chromatin condensation via an unidentified mechanism. Moreover, cells contain many nucleases (Liu et al., 1999) that may be activated after kinase inhibition by staurosporine treatment.
EndoG and AIF have been reported to induce caspase-independent nuclear apoptosis and thus it has been proposed that they are involved in caspase-independent cell death processes (Susin et al., 1999, 2000; L.Y.Li et al., 2001). Paradoxically, our results, based on the use of the broad caspase inhibitor zVAD-fmk or of Apaf-1 –/– MEFs, suggest that EndoG and AIF require caspase activation downstream of mitochondria to be released into the cytosol and thus to participate in the cell suicide. Our observations are consistent with the Caenorhabditis elegans model where the homologs of EndoG and AIF (Cps-6 and Wah-1, respectively) have been reported to act downstream of Ced-3 during C.elegans apoptosis (Parrish et al., 2001; Wang et al., 2002). Thus, EndoG and AIF seem to define a ‘caspase-dependent’ mitochondria-initiated apoptotic DNA degradation pathway that is conserved between mammals and nematodes (Figure 8). Further studies are required to understand how, in both models, active caspases trigger the mitochondrial release of AIF and EndoG.

Fig. 8. EndoG and AIF define a caspase-dependent mitochondria-initiated apoptotic DNA degradation pathway that is conserved between mammals and C.elegans. The genetic control of apoptosis leading to caspase activation is conserved between C.elegans and mammals (Horvitz, 1999; Hengartner, 2000). In both models, active caspases trigger the mitochondrial release of EndoG and AIF (Cps-6 and Wah-1 respectively in C.elegans) that are involved in DNA degradation. In mammals, CAD and Acinus, two other factors involved in nuclear apoptosis are activated by caspases (Sahara et al., 1999; Nagata, 2000). The dashed line indicates that the inhibition of Apaf-1 by Bcl-2 or Bcl-XL is not direct.
Surprisingly, in C.elegans, Wah-1 associates and cooperates with Cps-6 to promote DNA degradation (Wang et al., 2002). This is inconsistent with the reports that AIF and EndoG promote DNA degradation independently of each other (Susin et al., 1999; L.Y.Li et al., 2001). Indeed, recombinant AIF does not need EndoG to trigger chromatin condensation/fragmentation on isolated nuclei (Susin et al., 1999) and recombinant EndoG does not require AIF to trigger DNA degradation (L.Y.Li et al., 2001). Nevertheless, it has been reported that EndoG is stimulated by exonucleases and DNase I to generate double-stranded DNA cleavage products (Widlak et al., 2001) and AIF may also stimulate EndoG. AIF does not have nuclease activity per se but activates nuclease(s) present in the nucleus to trigger DNA condensation and fragmentation (Susin et al., 1999). This nuclease may be EndoG because it has been reported that a form of this nuclease is also present in the nucleus (Gerschenson et al., 1995).
Here, we have identified the mitochondrial response to the pro-apoptotic Bcl-2 members as a selective process leading to a hierarchical ordering of the effectors involved in cell death induction. Our findings also provide a paradigm for mitochondria-dependent cell death pathways since the intracytosolic release of the caspase-independent effectors EndoG and AIF requires caspase activation downstream of the co-release of cytochrome c, Smac/Diablo and HtrA2/Omi.
Materials and methods
Cell culture and transfection
HeLa cells and MEFs were cultured in Dulbecco’s modified Eagle’s medium (Gibco Brl) supplemented with 10% fetal calf serum, 2 mM l-glutamine, 50 IU penicillin and 50 µg/ml streptomycin under standard conditions. Transient transfection of HeLa cells was performed using Fugene 6 (Roche).
Isolation of mitochondria and in vitro assays for the release of apoptogenic factors
Mitochondria were isolated from HeLa cells or mouse liver by sucrose density gradient centrifugation and the in vitro assays for the release of apoptogenic factors were performed as previously described (Eskes et al., 1998; Desagher et al., 1999; Arnoult et al., 2002). Full-length oligomeric and monomeric recombinant Bax was produced as previously described (Antonsson et al., 2000; Suzuki et al., 2000). Recombinant tBid was purchased in R&D system. Preparation of mitoplasts was performed as previously described (Arnoult et al., 2002).
Subcellular fractionation
HeLa cells were harvested in isotonic mitochondrial buffer (MB) (210 mM mannitol, 70 mM sucrose, 1 mM EDTA and 10 mM HEPES pH 7.5), supplemented with protease inhibitor cocktail Complete (Boehringer Mannheim), and homogenized for 30–40 strokes with a Dounce homogenizer. Samples were transferred to Eppendorf centrifuge tubes and centrifuged at 500 g for 5 min at 4°C to eliminate nuclei and unbroken cells. The resulting supernatant was centrifuged at 10 000g for 30 min at 4°C to obtain the heavy membrane pellet enriched for mitochondria, and the resulting supernatant was stored as the cytosolic fraction.
Protein studies
Preparation of cellular lysates, immunoblotting and immunofluorescence were performed as described previously (Desagher et al., 1999; Finucane et al., 1999; Arnoult et al., 2002). Antibodies used in immunoblotting were as follows: anti-cytochrome c mAb (clone 7H8.2C12, Pharmingen) (1/2000), rabbit polyclonal anti-Endo G (Pro-Sci Inc.) (1/1000), rabbit polyclonal anti-Smac/Diablo (Pro-Sci Inc.) (1/1000), a purified rabbit polyclonal anti-HtrA2/Omi antibody that we obtained from a rabbit immunized against a mixture of two different human HtrA2/Omi peptides (amino acids 65–80 and 203–220) (1/1000), anti-AIF mAb (clone E1, Santa Cruz) (1/2000), anti-PARP mAb (clone C2-10, Pharmingen) (1/2000), anti-VDAC mAb (clone Ab4, Calbiochem) (1/6000), anti-Cox IV mAb (clone 10G8, Molecular Probe) (1/1000), anti-actin mAb (Sigma) (1/5000) and rabbit polyclonal antibodies specific for caspase-9 (Cayman Chemicals) (1/1000) or caspase-3 (Stressgen) (1/2000). Primary antibodies were then visualized using horseradish-peroxidase-conjugated secondary antibodies (Amersham), followed by enhanced chemiluminescence (Amersham).
Antibodies used in immunofluorescence were as follows: anti-AIF mAb (clone E1, Santa Cruz) (1/100), anti-cytochrome c mAb (clone 6H2.B4, Pharmingen) (1/800) or a sheep polyclonal anti-cytochrome c (Sigma) (1/800), rabbit polyclonal anti-Endo G (Pro-Sci Inc.) (1/400) and rabbit polyclonal anti-Smac/Diablo (Pro-Sci Inc.) (1/600). Antibodies were then labeled with secondary anti-mouse, anti-rabbit and anti-sheep antibodies (Alexa, Molecular Probes) (1/800). The cells were examined under a Zeiss LSM 510 confocal microscope.
In vitro assay for DNA degradation
Isolated CEM nuclei (2 × 103) were incubated for 6 h with 40 µl of supernatant from isolated mitochondria incubated or not with recombinant Bax as described above and with mitochondrial lysis extract (lysis of 100 µg of mitochondria by 10× 10 s of sonication) as positive control. Relative DNA content was assessed by flow cytometry. CEM nuclei were prepared as follows. CEM cells were washed twice in phosphate-buffered saline and once with nuclei isolation buffer (NB) [10 mM PIPES pH 7.4, 10 mM KCl, 2 mM MgCl2, 1 mM dithiothreitol (DTT), 10 µM cytochalasin B and 1 mM phenyl-methylsulfonyl fluoride (PMSF)]. Next, they were suspended in NB, allowed to swell on ice for 20 min and gently lysed with a Dounce homogenizer. Liberated nuclei were then layered over 30% sucrose in NB and centrifuged at 800 g for 10 min, followed by washing in NB and suspension in nucleus storage buffer (10 mM PIPES pH 7.4, 80 mM KCl, 20 mM NaCl, 250 mM sucrose, 5 mM EGTA, 1 mM DTT, 0.5 mM spermidine, 0.2 mM spermine, 1 mM PMSF and 50% glycerol) at 2 × 108 nuclei/ml.
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We are very grateful to Dr X.Wang (Dallas, Texas) for the provision of the Smac/Diablo and EndoG expression vectors and to Dr A.M.Verhagen (Melbourne, Australia) for the HtrA2/Omi plasmids. Special thanks are due to Dr J.Estaquier and Dr F.Petit (Institut Pasteur, France). This work was supported by ARC (postdoctoral fellowship to D.A.) and by AIRC (grants to F.C.).
References
- Adrain C., Creagh,E.M. and Martin,S.J. (2001) Apoptosis-associated release of Smac/DIABLO from mitochondria requires active caspases and is blocked by Bcl-2. EMBO J., 20, 6627–6636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonsson B., Montessuit,S., Lauper,S., Eskes,R. and Martinou,J.C. (2000) Bax oligomerization is required for channel-forming activity in liposomes and to trigger cytochrome c release from mitochondria. Biochem. J., 345, 271–278. [PMC free article] [PubMed] [Google Scholar]
- Arnoult D., Parone,P., Martinou,J.C., Antonsson,B., Estaquier,J. and Ameisen,J.C. (2002) Mitochondrial release of apoptosis-inducing factor occurs downstream of cytochrome c release in response to several proapoptotic stimuli. J. Cell Biol., 159, 923–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cecconi F., Alvarez-Bolado,G., Meyer,B.I., Roth,K.A. and Gruss,P. (1998) Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell, 94, 727–737. [DOI] [PubMed] [Google Scholar]
- Desagher S. and Martinou,J.C. (2000) Mitochondria as the central control point of apoptosis. Trends Cell. Biol., 10, 369–377. [DOI] [PubMed] [Google Scholar]
- Desagher S., Osen-Sand,A., Nichols,A., Eskes,R., Montessuit,S., Lauper,S., Maundrell,K., Antonsson,B. and Martinou,J.C. (1999) Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J. Cell Biol., 144, 891–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doran E. and Halestrap,A.P. (2000) Cytochrome c release from isolated rat liver mitochondria can occur independently of outer-membrane rupture: possible role of contact sites. Biochem. J., 348, 343–350. [PMC free article] [PubMed] [Google Scholar]
- Du C., Fang,M., Li,Y., Li,L. and Wang,X. (2000) Smac, a mitochondrial protein that promotes cytochrome-c-dependent caspase activation by eliminating IAP inhibition. Cell, 102, 33–42. [DOI] [PubMed] [Google Scholar]
- Eskes R., Antonsson,B., Osen-Sand,A., Montessuit,S., Richter,C., Sadoul,R., Mazzei,G., Nichols,A. and Martinou,J.C. (1998) Bax-induced cytochrome c release from mitochondria is independent of the permeability transition pore but highly dependent on Mg2+ ions. J. Cell Biol., 143, 217–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finucane D.M., Bossy-Wetzel,E., Waterhouse,N.J., Cotter,T.G. and Green,D.R. (1999) Bax-induced caspase activation and apoptosis via cytochrome c release from mitochondria is inhibitable by Bcl-xL. J. Biol. Chem., 274, 2225–2233. [DOI] [PubMed] [Google Scholar]
- Gerschenson M., Houmiel,K.L. and Low,R.L. (1995) Endonuclease G from mammalian nuclei is identical to the major endonuclease of mitochondria. Nucleic Acids Res., 23, 88–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein J.C., Waterhouse,N.J., Juin,P., Evan,G.I. and Green,D.R. (2000) The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nat. Cell Biol., 2, 156–162. [DOI] [PubMed] [Google Scholar]
- Gottlieb E., Armour,S.M., Harris,M.H. and Thompson,C.B. (2003) Mitochondrial membrane potential regulates matrix configuration and cytochrome c release during apoptosis. Cell Death Differ., 10, 709–717. [DOI] [PubMed] [Google Scholar]
- Gross A., McDonnell,J.M. and Korsmeyer,S.J. (1999) Bcl-2 family members and the mitochondria in apoptosis. Genes Dev., 13, 1899–1911. [DOI] [PubMed] [Google Scholar]
- Hegde R. et al. (2002) Identification of Omi/HtrA2 as a mitochondrial apoptotic serine protease that disrupts inhibitor of apoptosis protein-caspase interaction. J. Biol. Chem., 277, 432–438. [DOI] [PubMed] [Google Scholar]
- Hengartner M.O. (2000) The biochemistry of apoptosis. Nature, 407, 770–776. [DOI] [PubMed] [Google Scholar]
- Horvitz H.R. (1999) Genetic control of programmed cell death in the nematode Caenorhabditis elegans. Cancer Res., 59, 1701s–1706s. [PubMed] [Google Scholar]
- Jacobson M.D., Weil,M. and Raff,M.C. (1997) Programmed cell death in animal development. Cell, 88, 347–354. [DOI] [PubMed] [Google Scholar]
- Jurgensmeier J.M., Xie,Z., Deveraux,Q., Ellerby,L., Bredesen,D. and Reed,J.C. (1998) Bax directly induces release of cytochrome c from isolated mitochondria. Proc. Natl Acad. Sci. USA, 95, 4997–5002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluck R.M., Bossy-Wetzel,E., Green,D.R. and Newmeyer,D.D. (1997) The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science, 275, 1132–1136. [DOI] [PubMed] [Google Scholar]
- Kroemer G. and Reed,J.C. (2000) Mitochondrial control of cell death. Nat. Med., 6, 513–519. [DOI] [PubMed] [Google Scholar]
- Kuwana T., Mackey,M.R., Perkins,G., Ellisman,M.H., Latterich,M., Schneiter,R., Green,D.R. and Newmeyer,D.D. (2002) Bid, bax and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell, 111, 331–342. [DOI] [PubMed] [Google Scholar]
- Li L.Y., Luo,X. and Wang,X. (2001) Endonuclease G is an apoptotic DNase when released from mitochondria. Nature, 412, 95–99. [DOI] [PubMed] [Google Scholar]
- Li P., Nijhawan,D., Budihardjo,I., Srinivasula,S.M., Ahmad,M., Alnemri,E.S. and Wang,X. (1997) Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell, 91, 479–489. [DOI] [PubMed] [Google Scholar]
- Liu Q.Y., Ribecco,M., Pandey,S., Walker,P.R. and Sikorska,M. (1999) Apoptosis-related functional features of the DNaseI-like family of nucleases. Ann. N.Y. Acad. Sci., 887, 60–76. [DOI] [PubMed] [Google Scholar]
- MacFarlane M., Merrison,W., Bratton,S.B. and Cohen,G.M. (2002) Proteasome-mediated degradation of Smac during apoptosis: XIAP promotes Smac ubiquitination in vitro. J. Biol. Chem., 277, 36611–36616. [DOI] [PubMed] [Google Scholar]
- Martinou J.C. and Green,D.R. (2001) Breaking the mitochondrial barrier. Nat. Rev. Mol. Cell. Biol., 2, 63–67. [DOI] [PubMed] [Google Scholar]
- Marzo I. et al. (1998) Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science, 281, 2027–2031. [DOI] [PubMed] [Google Scholar]
- Mikhailov V., Mikhailova,M., Pulkrabek,D.J., Dong,Z., Venkatachalam,M.A. and Saikumar,P. (2001) Bcl-2 prevents bax oligomerization in the mitochondrial outer membrane. J. Biol. Chem., 276, 18361–18374. [DOI] [PubMed] [Google Scholar]
- Nagata S. (2000) Apoptotic DNA fragmentation. Exp. Cell Res., 256, 12–18. [DOI] [PubMed] [Google Scholar]
- Ott M., Robertson,J.D., Gogvadze,V., Zhivotovsky,B. and Orrenius,S. (2002) Cytochrome c release from mitochondria proceeds by a two-step process. Proc. Natl Acad. Sci. USA, 99, 1259–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish J., Li,L., Klotz,K., Ledwich,D., Wang,X. and Xue,D. (2001) Mitochondrial endonuclease G is important for apoptosis in C.elegans. Nature, 412, 90–94. [DOI] [PubMed] [Google Scholar]
- Ricci J.E., Gottlieb,R.A. and Green,D.R. (2003) Caspase-mediated loss of mitochondrial function and generation of reactive oxygen species during apoptosis. J. Cell Biol., 160, 65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahara S., Aoto,M., Eguchi,Y., Imamoto,N., Yoneda,Y. and Tsujimoto,Y. (1999) Acinus is a caspase-3-activated protein required for apoptotic chromatin condensation. Nature, 401, 168–173. [DOI] [PubMed] [Google Scholar]
- Scorrano L., Ashiya,M., Buttle,K., Weiler,S., Oakes,S.A., Mannella,C.A. and Korsmeyer,S.J. (2002) A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev. Cell, 2, 55–67. [DOI] [PubMed] [Google Scholar]
- Susin S.A., et al. (1999) Molecular characterization of mitochondrial apoptosis-inducing factor. Nature, 397, 441–446. [DOI] [PubMed] [Google Scholar]
- Susin S.A., et al. (2000) Two distinct pathways leading to nuclear apoptosis. J. Exp. Med., 192, 571–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M., Youle,R.J. and Tjandra,N. (2000) Structure of Bax: coregulation of dimer formation and intracellular localization. Cell, 103, 645–654. [DOI] [PubMed] [Google Scholar]
- Suzuki Y., Imai,Y., Nakayama,H., Takahashi,K., Takio,K. and Takahashi,R. (2001) A serine protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death. Mol. Cell, 8, 613–621. [DOI] [PubMed] [Google Scholar]
- Thompson C.B. (1995) Apoptosis in the pathogenesis and treatment of disease. Science, 267, 1456–1462. [DOI] [PubMed] [Google Scholar]
- Thornberry N.A. and Lazebnik,Y. (1998) Caspases: enemies within. Science, 281, 1312–1316. [DOI] [PubMed] [Google Scholar]
- Van Loo G., Saelens,X., Van Gurp,M., MacFarlane,M., Martin,S.J. and Van den abeele,P. (2002) The role of mitochondrial factors in apoptosis: a Russian roulette with more than one bullet. Cell Death Differ., 9, 1031–1042. [DOI] [PubMed] [Google Scholar]
- Van der Heiden M.G. and Thompson,C.B. (1999) Bcl-2 proteins: regulators of apoptosis or of mitochondrial homeostasis? Nat. Cell Biol., 1, E209–216. [DOI] [PubMed] [Google Scholar]
- Verhagen A.M., Ekert,P.G., Pakusch,M., Silke,J., Connolly,L.M., Reid,G.E., Moritz,R.L., Simpson,R.J. and Vaux,D.L. (2000) Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell, 102, 43–53. [DOI] [PubMed] [Google Scholar]
- Verhagen A.M., et al. (2002) HtrA2 promotes cell death through its serine protease activity and its ability to antagonize inhibitor of apoptosis proteins. J. Biol. Chem., 277, 445–454. [DOI] [PubMed] [Google Scholar]
- Wang X. (2001) The expanding role of mitochondria in apoptosis. Genes Dev., 15, 2922–2933. [PubMed] [Google Scholar]
- Wang X., Yang,C., Chai,J., Shi,Y. and Xue,D. (2002) Mechanisms of AIF-mediated apoptotic DNA degradation in Caenorhabditis elegans. Science, 298, 1587–1592. [DOI] [PubMed] [Google Scholar]
- Waterhouse N.J., Goldstein,J.C., von Ahsen,O., Schuler,M., Newmeyer,D.D. and Green,D.R. (2001) Cytochrome c maintains mitochondrial transmembrane potential and ATP generation after outer mitochondrial membrane permeabilization during the apoptotic process. J. Cell Biol., 153, 319–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei M.C., et al. (2001) Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science, 292, 727–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widlak P., Li,L.Y., Wang,X. and Garrard,W.T. (2001) Action of recombinant human apoptotic endonuclease G on naked DNA and chromatin substrates: cooperation with exonuclease and DNase I. J. Biol. Chem., 276, 48404–48409. [DOI] [PubMed] [Google Scholar]
- Yoshida H., Kong,Y.Y., Yoshida,R., Elia,A.J., Hakem,A., Hakem,R., Penninger,J.M. and Mak,T.W. (1998) Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell, 94, 739–750. [DOI] [PubMed] [Google Scholar]
- Zamzami N. and Kroemer,G. (2001) The mitochondrion in apoptosis: how Pandora’s box opens. Nat. Rev. Mol. Cell. Biol., 2, 67–71. [DOI] [PubMed] [Google Scholar]