

Abstract
The 20S proteasome is a large multisubunit assembly that performs most of the intracellular non-lysosomal proteolysis of eukaryotes. Substrates access the proteasome active sites, which are sequestered in the interior of the barrel-shaped structure, through pores that are opened by binding of activator complexes. The crystal structure of yeast proteasome in complex with an 11S activator suggested that activation results from disordering of the proteasome gate residues. Here we report further analysis of this structure, which demonstrates that, in contrast to earlier models, the activated proteasome adopts an ordered 7-fold symmetric pore conformation that is stabilized by interactions formed by a cluster of highly conserved proteasome residues (Tyr8, Asp9, Pro17 and Tyr26). One non-canonical cluster, which appears to be mandated by the requirement that eukaryotic proteasomes also form an ordered closed conformation, explains all deviations from perfect conservation of these residues. We also demonstrate the importance of these conserved residues for proteolysis by an archaeal proteasome. Evolutionary considerations suggest that other activators might induce the same open proteasome conformation as seen with the 11S activator.
Keywords: activation/7-fold symmetry/macromolecular crystal structure/multisubunit complex/proteasome
Introduction
The 20S proteasome is an abundant protease that performs most of the proteolysis that occurs in the cytosol and nucleus of eukaryotes (Rock et al., 1994; Bochtler et al., 1999; Voges et al., 1999; Zwickl et al., 2000; Zwickl, 2002). This activity functions to regulate many critical facets of cellular metabolism including homeostasis, signaling pathways, cell cycle progression and the production of antigenic peptides (Hershko and Ciechanover, 1998). The mechanism of substrate selection is of critical importance for proteasome function, and crystal structures of 20S proteasomes from archaea (Löwe et al., 1995) and eukaryotes (Groll et al., 1997) explain how this ∼700 kDa, 28-subunit complex is normally maintained in a repressed conformation by sequestration of the catalytic centers within a central chamber (Bochtler et al., 1999; Zwickl et al., 2001).
The structure of the 20S proteasome from the archaeon Thermoplasma acidophilum revealed an exactly 7-fold symmetric assembly of four stacked rings, in which the two outer rings are each composed of seven α-subunits and the two inner rings are composed of seven β-subunits (Löwe et al., 1995). The 14 active sites of this complex are located at the N-termini of β-subunits and face the central chamber (Seemüller et al., 1995), which is accessed via an opening (α-annulus) through the end rings of α-subunits that is too narrow (13 Å diameter) to allow passage of folded proteins (Wenzel and Baumeister, 1995). The archaeal (Löwe et al., 1995), yeast (Groll et al., 1997) and bovine (Unno et al., 2002) 20S proteasomes have essentially identical architectures, except that the exact 7-fold symmetry is broken in eukaryotic proteasomes by the presence of seven different α-subunits (α1–α7) and seven different β-subunits (β1–β7) that occupy unique positions in their respective rings. This asymmetry allows for a more complete isolation of the eukaryotic 20S proteasome’s internal catalytic chamber, since residues near the N-terminus of α-subunits, especially α2, α3 and α4, pack closely against each other in a highly asymmetric fashion to close a gate that is formed above the α-annulus. In contrast, the archaeal proteasome gate appears to be more open because the first 12 residues of its α-subunits are disordered in the crystal structure (Löwe et al., 1995).
Proteasomes are stimulated by association of their α-subunits with activators, including members of the 11S activator family. Our previously reported 3.2 Å resolution crystal structure of a complex between yeast 20S proteasome and PA26, the 11S activator from Trypanosoma brucei, showed how 11S activators open the proteasome’s entrance port by imposing their own 7-fold symmetry on the proteasome (Whitby et al., 2000; Hill et al., 2002). This analysis appeared to show that the port is opened by disordering of the N-terminal residues of proteasome α-subunits. Further support for this disordering model of proteasome activation was provided by a nine-residue deletion at the N-terminus of the α3 subunit of the yeast proteasome, which resulted in stimulation of proteasomal peptidase activity and disordering of the remaining pore residues (Groll et al., 2000).
In an effort to understand better the mechanism of proteasome activation, we have analyzed the PA26–20S proteasome crystal structure further to reveal that the N-terminal tails of α-subunits do in fact adopt an ordered conformation in this complex. The role observed for highly conserved residues in stabilizing this open conformation and the apparent absence of 11S activators in yeast and archaea suggest that the same open conformation might also exist in other active proteasomes. We have tested this hypothesis by biochemical analyses that demonstrate the importance of these conserved residues for proteolysis by an archaeal proteasome.
Results
Crystallographic refinement
We previously reported the crystal structure of yeast 20S proteasome with the 11S (PA26) activator from the trypanosome T.brucei (Whitby et al., 2000). In common with other 11S activators (Mykles, 1996), PA26 stimulates hydrolysis of peptide substrates by proteasomes from several diverse species (Yao et al., 1999), including yeast (Whitby et al., 2000). Consistent with previous biochemical analysis (Song et al., 1997; Li et al., 2000), the crystal structure revealed that the PA26 C-terminal residues provide binding affinity by docking into pockets formed between the proteasome α-subunits. This positions a nine-residue segment known as the activation loop (Knowlton et al., 1997; Zhang et al., 1998) over a reverse turn of the proteasome α-subunits that contains Pro17, thereby causing this residue in each of the seven α-subunits to move as much as 2.5 Å to match the 7-fold symmetry of the activator. The extended conformation ensures that these displacements are propagated to more N-terminal residues and therefore disrupt the numerous hydrogen bond and van der Waals interactions that stabilize the closed conformation. Correspondingly, the α-subunit N-terminal tails were seen to rearrange and project away from the gate region into the channel of the 11S activator, although well-defined conformations were only discernible after residue 12 (Whitby et al., 2000).
The current structural analysis (Table I), which was performed against the original diffraction data (Whitby et al., 2000) and gave an Rfree value of 30.8%, compared with 32.5% from the original analysis, was prompted by the realization that the exactly 7-fold symmetric PA26 functions by imposing its own symmetry on the 20S proteasome. Furthermore, residues that have been strongly conserved through evolution and are also conserved between all seven of the α-subunits appeared to be disordered in the structure and to lack a functional role. Alignment of sequences of α-subunit N-terminal tails (Figure 1) showed that the apparently disordered Tyr8 and Asp9 residues were highly conserved between all seven α-subunits and between all species from archaea to human. (Residue numbering according to the T.acidophilum proteasome α-subunits is used throughout.) The basis for this conservation was unclear because, although in some subunits these residues make important contacts in the closed conformation, in other subunits they do not (Groll et al., 1997, 2000). Because refinement of the PA26–proteasome crystal structure suffers from an inherently high B-factor and limited resolution, we considered the possibility that an ordered and functionally important 7-fold symmetric conformation of these proteasome residues had been missed in the original analysis (Whitby et al., 2000). We therefore extended the crystallographic study to include 7-fold non-crystallographic symmetry averaging over the region of the proteasome gate. Despite only limited conservation of most N-terminal residues between proteasome α-subunits, this process revealed ordered conformations for the α-subunits after residue 7, with additional N-terminal residues also being ordered for some of the subunits (Figure 2). Some minor improvements were also made to the PA26 part of the model, although no other significant changes were made to the proteasome structure.
Table I. Crystallographic statistics.
| Resolution range | 100–3.2 Å | (3.26–3.2 Å) |
| No. of reflections measured | 503 540 | |
| No. of unique reflections | 189 615 | (6632) |
| Completeness | 89.1% | (62.9%) |
| Rmergea | 0.112 | (0.331) |
| I/σ(I) | 7.0 | (1.9) |
| R-factorb | 0.263 | (0.345) |
| Rfreec | 0.308 | (0.411) |
| R.m.s. bond lengthsd | 0.009 Å | |
| R.m.s. bond anglesd | 1.391° | |
| Average B-factor: proteasome | 64.7 Å2 | |
| Average B-factor: PA26 | 95.9 Å2 |
The data used are those of Whitby et al. (2000). Slight improvements have been made throughout the model by further refinement, especially for the N-terminal residues of α-subunits, which were visualized following non-crystallographic symmetry averaging in the proteasome gate region as described in the text. Values in parentheses refer to the highest resolution shell.
aRmerge = Σ|I – <I>|/ΣI, where I is the intensity of an individual measurement and <I> is the corresponding mean value.
bR-factor = Σ||Fo| – |Fc||/Σ|Fo|, where |Fo| is the observed and |Fc| the calculated structure factor amplitude.
cRfree is the same as Rfactor calculated with a randomly selected test set of 1264 reflections that were never used in refinement calculations.
dThe r.m.s. deviation in bond lengths and bond angles from ideal values.

Fig. 1. Sequence alignment of 20S proteasome α-subunit N-terminal residues. The seven (α1–α7) subunits of yeast (Sc) and human (Hs) proteasomes are shown with the single α-subunit of the T.acidophilum proteasome. The four conserved residues that stabilize the open gate conformation are shown on a yellow background. Other conserved residues generally perform obvious structural roles that do not change between open and closed proteasome conformations.

Fig. 2. Unbiased map and refined model stereoview. N-terminal residues of the proteasome α6 subunit are shown with the 14-fold averaged simulated annealing omit 2Fo – Fc map contoured at 2.2 times the r.m.s.d. and shown with a 1.5 Å cover. All 14 N-terminal tails in the asymmetric unit through residue 26 (T.acidophilum numbering) were omitted from the model. The view direction is from the middle of the pore with the 7-fold axis vertical.
Tyr8, Asp9, Pro17 and Tyr26 stabilize the open conformation
The refined structure (Figure 3) now shows that PA26 induces all seven of the α-subunit N-terminal tails to adopt an ordered conformation for residues 7–12, with some more N-terminal residues also ordered for some of the subunits. This results in formation of clusters of four highly conserved residues, Tyr8, Asp9, Pro17 and Tyr26, that pack against each other at the interface between each of the adjacent α-subunits (Figure 4). Thus, in addition to destabilizing the closed conformation, as described earlier (Whitby et al., 2000), repositioning of Pro17 by the PA26 activation loop allows stabilizing interactions to form between the four conserved proteasome residues.
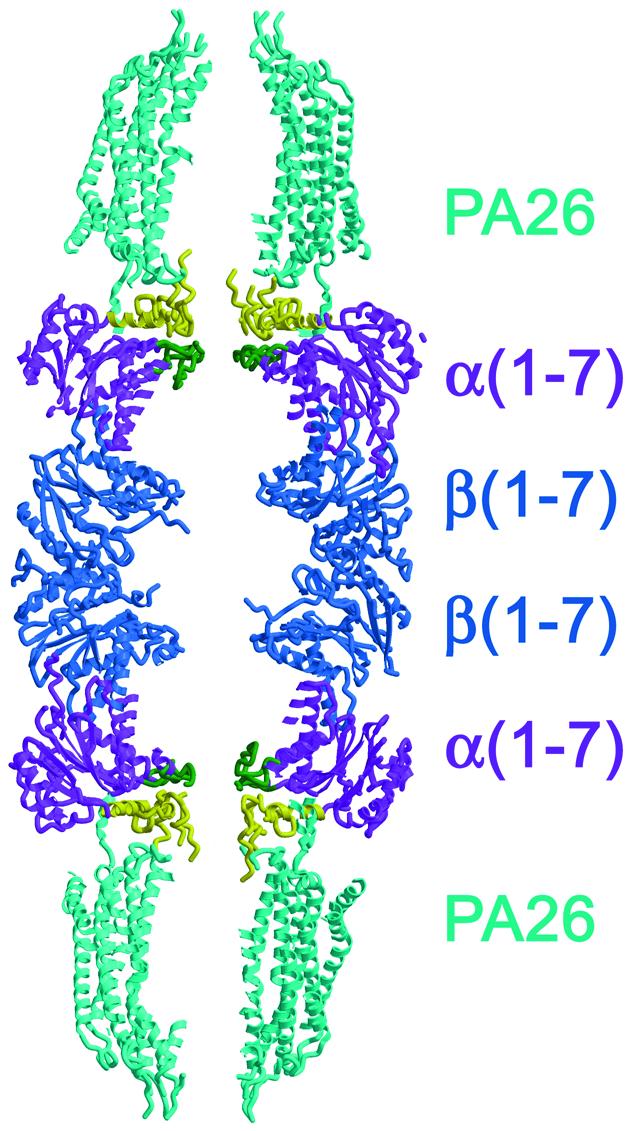
Fig. 3. Structure of the PA26–yeast 20S proteasome complex. Cut-away ribbon representation. Ordered N-terminal residues of proteasome α-subunits are colored yellow up to position 34. Disordered residues at the α-subunit N-termini (typically before residue 7) appear to project further into the central channel of PA26, but are not shown on this figure. Residues of proteasome α-subunits that form the α-annulus are colored green.

Fig. 4. Interactions of conserved residues that stabilize the open proteasome conformation. The clusters of Tyr8, Asp9, Pro17 and Tyr26 side chains that stabilize the open pore structure are shown explicitly with yellow carbon atoms. (A) Top view of the yeast proteasome α-subunits as seen in the complex with PA26. (B) Enlarged view of the central region of (A). (C) Close up view of the cluster outlined in (B). Hydrogen bonding interactions are shown by dotted lines.
Residues of the conserved cluster make only limited contact with PA26. Asp9 does not contact PA26, while Tyr8 and Tyr26 make one or two van der Waals contacts that do not appear to constrain their conformations significantly. The location of Pro17 is defined by contacts to the PA26 activator, and repositioning of this residue appears to drive formation of the open conformation. The Pro17–PA26 interaction is indirect, however, since the dominant interaction is between PA26 Glu102 and residues adjacent to Pro17 (Figure 5), while contacts of PA26 Glu102 with Pro17 are minor and Pro17 does not contact any other PA26 residue. This suggests that the clusters of conserved residues seen in the open gate complex with PA26 are an inherent property of 20S proteasomes, and do not depend critically upon the specific details of interactions with an activator.
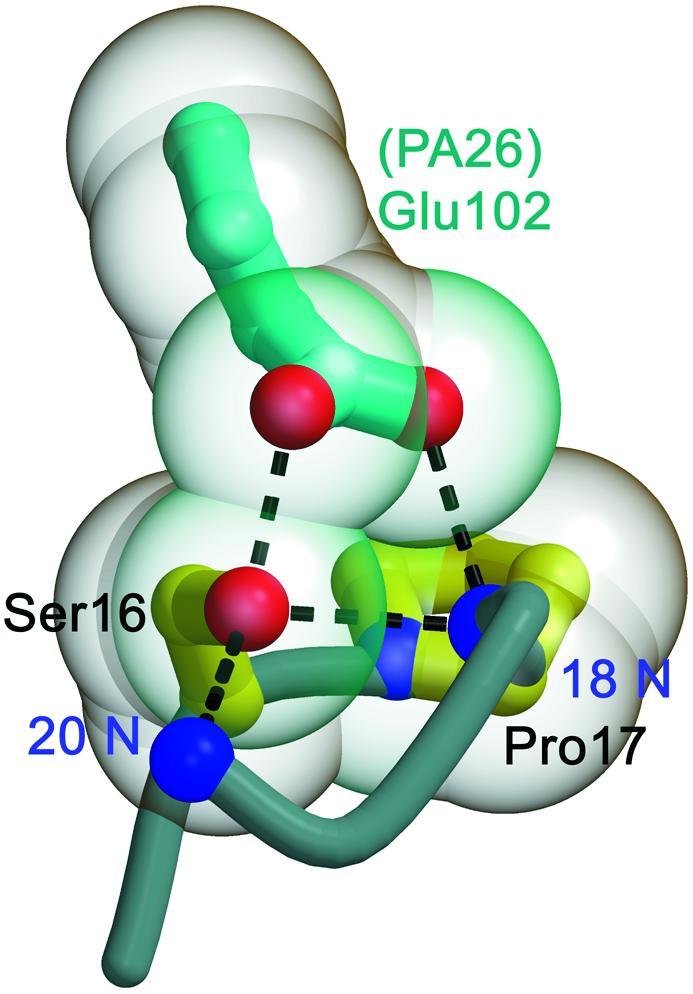
Fig. 5. Interactions with PA26 Glu102 and the position of Pro17 reverse turns. PA26 makes contacts with residues adjacent to Pro17, but does not significantly contact residues of the conserved cluster directly. Hydrogen bonds are shown as dashed lines.
Tyr8, Asp9, Pro17 and Tyr26 are also important for proteolysis by an archaeal proteasome
The role of proteasome α-subunit residues Tyr8, Asp9, Pro17 and Tyr26 in stabilizing the open conformation explains their conservation in all species that possess 11S activators. It is less obvious, however, why these residues are also conserved in species, such as yeast and the archaea, that do not seem to encode related activators. One possible explanation for the evolutionary data is that the residues seen to stabilize the open conformation in the 11S activator complex might also be important for the activity of other proteasomes. We have tested this possibility using a biochemical assay with an archaeal 20S proteasome and the activator PAN (Zwickl et al., 1999), which is generally considered to be the archaeal homolog of the 19S activator. Although this proteasome and activator are from different archaeal species (T.acidophilum and Methanococcus jannaschii, respectively), they provide a tractable system and are used routinely together in studies of proteasome mechanism (for example see Benaroudj et al., 2003). This system has the advantage of simplicity, since the symmetric archaeal proteasome has just one type of α- and one type of β-subunit (each repeated seven times in a ring), and PAN is a homo-oligomer. Furthermore, the archaeal proteasome and PAN can be expressed in bacteria.
We have prepared a series of 13 mutant T.acidophilum 20S proteasomes and tested their activity in an assay that records the rate of degradation of a His6-tagged green fluorescent protein (GFP) substrate (Navon and Goldberg, 2001). This substrate has the advantages of thermal stability (assays for the thermophilic proteasome/PAN system are done at 57°C), ease of detection via western blot, and a C-terminal ssrA tag that targets substrates to PAN. All of the recombinant proteasomes used in these assays were highly pure as judged by Coomassie-stained SDS–PAGE, correctly assembled as indicated by sizing chromatography, and showed comparable basal activity against the small fluorogenic peptide substrate N-succinyl-Leu-Leu-Val-Tyr-7-amido-4-methylcoumarin (Seemüller et al., 1995) (data not shown).
Whereas the wild-type archaeal proteasome degrades substrate rapidly in the assay with PAN, all 10 proteasome mutants with substitutions in Tyr8, Asp9, Pro17 or Tyr26 show greatly reduced activity (Figure 6 and Supplemen tary data available at The EMBO Journal Online). The three mutant proteasomes that show similar activity to wild type are also consistent with the model. The high activity observed for the Δ(2–7) proteasome is expected because these N-terminal residues are not generally well ordered in the PA26–proteasome structure and therefore do not appear to contribute to the stability of the open conformation. The high activity of the Δ(2–12) proteasome is expected since this truncation deletes all residues of the axial pore. Finally, the relatively high activity of the Arg10Ala proteasome is consistent with the lack of conservation of this residue in most eukaryotic α-subunits (Figure 1) and the apparent lack of strongly stabilizing interactions of these side chains in the open PA26-activated proteasome. There is some relatively small but significant decrease in activity for this mutant, however, which, as discussed more fully below, is consistent with a recently published crystal structure (Groll et al., 2003) that suggests that this residue makes a peripheral contact that stabilizes the open gate conformation of archaeal proteasomes to some extent. None of the proteasomes degrade GFP in the absence of PAN or ATP (data not shown).

Fig. 6. GFP degradation by T.acidophilum 20S proteasome mutants activated by M.jannaschii PAN. Proteasomes mutated at a single residue are denoted r10a for the Arg10 to Ala proteasome, etc. Proteasomes mutated in both Tyr8 and Asp9 are denoted aa, gg and dy, for the AlaAla, GlyGly and AspTyr substitutions. Proteasomes for which residues 2–7 or 2–12 were deleted are denoted Δ(2–7) and Δ(2–12). (A) Representative data for wild-type and mutant proteasomes that show high activity. The r10a mutant is included in this panel because, although it shows somewhat reduced activity compared with wild type, it is still much more active than mutations of the invariant cluster residues. Degradation of His6-GFP–ssrA is followed by western blot using anti-His antibodies. The top band in each panel is the substrate. The lower band is the His6-tagged proteasome β-subunit. Time in minutes is indicated above. (B) Same as (A) for proteasomes that show low activity.
We have demonstrated that these effects are not specific to the GFP substrate by repeating the assay with casein, another widely used model substrate that requires PAN for efficient degradation by T.acidophilum 20S proteasome (Supplementary data). This time the substrate was visualized by Coomassie-stained SDS–PAGE, rather than by western blot, and so the quantification is less precise. Nevertheless, it is clear that the mutant and wild-type T.acidophilum 20S proteasomes show the same relative efficiency in degradation of casein as they do for GFP, further substantiating the finding that these conserved residues are important for proteolysis by the archaeal proteasome. These data also demonstrate that proteolysis proceeds without formation of detectable levels of intermediately processed substrate (Supplementary data).
Discussion
The non-canonical cluster
Binding of PA26 activates yeast 20S proteasome by inducing a more symmetric open conformation of the entrance gate that is stabilized by contacts between residues that are highly conserved between all seven α-subunits and across species. All 18 of the available archaeal 20S proteasome sequences were found to be invariant for the cluster residues Tyr8, Asp9, Pro17 and Tyr26 (data not shown). For eukaryotes, however, the conservation of these residues between subunits is not absolute, with the few exceptions from perfect conservation of the cluster residues all being associated with a non-canonical cluster that is formed between subunits α1 and α2 (Figure 7).

Fig. 7. The non-canonical cluster. (A) Stereoview showing overlap of a canonical (yellow) and the α1/α2 non-canonical (cyan) clusters. Overlap was performed according to the 7-fold symmetry of the PA26 heptamer. All six canonical clusters overlap closely with the α6/α7 cluster shown here. View direction is from the position of the 7-fold axis, slightly above the top of the proteasome α-subunits. Hydrogen bonds of canonical clusters are shown as dotted lines. Potential steric clashes with α-2 Phe10 that exclude aspartate from position 9 of this subunit and displace Tyr8 of subunit α-1 are indicated with black lines. (B) Deviations from perfect conservation of cluster residues. All cluster residues of all subunits are invariant unless indicated here. Hs, Homo sapiens; Mm, Mus musculus; Rn, Rattus norvegicus; Dm, Drosophila melanogaster; Ce, Caenorhabditis elegans; At, Arabidopsis thaliana; Sp, Schizosaccharomyces pombe; Sc, Saccharomyces cerevisiae. Residue identities corresponding to the canonical sequences are shown on a yellow background.
The non-canonical cluster of the open conformation appears to be mandated by the requirement for eukaryotic proteasomes to form a precisely closed conformation (Groll et al., 1997; Unno et al., 2002). Whereas all other subunits have an aspartate at position 9, this residue is always serine or a smaller residue in α2. This substitution is required by the buried environment of α2 Ser9 in the closed conformation, and explains why α2 is the only subunit that shows deviation from perfect conservation of Tyr26, because the α2 Ser9 OG–α2 Tyr26 OH interaction is, at best, very weak (distance 3.5 Å compared with ∼3.0 Å for canonical Asp9–Tyr26 hydrogen bonds). Similarly, residue 10 is always a phenylalanine in α2, apparently because of its buried environment in the closed conformation. This residue is never a phenylalanine in other subunits, and its presence in α2 explains why α1 Tyr8 is not invariant, since α2 Phe10 displaces α1 Tyr8 from the canonical location and prevents its hydrogen bonding interaction with α2 Ser9. Thus, all deviations from perfect conservation of the cluster residues are explained by the requirement for eukaryotic proteasomes to form an ordered closed conformation that buries Ser9 and Phe10 of α2. This emphasizes the two-state character of eukaryotic proteasomes, which have precisely defined open and closed conformations, each of which place evolutionary constraints upon the allowable amino acid sequence.
The apparent inability of archaeal proteasomes to adopt the precisely closed conformation of their eukaryotic counterparts is explained by their perfect 7-fold symmetry. Because Ser9 and Phe10 of the non-canonical cluster are required for formation of the closed conformation of the eukaryotic proteasome, the ability to form the closed conformation in archaeal proteasomes would require that all seven of the identical archaeal α-subunits adopt the non-canonical cluster conformation. Our observation that the Asp9Ser mutation severely impairs the proteolytic activity of the T.acidophilum proteasome suggests that whereas the eukaryotic proteasome has one suboptimal cluster, seven non-canonical clusters are incompatible with a stable open conformation. The biological basis for a precisely closed conformation in eukaryotes but not in archaea is mysterious and merits further study.
The open conformation of archaeal proteasomes
The PA26–20S proteasome structure explains why Tyr8, Asp9, Pro17 and Tyr26 are conserved in species that possess 11S activators, but does not explain why these residues are also conserved in species, such as yeast and the archaea, that appear to lack 11S activators. We therefore performed biochemical experiments that demonstrated that these residues are important for proteolysis by an archaeal proteasome, and we propose that the reason these residues are important in archaea is because they stabilize the same open gate conformation that is observed in the structure of the yeast proteasome with PA26.
The crystal structure of the T.acidophilum 20S proteasome revealed that the first 12 residues of the α-subunits are disordered (Löwe et al., 1995), although they nevertheless occupy the region of the pore and provide a barrier to passage of protein substrates (Benaroudj et al., 2003). Our proposal that protein substrates enter the proteasome most efficiently while the gate is in an ordered open conformation, rather than in the flexible disordered state of the T.acidophilum 20S crystal structure, is most strongly supported by our observation that the Tyr8Gly Asp9Gly double mutant shows considerably reduced activity against protein substrates. These substitutions greatly reduce the mass of protein in the gate region and must greatly increase the inherent flexibility of the N-terminal tails. Thus, the model that substrates can enter the proteasome because these regions are flexible (Löwe et al., 1995; Groll et al., 2000; Whitby et al., 2000) would predict that this mutation should have increased activity, whereas the observed reduction in activity is consistent with our updated model for the open pore in which Tyr8 and Asp9 are ordered and participate in stabilizing interactions.
The possibility that binding of PAN induces the open conformation of the α-subunit N-terminal residues motivated us to detect a physical interaction between T.acidophilum 20S proteasome and M. jannaschii PAN. To this end, we have made determined efforts using gel filtration, native gel electrophoresis, surface plasmon resonance and analytical ultracentrifugation, but have failed to detect an interaction (data not shown). Similarly, although it is generally assumed in the literature that PAN and proteasome contact each other, we are unaware of a report that establishes this as a reliable observation, even for a cognate PAN–proteasome interaction (note that T.acidophilum does not appear to possess a PAN homolog). We therefore conclude that either the PAN–proteasome interaction is very transient, or that PAN makes substrates competent for proteasome entry through its unfolding activity but does not contact the proteasome directly. Our inability to detect a PAN–proteasome interaction argues against the possibility that the conserved cluster residues mediate direct contacts with the PAN activator. Further evidence against this possibility comes from the Δ(2–12) mutant, which completely lacks the cluster residues Tyr8 and Asp9, yet shows greater than wild-type activity in the GFP degradation assay (Figure 6). This increased activity presumably results from the greater ease of entry of unfolded substrates through the larger pore of Δ(2–12) proteasomes (Benaroudj et al., 2003) (Supplementary data). Nevertheless, the failure of the Δ(2–12) truncation to inhibit PAN-dependent proteolysis strongly implies that any direct contact that might be made between these residues and PAN is insignificant.
The ability of archaeal proteasomes to adopt the ordered open conformation in the absence of an activator is demonstrated by a recent crystal structure of a ring of α-subunits from Archaeoglobus fulgidus (Groll et al., 2003). This structure superimposes very closely with the structure of the T.acidophilum 20S proteasome (Löwe et al., 1995), except that the N-terminal residues of α-subunits that are disordered in the earlier structure are now ordered and superimpose almost exactly with the open gate conformation that we see in PA26-bound yeast proteasome (Figure 8). Groll et al. (2003) discuss the ordering of the N-terminal residues as being relevant for proteasome assembly. We do not favor this proposal, however, because all of our mutant proteasomes, including those for which residues 2–12 are deleted, appear to assemble like wild type, as judged by sizing chromatography and peptidase activity (data not shown). Furthermore, a recent investigation reported that the T.acidophilum proteasome for which residues 2–12 were deleted appears identical to wild type by electron microscopy, except for the removal of mass from the pore region (Benaroudj et al., 2003). Nevertheless, the demonstration that archaeal proteasome α-subunits can adopt the ordered open conformation in at least some crystallographic environments is consistent with our suggestion that this conformation is important for efficient entry of protein substrates.

Fig. 8. Superposition of yeast 20S proteasome open conformation with A.fulgidus α-subunits (Groll et al., 2003). (A) Same as Figure 4B, with superposition of A.fulgidus α-subunits in blue. (B) Same as Figure 4C, with A.fulgidus α-subunits. The side chain for A.fulgidus Arg10 is shown. It forms a hydrogen bonding interaction with the main chain oxygen atom of residue 7.
The structure of A.fulgidus α-subunits (Groll et al., 2003) also reveals that Arg10 makes a hydrogen bonding interaction with the main chain oxygen atom of residue 7 (Figure 8). This explains why the Arg10Ala mutation shows partially impaired activity (Figure 6), because although Arg10 appears less important than the four principal residues, it is expected to stabilize the open conformation of archaeal proteasomes to some extent. The equivalent residue is an arginine in only three of the seven yeast proteasome α-subunits, and these side chains move upon binding PA26 to form similar interactions to those seen in the A.fulgidus α-subunit structure, suggesting that these residues may also contribute somewhat to gate opening of eukaryotic proteasomes. The net effect that these arginine side chains have on gate opening in eukaryotic proteasomes is unclear, however, since some of these residues may also be important for formation of the closed conformation (Groll et al., 2000).
The open conformation of 26S proteasomes?
Although PAN might not bind directly to the archaeal 20S proteasome, stable binding of the 19S activator is clearly required for gate opening of the eukaryotic 20S component of the 26S proteasome. Unlike PA26, the 19S activator is asymmetric, with a proteasome-binding surface that is probably comprised of six different ATPase subunits that make distinct contributions to activation (Glickman et al., 1998; Rubin et al., 1998; Köhler et al., 2001; Lam et al., 2002). Despite this asymmetry, we speculate that the open gate induced by the 19S activator may adopt the same symmetric conformation as seen in the complex with PA26. This could occur, in principle, because α-subunits are packed intimately against each other and a conformational change induced in one subunit could therefore propagate through all subunits in a cooperative transition. Thus, provided an asymmetric activator, such as the 19S activator, repositioned one α-subunit and did not restrict movement of the other subunits, a symmetric open conformation of the 20S proteasome could be induced.
Deletion of ∼10 residues from one or two α-subunit N-termini does not have a debilitating effect upon proteolysis by the yeast 26S proteasome (Groll et al., 2000; Köhler et al., 2001). This observation merits some discussion, since these deletions are expected to destabilize the open conformation to some extent. As expected from the crystal structure of unliganded proteasomes (Groll et al., 1997; Unno et al., 2002), the α3 deletion, in particular, greatly destabilizes the closed conformation (Groll et al., 2000). In comparison, the deletions are expected to destabilize the open conformation to a far lesser degree. Whereas α3 residues make many buried interactions in the closed conformation and are required for ordering of neighboring subunits, these residues lie along a solvent-exposed surface in the open conformation, make fewer interactions, and their absence is therefore not expected to prevent neighboring subunits from adopting the open conformation.
The observation that the α3 deletion greatly destabilizes the closed conformation, but results in disordering of the gate residues rather than formation of the open conformation (Groll et al., 2000), supports our conclusion that repositioning of the Pro17 reverse turns is required to allow packing interactions of the cluster residues. This rationalizes why PA26, and we suspect 19S, activates proteasomes by repositioning the Pro17 turn, since this both accomplishes destabilization of the closed conformation and allows formation of the open conformation. It is also important to note that proteasomes with some subunits lacking the first ∼12 residues are expected to retain a cooperative closed/open transition, since intersubunit communications appear to depend primarily upon interactions between more C-terminal residues. Finally, the ability of the open conformation to accommodate defective or absent clusters is supported by the observation that eukaryotic proteasomes have a non-canonical cluster.
In summary, there are several factors that cause us to suggest that the open conformation of the yeast 20S proteasomes seen in complex with PA26 might also be formed in other activated proteasomes, including: (i) the open conformation of the PA26 complex is stabilized by interactions between residues that are invariant from archaea to human, including species that appear to lack an 11S (PA26) activator; (ii) whereas some of these invariant residues stabilize the closed conformation, others lack an obvious alternative explanation for their conservation; (iii) archaeal proteasome α-subunits adopt the open conformation in at least one crystalline environment; (iv) the invariant residues are important for proteolysis by an archaeal proteasome, even though they do not appear to mediate interactions with the PAN activator; and (v) the lack of proteolytic activity by an archaeal proteasome that includes glycine residues in place of Tyr8 and Asp9 suggests that flexibility is not an important factor for entry of protein substrates. Although these considerations cause us to suggest that the same ordered, open conformation will be induced by the 19S activator, this proposal remains speculative, and further structural studies will be required to determine the mechanism of proteolysis by 26S proteasomes.
Materials and methods
Crystallographic analysis
The current refinement started from the previously reported 3.2 Å PA26–20S proteasome crystal structure (Whitby et al., 2000), which contains a full 1.1 MDa PA26:20S:PA26 complex in the asymmetric unit. The same data were used in the current analysis. As before, map and refinement calculations benefited from 2-fold non-crystallographic symmetry (NCS) averaging over the 20S proteasome model and 14-fold averaging over PA26. The key difference in the current analysis is that the mask for application of the 7-fold PA26 symmetry in map calculations was extended to cover regions of the 20S proteasome α-subunits. This resulted in well-defined electron density for the α-subunit N-terminal sequences, including Tyr8 and Asp9. Averaging was performed with AVE (http://alpha2.bmc.uu.se/~gerard/manuals/) and model building was performed using O (Jones et al., 1991). The rebuilt model was refined using CNS (Brünger et al., 1998) with NCS restraints additionally applied to equivalent residues of the α-subunit N-terminal residues. Symmetry-averaged and unaveraged simulated annealing omit maps confirmed the correctness of the model and were used for subsequent rebuilding. Improved Fourier maps also allowed correction of a four-residue misalignment in the first helix of PA26, inclusion of more residues at the PA26 N-terminus, rebuilding of the loop connecting PA26 helices 1 and 2, and other relatively minor improvements throughout the structure.
Cloning of proteasome mutants
Bicistronic expression constructs for α-subunit and C-terminally His6-tagged β-subunit of the T.acidophilum 20S proteasome [wild-type and Δ(2–12)] were gifts from Drs Erika Seemüller and Peter Zwickl of the Max-Planck-Institute for Biochemistry, Martinsried, Germany. The other 12 mutant proteasomes were generated following Stratagene’s QuikChange protocol with the minor exception that RbCl-competent DH-5α cells were used for transformations instead of XL1-Blue supercompetent cells.
Protein purification
Recombinant T.acidophilum proteasomes were expressed at 37°C in Escherichia coli BL21 (DE3) cells essentially as described (Seemüller et al., 1995). Cells were induced at an OD600 of 0.7–0.9 with 1 mM isopropyl-1-thio-β-d-galactopyranoside (IPTG), and harvested 8–12 h later. Cell pellets were resuspended in lysis buffer (50 mM NaPi pH 8.0, 300 mM NaCl) and lysed by French press and sonication. The lysate was cleared by centrifugation at 26 000 g for 10 min and heat-precipitated by addition of three volumes of boiling lysis buffer. The lysate was kept at 80°C for 15 min, rapidly cooled on ice and centrifuged at 26 000 g for 25 min. The supernatant was loaded onto an Ni-NTA column (Qiagen), washed with 7% elution buffer (50 mM NaPi pH 5.5, 300 mM NaCl, 500 mM imidazole), and eluted with 100% elution buffer. Fractions active against Suc-LLVY-AMC (Seemüller et al., 1995) were pooled and dialyzed against S400 high salt buffer (50 mM Tris pH 7.5, 500 mM NaCl, 1 mM EDTA). After concentration, the sample was run on a Sephacryl 26/60 S400 column (Amersham Pharmacia Biotech) equilibrated in S400 high salt buffer. The preparations were concentrated to 0.3 mg/ml and were stable for several months when stored at 4°C.
The expression plasmid for Met74Ala M.jannaschii PAN was a gift from Dr Peter Zwickl, and is essentially identical to that described (Zwickl et al., 1999), except that it lacks the N-terminal His6 tag of the published construct. Cells were grown as described (Zwickl et al., 1999). Cell pellets were resuspended in Q running buffer (20 mM Tris pH 8.0, 1 mM EDTA) and lysed and heat-precipitated as described above for the 20S proteasomes. The supernatant was loaded onto a HiLoad Q column equilibrated in Q running buffer and eluted with a linear 0–500 mM NaCl gradient. Pooled fractions were dialyzed against HyAp running buffer (10 mM KPi pH 7.5), diluted 3-fold, loaded onto a hydroxyapatite column and eluted with a linear 10–100 mM KPi gradient in HyAp running buffer. Pooled fractions were dialyzed against S400 high salt buffer (50 mM Tris pH 7.5, 500 mM NaCl, 1 mM EDTA), concentrated, and run on a Sephacryl S400 column. Pure PAN was concentrated to 0.3 mg/ml and stored at 4°C, where it was stable for several months.
GFP degradation assay
The PAN-dependent His6-GFP–ssrA substrate was a gift from Drs Ami Navon and Alfred Goldberg of Harvard Medical School. The assay was done essentially as described (Navon and Goldberg, 2001). Concentrations of the various proteasome solutions were confirmed by SDS–PAGE. Tubes were pre-incubated for at least 2 h with a 5 mg/ml solution of bovine serum albumin that was discarded immediately prior to setting up the reaction. The total reaction volume was 0.2 ml and contained GFP substrate (450 nM), proteasome (20 nM), PAN (80 nM, assuming a homohexamer) and ATP (5 mM). Buffer, proteasome and PAN were heated to 57°C, and the reaction was started by the addition of GFP (4 µl) and ATP (10 µl) stock solutions. At the indicated time points, aliquots were quenched by pipetting into SDS loading dye and stored on ice. Samples were separated on SDS–polyacrylamide gels, blotted to Sequi-blot PVDF membrane (Bio-Rad) and incubated with a horseradish peroxidase-conjugated anti-his tag antibody (Qiagen). After development with ECL solution (Amersham Pharmacia), bands were visualized on film (Kodak X-OMAT) and quantified using NIH ImageJ (http://rsb.info.nih.gov/ij/).
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We thank Peter Zwickl and Erika Seemüller for expression constructs, Ami Navon and Alfred Goldberg for the GFP substrate and advice on the activity assay, Eugene Masters and other members of the Hill lab for valuable discussions, and Carlos Gorbea and Marty Rechsteiner for critical comments on the manuscript. Supported by a University of Utah Graduate Research Fellowship to A.F. and NIH RO1-GM59135. The protein–DNA core facility at the University of Utah receives support from the National Cancer Institute (5P30 CA42014).
References
- Benaroudj N., Zwickl,P., Seemüller,E., Baumeister,W. and Goldberg,A.L. (2003) ATP hydrolysis by the proteasome regulatory complex PAN serves multiple functions in protein degradation. Mol. Cell, 11, 69–78. [DOI] [PubMed] [Google Scholar]
- Bochtler M., Ditzel,L., Groll,M., Hartmann,C. and Huber,R. (1999) The proteasome. Annu. Rev. Biophys. Struct., 28, 295–317. [DOI] [PubMed] [Google Scholar]
- Brünger A.T. et al. (1998) Crystallography and NMR system: a new software system for macromolecular structure determination. Acta Crystallogr. D, 54, 905–921. [DOI] [PubMed] [Google Scholar]
- Glickman M.H., Rubin,D.M., Fried,V.A. and Finley,D. (1998) A regulatory particle of the Saccharomyces cerevisiae proteasome. Mol. Cell. Biol., 18, 3149–3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groll M., Ditzel,L., Löwe,J., Stock,D., Bochtler,M., Bartunik,H.D. and Huber,R. (1997) Structure of 20S proteasome from yeast at 2.4 Å resolution. Nature, 386, 463–471. [DOI] [PubMed] [Google Scholar]
- Groll M., Bajorek,M., Köhler,A., Moroder,L., Rubin,D.M., Huber,R., Glickman,M.H. and Finley,D. (2000) A gated channel into the proteasome core particle. Nat. Struct. Biol., 7, 1062–1067. [DOI] [PubMed] [Google Scholar]
- Groll M., Brandstetter,H., Bartunik,H., Bourenkow,G. and Huber,R. (2003) Investigations on the maturation and regulation of archaebacterial proteasomes. J. Mol. Biol., 327, 75–83. [DOI] [PubMed] [Google Scholar]
- Hershko A. and Ciechanover,A. (1998) The ubiquitin system. Annu. Rev. Biochem., 67, 425–479. [DOI] [PubMed] [Google Scholar]
- Hill C.P., Masters,E.I. and Whitby,F.G. (2002) The 11S regulators of 20S proteasome activity. Curr. Top. Microbiol. Immunol., 268, 73–89. [DOI] [PubMed] [Google Scholar]
- Jones T.A., Zou,J.-Y., Cowan,S.W. and Kjeldgaard,M. (1991) Improved methods for building protein models in electron density maps and location of errors in these models. Acta Crystallogr. A, 47, 110–119. [DOI] [PubMed] [Google Scholar]
- Knowlton J.R., Johnston,S.C., Whitby,F.G., Realini,C.R., Zhang,Z., Rechsteiner,M.C. and Hill,C.P. (1997) Structure of the proteasome activator REGα (PA28α). Nature, 390, 639–643. [DOI] [PubMed] [Google Scholar]
- Köhler A., Cascio,P., Leggett,D.S., Woo,K.M., Goldberg,A.L. and Finley,D. (2001) The axial channel of the proteasome core particle is gated by the Rpt2 ATPase and controls both substrate entry and product release. Mol. Cell, 7, 1143–1152. [DOI] [PubMed] [Google Scholar]
- Lam Y.A., Lawson,T.G., Velayutham,M., Zweier,J.L. and Pickart,C.M. (2002) A proteasomal ATPase subunit recognizes the polyubiquitin degradation signal. Nature, 416, 763–767. [DOI] [PubMed] [Google Scholar]
- Li J., Gao,X., Joss,L. and Rechsteiner,M. (2000) The proteasome activator 11S REG or PA28: chimeras implicate carboxyl-terminal sequences in oligomerization and proteasome binding but not in the activation of specific proteasome catalytic subunits. J. Mol. Biol., 299, 641–654. [DOI] [PubMed] [Google Scholar]
- Löwe J., Stock,D., Jap,B., Zwickl,P., Baumeister,W. and Huber,R. (1995) Crystal structure of the 20S proteasome from the archaeon T.acidophilum at 3.4 Å resolution. Science, 268, 533–539. [DOI] [PubMed] [Google Scholar]
- Mykles D.L. (1996) Differential effects of bovine PA28 on six peptidase activities of the lobster muscle proteasome (multicatalytic proteinase). Arch. Biochem. Biophys., 325, 77–81. [DOI] [PubMed] [Google Scholar]
- Navon A. and Goldberg,A.L. (2001) Proteins are unfolded on the surface of the ATPase ring before transport into the proteasome. Mol. Cell, 8, 1339–1349. [DOI] [PubMed] [Google Scholar]
- Rock K.L., Gramm,C., Rothstein,L., Clark,K., Stein,R., Dick,L., Hwang,D. and Goldberg,A.L. (1994) Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell, 78, 761–771. [DOI] [PubMed] [Google Scholar]
- Rubin D.M., Glickman,M.H., Larsen,C.N., Dhruvakumar,S. and Finley,D. (1998) Active site mutants in the six regulatory particle ATPases reveal multiple roles for ATP in the proteasome. EMBO J., 17, 4909–4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seemüller E., Lupas,A., Stock,D., Löwe,J., Huber,R. and Baumeister,W. (1995) Proteasome from Thermoplasma acidophilum: a threonine protease. Science, 268, 579–582. [DOI] [PubMed] [Google Scholar]
- Song X., von Kampen,J., Slaughter,C.A. and DeMartino,G.N. (1997) Relative functions of the α and β subunits of the proteasome activator, PA28. J. Biol. Chem., 272, 27994–28000. [DOI] [PubMed] [Google Scholar]
- Unno M., Mizushima,T., Morimoto,Y., Tomisugi,Y., Tanaka,K., Yasuoka,N. and Tsukihara,T. (2002) The structure of the mammalian 20S proteasome at 2.75 Å resolution. Structure, 10, 609–618. [DOI] [PubMed] [Google Scholar]
- Voges D., Zwickl,P. and Baumeister,W. (1999) The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu. Rev. Biochem., 1015–1068. [DOI] [PubMed] [Google Scholar]
- Wenzel T. and Baumeister,W. (1995) Conformational constraints in protein degradation by the 20S proteasome. Nat. Struct. Biol., 2, 199–204. [DOI] [PubMed] [Google Scholar]
- Whitby F.G., Masters,E.I., Kramer,L., Knowlton,J.R., Yao,Y., Wang,C.C. and Hill,C.P. (2000) Structural basis for the activation of 20S proteasomes by 11S regulators. Nature, 408, 115–120. [DOI] [PubMed] [Google Scholar]
- Yao Y., Huang,L., Krutchinsky,A., Wong,M.L., Standing,K.G., Burlingame,A.L. and Wang,C.C. (1999) Structural and functional characterization of the proteasome-activating protein PA26 from Trypanosoma brucei. J. Biol. Chem., 274, 33921–33930. [DOI] [PubMed] [Google Scholar]
- Zhang Z., Clawson,A., Realini,C., Jensen,C.C., Knowlton,J.R., Hill,C.P. and Rechsteiner,M. (1998) Identification of an activation region in the proteasome activator REGα. Proc. Natl Acad. Sci. USA, 95, 2807–2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwickl P. (2002) The 20S proteasome. Curr. Top. Microbiol. Immunol., 268, 23–41. [DOI] [PubMed] [Google Scholar]
- Zwickl P., Ng,D., Woo,K.M., Klenk,H.P. and Goldberg,A.L. (1999) An archaebacterial ATPase, homologous to ATPases in the eukaryotic 26S proteasome, activates protein breakdown by 20S proteasomes. J. Biol. Chem., 274, 26008–26014. [DOI] [PubMed] [Google Scholar]
- Zwickl P., Baumeister,W. and Steven,A. (2000) Dis-assembly lines: the proteasome and related ATPase-assisted proteases. Curr. Opin. Struct. Biol., 10, 242–250. [DOI] [PubMed] [Google Scholar]
- Zwickl P., Seemüller,E., Kapelari,B. and Baumeister,W. (2001) The proteasome: a supramolecular assembly designed for controlled proteolysis. Adv. Protein Chem., 59, 187–222. [DOI] [PubMed] [Google Scholar]